
Current and Future Treatments for Classic Galactosemia

1
Department of Pediatrics, Maastricht University Medical Centre, 6229 HX Maastricht, The Netherlands
2
Department of Clinical Genetics, Maastricht University Medical Centre+, 6229 HX Maastricht, The Netherlands
3
GROW-School for Oncology and Developmental Biology, Maastricht University, 6229 HX Maastricht, The Netherlands
*
Author to whom correspondence should be addressed.
J. Pers. Med. 2021, 11(2), 75; https://doi.org/10.3390/jpm11020075
Submission received: 28 December 2020
/
Revised: 23 January 2021
/
Accepted: 24 January 2021
/
Published: 28 January 2021
(This article belongs to the Special Issue Genotype–Phenotype Relationships, Molecular Mechanisms and Personalized Therapy of Metabolic Diseases)
{kind=link}
Abstract
:Type I (classic) galactosemia, galactose 1-phosphate uridylyltransferase (GALT)-deficiency is a hereditary disorder of galactose metabolism. The current therapeutic standard of care, a galactose-restricted diet, is effective in treating neonatal complications but is inadequate in preventing burdensome complications. The development of several animal models of classic galactosemia that (partly) mimic the biochemical and clinical phenotypes and the resolution of the crystal structure of GALT have provided important insights; however, precise pathophysiology remains to be elucidated. Novel therapeutic approaches currently being explored focus on several of the pathogenic factors that have been described, aiming to (i) restore GALT activity, (ii) influence the cascade of events and (iii) address the clinical picture. This review attempts to provide an overview on the latest advancements in therapy approaches.
1. Introduction
The galactosemias are a rare group of hereditary disorders of galactose metabolism. To date, four types have been described, each affecting a different step in the main route of galactose disposal: type I, galactose 1-phosphate uridylyltransferase- (GALT, EC 2.7.7.12); type II, galactokinase- (GALK1, EC 2.7.1.6); type III UDP-galactose 4 epimerase- (GALE, EC 5.1.3.2) and the recently described type IV galactose mutarotase- (GALM, EC 5.1.3.3) deficiency, Figure 1 [1,2,3,4,5,6,7,8]. We focus on type I galactosemia and review the latest advancements in therapy approaches.
In type I, classic galactosemia (OMIM 230400), the conversion of -d-galactose-1-phosphate (Gal-1-P) to -d-glucose-1-phosphate (Glc-1-P) and uridine diphosphate-galactose (UDP-Gal) is hampered by the severe GALT deficiency. It is an autosomal recessive disorder with a prevalence of 1:16,000 to 1:50,000 live births in Western countries [9,10,11,12,13,14]. The current cornerstone of treatment, a galactose-restricted diet, resolves the neonatal syndrome but fails to prevent burdensome chronic impairments. Most affected patients develop brain impairments (85.0%), primary ovarian insufficiency (79.9%) and a diminished bone mineral density (26.5%) (data based on a large cohort from the Galactosemia network registry (Registry | GalNet (galactosemianetwork.org)). Additionally, data showed that a more favorable outcome among patients is achieved by onset of the diet in the first week of life and detection by newborn screening (NBS). Furthermore, a less strict diet (lactose free without further restrictions) was associated with less neurological complications [6,15]. There is a need for a more adequate treatment to prevent complications.
Classic galactosemia is caused by pathogenic genetic variants in the GALT gene, which is located on chromosome 9, leading to a severely diminished enzymatic activity [16]. At present, over 300 genetic variants in the GALT gene have been described, the most frequent disease-causing variant among people of European ancestry being the NM_000155.4: c.563A>G genetic variant (p.Gln188Arg) [17]. In addition to classic galactosemia, clinical variants with a higher residual activity are being detected through NBS in which a milder phenotype is anticipated. Structural and functional studies show that in the majority of genetic variants, the dysfunction of the enzyme is due to changes in protein stability and folding [4,5,7,18]. The most frequent genetic variants are NM_000155.4: c.563A>G (p.Gln188Arg); NM_000155.4: c.404C>T (p.Ser135Leu); and NM_000155.4: c.855G>T (p.Lys285Asn) [4,5,7,18].
The pathogenic mechanisms underlying the acute and long-term organ-specific complications of classic galactosemia are complex and remain to be fully elucidated. Currently described contributing factors include accumulation of galactose metabolites (galactitol, galactonate and Gal-1-P) [2,19,20,21,22,23,24,25,26,27,28,29,30], uridine diphosphate (UDP)-hexose alterations and impaired glycosylation [21,25,31,32,33,34,35,36], endoplasmic reticulum (ER) stress with subsequent unfolded protein response (UPR), induction and alteration of signaling pathways [2,3,4,37,38,39,40,41,42], and oxidative stress [4,29,43,44] (Figure 1).
In recent years, a number of animal models (mouse, fruit fly, zebrafish, rat) [24,29,45,46] of classic galactosemia have been developed that mimic, at least partly, the biochemical and clinical phenotypes and complement the cellular models, allowing us to advance our comprehension of the complex playing field of the metabolism of galactose. We have learned that there is different expression of the Leloir pathway components and alternative galactose disposal routes in the different tissues, that combined with specific tissue demands, epigenetic and environmental factors, urge to revisit our understanding. GALT activity levels in affected organs (brain, ovaries) do not seem to differ from the activity levels in organs not affected in classic galactosemia and distinct organ-specific levels of GALT activity have been found [19]. This is reflected in the tissue related differences in relative levels of galactose metabolites (galactitol, galactose and Gal-1-P) in the rat galactosemia model [45], as well as in the nucleotide sugar profiles variation through development and in the different tissues in the zebrafish galactosemia model [31]. Besides, activity differs across different stages in development [19].
The recent elucidation of the crystal structure of human GALT, with a resolution of 1.9 Å, has provided a framework for understanding the molecular consequences of genetic variants and can greatly contribute to the design of therapeutic compounds [5,7]. Novel therapeutic approaches for classic galactosemia currently (defined as: in the last 5 years) being explored aim either to (i) restore GALT activity, (ii) influence the cascade of events or (iii) address the clinical picture. For these strategies to be optimal, characterization of the window of opportunity for treatment is of outmost importance; e.g., neonatal and early infancy or through adolescence or lifelong treatment.
2. Potential Therapies
2.1. Restoring GALT Activity
Directly restoring GALT activity is the aim of multiple techniques currently being investigated: (i) GALT gene therapy, (ii) mRNA therapy and (iii) pharmacological chaperones. Notably, restoring GALT activity up to 10–15% is likely to rescue the phenotype in classic galactosemia. This is suggested by in the so-called “biochemical variant galactosemia” where GALT activity levels between 10% and 15% do not result in clinical disease [47,48].
2.1.1. Gene Therapy
Gene therapy and gene correction techniques aim to permanently compensate for the primary genetic defect [49,50]. In conventional gene therapy, this is achieved by providing patients with the correct coding DNA (cDNA) sequence of the defective gene leading to the expression of normal protein [50,51]. Delivery is accomplished by the use of vectors, either viral or non-viral, containing the genetic sequence of the protein [49,52,53]. In recent decades, significant advances have been made and gene therapy is showing great promise in animal models and clinical trials of inborn errors of metabolism [51,52,53].
In classic galactosemia, viral mediated in vivo gene therapy is currently being investigated using recombinant adeno-associated virus (AAV) vectors [54], which are emerging in the field of gene therapy as very promising [51,52,53]. Rasmussen et al. recently showed, in a pilot study in a galactosemia rat model (n = 7), that neonatal human GALT gene therapy with a AAV9 vector via tail vein injection led to GALT restoration in both liver and brain [54]. Relative to wildtype activity, GALT activity levels of 64–595% in liver and 2–42% in brain were found without significant adverse effects. Furthermore galactose metabolites in blood, liver and brain were lowered and a positive effect on cataract was observed [54]. Lai and Loiler expanded the serotypes of the AAV vectors to include AAV8 and AAVrh10, and showed that intravenous administration of AAV8-hGALT and AAVrh10-hGALT resulted in GALT (hGALT) protein expression and decreased levels of Gal-1-P in liver, brain, ovaries and red blood cells of Galt knockout mice [55]. In line with this data, Brophy et al. showed that a recombinant AAV vector encoding GALT was able to rescue GALT activity and reduce galactose-induced ER stress in fibroblasts from galactosemic patients [56].
Important future challenges will be focused at safety and efficacy. Safety concerns encompass elicited immune responses by the viral vector capsid proteins and the transgene product, and possible off target effects leading to insertional mutagenesis as well as genomic instability [49,52]. With regard to efficacy, Rasmussen et al. reported differences between liver and brain transgene activity. The authors hypothesized that these differences might stem from preferential perfusion of the liver and limited permeability of the blood–brain barrier and might reflect the differences in activity level of the organs [54]. These findings are consistent with prior reports on AAV9 transgene delivery by intravenous injection [57,58]. The reported studies illustrate the potential of gene therapy for the treatment of GALT-deficient galactosemia.
2.1.2. mRNA Therapy
Exogenous, intracellular delivery of mRNA is emerging as a new class of medicine [59,60,61,62]. Recent advances in mRNA technology have led to improvements in stability and translation by codon optimization and encapsulation within biodegradable delivery systems, preserving mRNA integrity and allowing targeted intracellular delivery [59,60,61,62,63,64,65,66,67,68,69,70]. The required repeated dosing of this therapeutic modality tends to raise concerns about immunogenicity and therapy burden for the patients. As with gene therapy targeting, potential long-term adverse effects therapeutic index and dosing interval of mRNA remain important challenges for the future [60,68,70]. The transient nature of the mRNA requiring repeated dosing to remain effective also bears an advantage, by allowing individual dose titration and cessation of therapy if necessary [60,68,70].
Several preclinical studies evaluating the potential of systemically administered mRNA in metabolic disorders have shown that mRNA provides transient, half-life dependent protein expression, while bearing a low risk of insertional mutagenesis and maintaining dose-responsiveness [67,68,69,71,72,73,74,75]. This therapeutic approach is being explored in classic galactosemia using lipid nanoparticles as delivery system. Balakrishnan et al. recently showed in adult Galt knockout mice that systemic administration of LNP-packaged mouse and hGALT mRNA resulted in hepatic expression of active, long-lasting GALT enzyme and effectively eliminated galactose 1-phosphate in liver and other peripheral tissues [68]. Using the galt knockout zebrafish model, Haskovic et al. also showed successful delivery and translation of functional hGALT after single dose injections with LNP-packaged hGALT mRNA (n = 3) delivered at day 0 and measured on day 5 post fertilization (dpf) [76]. The mRNA approach holds great promise as a potential therapy for classic galactosemia.
2.1.3. Pharmacological Chaperones
Pharmacological chaperones are small molecules that bind specifically to their targets, rescuing specific variant proteins by facilitating intracellular folding and trafficking, increasing cellular stability and activity, and/or preventing premature degradation [4,21]. Pharmacological chaperones present advantageous features: (i) low synthesis cost; (ii) oral availability with broad biodistribution; (iii) low molecular weight that can potentially, allow them to cross the blood–brain barrier (BBB) [77,78,79]. The high occurrence of missense mutations (>60%) [17] and experimental evidence of misfolding and decreased stability of GALT variants makes classic galactosemia highly amenable to this therapeutic approach [18,77,78,79,80,81].
Arginine supplementation, known for its effect as an aggregation inhibitor [82] has been studied as a potential therapeutic approach for classic galactosemia. Despite promising results of this chemical chaperone in a bacterial model [83], in a pilot study with 4 patients carrying the NM_000155.4: c.563A>G (p.Gln188Arg) mutation, arginine did not lead to improved GALT stability nor GALT activity levels as there was no improvement in galactose oxidation capacity [4,84].
Despite being a promising therapeutic approach, its specificity for genetic variants and compound heterozygosity in patient profiles as well as the existence of a broad range of genetic variants complicate the development of pharmacological chaperones as a therapy for all galactosemia patients.
2.2. Influence the Cascade of Events
These therapies focus on one of the downstream effects, and as such are less likely to provide treatment for the complete clinical picture of the disease. However, every approach can be helpful by tackling one of the aspects involved in pathophysiology and the combination of treatments could also be an option. The main approaches currently being explored to influence downstream phenomena are inhibitors of galactokinase 1 (GALK1)- and aldose reductase (AR), and endoplasmic reticulum (ER) stress reducers. Oxidative stress reducers have also been proposed in the past, but have not been studied in the last 5 years [43,44,85].
2.2.1. Galactokinase 1 (GALK1) Inhibitors
GALK1 inhibitors were proposed as treatment strategy many years ago [86], and have been thoroughly investigated over the years. The strategy aims to reduce the accumulation of Gal-1-P in GALT deficiency, as Gal-1-P is considered a major key player in the pathogenic mechanism in classic galactosemia [4,12,21]. The milder clinical picture of type II (GALK1 deficiency) galactosemia supported this view although standardized follow-up data are currently lacking [87,88]. Inhibition of GALK1 appears to be an attractive therapeutic target, since GALK1, as member of the GHMP kinase family, is highly substrate specific and no other undesired inhibitions are to be expected [89,90].
High-throughput screenings (HTS) have identified candidate ligands with affinity for GALK1 [12,91,92,93,94]. Promising compounds have been identified and studied to optimize the efficacy and improve the potency. Several compounds have also been tested in cell culture models of patients’ fibroblasts where their efficacy to reduce the biochemical phenotype by lowering Gal-1-P levels has been shown [89,90,91,92,94,95]. The effect of a possible increase in galactitol and galactonate levels remains to be investigated. More research with these compounds beyond cellular models is warranted.
2.2.2. Aldose Reductase (AR) Inhibitors
Aldose reductase (AR) inhibitors target the NADPH-dependent aldose reductase conversion of [2,21,61,96] galactose to galactitol, which cannot be transported across the cell membrane due to its poor diffusivity and cannot be further metabolized, leading to accumulation in cells. This leads to an osmotic phenomenon with subsequent cell swelling and ultimately apoptosis [97,98]. The high expression of aldose reductase in the epithelial cells of the lens leads to the formation of galactosemic cataracts [88,99]. Galactitol involvement in cognitive and neurological symptoms has been suggested [61,100]. Pseudotumor cerebri, or elevated intracranial pressure, has also been reported and is postulated to be the result of accumulation of galactitol in the brain cells leading to cerebral edema [101,102].
AR inhibitors have shown to decrease galactitol levels and prevent cataract formation in hypergalactosemia animal models (rats and dogs) [103,104]. By blocking the galactitol accumulation in rats, the increased osmolarity, UPR and ER stress as well as cell death were prevented [105]. In a hypergalactosemia rat model, Mizisin et al. have, furthermore, shown increased sodium concentrations in endoneurinal fluid in rats that are reversible by AR inhibitors indicating a role for galactitol [106,107,108]. Since the exact role of galactitol in the pathophysiological mechanism of galactosemia remains unknown, other than for cataract, decreasing galactitol levels in other organs might show additional beneficial effects. However, caution needs to be taken since the effect of blocking the polyol pathway that converts galactose into the polyol (sugar alcohol) galactitol, is unknown. An increase in Gal-1-P and galactonate might occur, of which the effects remain to be investigated. AR inhibitors are currently being investigated by the biopharmaceutical company Applied Therapeutics [109].
2.2.3. Endoplasmic Reticulum (ER) Stress Reducers
ER stress has been shown to be involved in the pathology of classic galactosemia, leading to altered signaling pathways, such as the PI3K/Akt pathway [38,41,42]. Balakrishnan et al. have shown that the downregulation of the PI3K/Akt signaling pathway plays an important role in subfertility and cerebellar ataxia in galactosemic mice (n = 6) [38]. This indicates that reduced ER stress might be of therapeutic interest for the brain and ovary, suggesting a possible role for eukaryotic initiation factor 2α (eIF2α) inhibitors that reverse the downregulated pathway in fertility preservation and neurological complications. Positive effects of the eIF2α inhibitor salubrinal have been reported in a mouse model of classic galactosemia without detectable adverse effects [39,110]. Moreover, a protective effect on primordial follicle loss, and an increase in fertility, was found [39]. Although salubrinal is an experimental compound, these findings indicate that the downregulation of the PI3K/Akt pathway is a valid potential treatment target in classic galactosemia.
2.3. Address the Clinical Picture
2.3.1. Neonatal Period
A galactose-restricted diet resolves the neonatal clinical picture [111]. Newborn screening (NBS) for galactosemia has been implemented in many countries [112,113,114,115,116], and NBS and introduction of diet in the first week of life have shown a beneficial effect [6]. Over the years the strictness of the diet has been questioned and the current recommendation is to eliminate sources of lactose and galactose from dairy products, but permit small amounts of galactose from non-milk sources [111].
2.3.2. Beyond the Neonatal Period
Approaches specifically aimed in tackling the clinical consequences of galactosemia beyond the neonatal period cover the many aspects of the plethora of signs and symptoms associated with GALT-deficiency. In the next paragraphs we touch upon several recent developments regarding (i) fertility preservation, and (ii) neurological and cognitive complications.
Fertility Preservation
Primary ovarian insufficiency (POI) with ovarian follicular depletion leading to subfertility is reported in at least 80% of female patients despite a galactose-restricted diet, representing a heavy burden for female patients [6,117,118,119,120,121]. The etiology of ovarian failure and timing of ovarian damage (pre- or postnatal) have not yet been resolved, although studies suggest that follicular depletion has an early initiation [117,120,122,123,124]. Ovarian imaging results show an early onset of ovarian failure (n = 14) [125]. Moreover, Mamsen et al. have shown normal follicle morphology and follicle densities in galactosemic girls under 5 years (n = 5) [122]. This is supported by the finding of low anti-Mullerian hormone (AMH) levels and low antral follicle counts relative to age-matched controls, suggesting that follicular dysfunction occurs early in life [126,127]. Although spontaneous conception despite POI occurs in classic galactosemia [125,128,129], the chances of pregnancy are severely reduced and fertility preservation options are important. Nowadays, two options are available, cryopreservation and oocyte donation.
The early occurrence of damage supports cryopreservation of ovarian tissue at a very young age as a treatment option [122,125]. Cryopreservation of ovarian tissue has been applied for several years to preserve fertility in patients with (mostly) malignant pathologies that undergo treatments with a detrimental effect on fertility. In recent years, the optimization of cryopreservation strategies and thawing/warming protocols has been achieved, expanding the opportunities for females with various pathologies [130,131]. In classic galactosemia, cryopreservation has to be performed at a very young age, because of the early ovarian damage. Even though this procedure is traditionally associated with a reduction in the ovarian reserve, the technology has improved tremendously over the years [131], and it is now associated with a complication rate of merely 0.2% and an increasing success rate [122,132].
Another approach is intrafamilial oocyte donation (mother-to-daughter or sister-to-sister). In the last five years, a group of experts provided recommendations required to optimally address this option taking into account the professionals as well as the patients and family members views. Topics recommended to be discussed are: family relations, medical impact, patient’s cognitive level, agreements to be made in advance and organization of counseling, disclosure to the child, and need for follow-up [133].
The broad phenotypic spectrum of cognitive and neurological impairments that these female patients might develop make decisions around these complex matters challenging.
Neurological and Cognitive Complications
Brain impairments in classic galactosemia, consisting of neurological, cognitive and behavioral complications, are common and are known to have a significant impact on quality of life and general performance [6,134,135,136,137,138,139,140]. Brain impairments have been reported in 85% of patients, including developmental and language delay, neurological complications, language and speech disorders as well as mental and behavioral problems [6]. Structural changes in white and grey matter and functional alterations have been reported [134,135,136,137,138,139].
In addition to the current battery of approaches that are used to help the patients that develop these complications [111], two interesting approaches are currently under study to positively influence language and motor function. One of them is transcranial Alternating Current Stimulation (tACS), a form of non-invasive brain stimulation (NIBS). The rationale behind tACS is to stimulate the naturally occurring rhythmic patterns of electrophysiological activity, also known as brain oscillations, which have been found to be different in classic galactosemia (manuscript in preparation), and modify the oscillations through the delivery of alternating electric currents to the scalp inducing long-term synaptic plasticity [141,142,143,144,145]. Functional networks rely on the synchronization of brain oscillations between the different components of the network, making it an appealing approach for restoring affected functional networks in classic galactosemia [142]. This approach is widely used in cognitive neuroscience, and proven to be efficient in Parkinson and dyslexia and is emerging within the field of psychiatry [141,142,144]. Whether there is proof of concept for this approach is currently under investigation.
The other interesting approach is the Babble Boot Camp (BBC), which is focused on a proactive speech and language intervention program [146]. Current guidelines recommend regular extensive screening for speech and language delay, if children do not meet appropriate levels of speech or language treatment is initiated preferably during the first year of life [111]. However, since speech and language can be regarded as later-developing skill, deficits and signs of delay will only become apparent after an age greater than 24 and 36 months, respectively [111,146]. This approach focuses on a preventive intervention program starting at 2 to 4 months of age and consisting of active parental involvement guided by an expert speech language pathologist. Although the results are preliminary and generalizations cannot be made, a trend to beneficial effects on speech and language development was shown [146].
3. Conclusions
In recent years, several therapeutic approaches intended to provide more adequate treatment for classic galactosemia and prevent burdensome long-term complications have been researched. These approaches aim to (i) restore GALT activity, (ii) influence the cascade of events and (iii) address the clinical picture. Gene therapy and mRNA therapy are emerging therapies within the field of medicine, which show great potential in restoring GALT activity levels in animal models. Pharmacological chaperones form an interesting therapeutic approach for rescuing GALT protein, although this approach is genetic variant-specific as opposed to gene therapy or mRNA therapy. Downstream phenomena that are currently being explored aim to reduce ER stress and inhibit GALK1 or AR. Therapeutic strategies specifically tackling the clinical consequences of classic galactosemia are also evolving. For optimal treatment, the window of opportunity is urged to be characterized, defining when treatment is best given for the different complications.
Author Contributions
Conceptualization B.D., A.I.C. and M.E.R.-G.; writing—original draft preparation B.D., A.I.C. and M.E.R.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAV | Adeno-associated virus |
| AR | Aldose reductase |
| BBB | Blood–brain barrier |
| BBC | Babble Boot Camp |
| Cas13 | CRISPR-associated protein |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DHEA | Dehydroepiandrosterone |
| Dpf | Days post fertilization |
| eIF2α | Eukaryotic initiation factor 2α |
| ER | Endoplasmic reticulum |
| Gal-1-P | Galactose-1-phosphate |
| GALE | UDP-galactose 4-epimerase |
| GALK1 | Galactokinase 1 |
| GALM | Galactose mutarotase |
| GalNet | Galactosemias Network |
| GALT | Galactose 1-phosphate uridylyltransferase |
| GDH | Galactose dehydrogenase |
| GHMP | Galacto-, homoserine-, mevalonate- and phosphomevalonate kinase |
| Glc-1-P | Glucose-1-phosphate |
| hGALT | human GALT |
| HTS | High throughput screening |
| LNP | Lipid nanoparticle |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NBS | Newborn screening |
| NIBS | Non-invasive brain stimulation |
| PI3K/Akt | Phosphoinositide 3-kinase/protein kinase B |
| POI | Primary ovarian insufficiency |
| ROS | Reactive oxygen species |
| tACS | Transcranial alternating current stimulation |
| UGP | UDP-glucose pyrophosphorylase |
| UDP | Uridine diphosphate |
| UDP-Gal | Uridine diphosphate-galactose |
| UDP-Glc | Uridine diphosphate-glucose |
| UDP-GalNAc | UDP-N-acetylgalactosamine |
| UDP-GlcNAc | UDP-N-acetylglucosamine |
| UPR | Unfolded protein response |
References
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef]
- Coelho, A.I.; Rubio-Gozalbo, M.E.; Vicente, J.B.; Rivera, I. Sweet and sour: An update on classic galactosemia. J. Inherit. Metab. Dis. 2017, 40, 325–342. [Google Scholar] [CrossRef] [Green Version]
- Demirbas, D.; Coelho, A.I.; Rubio-Gozalbo, M.E.; Berry, G.T. Hereditary galactosemia. Metab. Clin. Exp. 2018, 83, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Haskovic, M.; Coelho, A.I.; Bierau, J.; Vanoevelen, J.M.; Steinbusch, L.K.M.; Zimmermann, L.J.I.; Villamor-Martinez, E.; Berry, G.T.; Rubio-Gozalbo, M.E. Pathophysiology and targets for treatment in hereditary galactosemia: A systematic review of animal and cellular models. J. Inherit. Metab. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Kopec, J.; Pey, A.L.; Fitzpatrick, F.; Patel, D.; Chalk, R.; Shrestha, L.; Yue, W.W. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum. Mol. Genet. 2016, 25, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gozalbo, M.E.; Haskovic, M.; Bosch, A.M.; Burnyte, B.; Coelho, A.I.; Cassiman, D.; Couce, M.L.; Dawson, C.; Demirbas, D.; Derks, T.; et al. The natural history of classic galactosemia: Lessons from the GalNet registry. Orphanet J. Rare Dis. 2019, 14, 86. [Google Scholar] [CrossRef] [Green Version]
- Timson, D.J. The molecular basis of galactosemia—Past, present and future. Gene 2016, 589, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Wada, Y.; Kikuchi, A.; Arai-Ichinoi, N.; Sakamoto, O.; Takezawa, Y.; Iwasawa, S.; Niihori, T.; Nyuzuki, H.; Nakajima, Y.; Ogawa, E.; et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1286–1294. [Google Scholar] [CrossRef]
- Antshel, K.M.; Epstein, I.O.; Waisbren, S.E. Cognitive strengths and weaknesses in children and adolescents homozygous for the galactosemia Q188R mutation: A descriptive study. Neuropsychology 2004, 18, 658–664. [Google Scholar] [CrossRef]
- Bosch, A.M.; Grootenhuis, M.A.; Bakker, H.D.; Heijmans, H.S.; Wijburg, F.A.; Last, B.F. Living with classical galactosemia: Health-related quality of life consequences. Pediatrics 2004, 113, e423–e428. [Google Scholar] [CrossRef] [Green Version]
- Lambert, C.; Boneh, A. The impact of galactosaemia on quality of life—A pilot study. J. Inherit. Metab. Dis. 2004, 27, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Odejinmi, S.I.; Vankayalapati, H.; Wierenga, K.J.; Lai, K. Innovative therapy for Classic Galactosemia—Tale of two HTS. Mol. Genet. Metab. 2012, 105, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waisbren, S.E.; Albers, S.; Amato, S.; Ampola, M.; Brewster, T.G.; Demmer, L.; Eaton, R.B.; Greenstein, R.; Korson, M.; Larson, C.; et al. Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress. JAMA 2003, 290, 2564–2572. [Google Scholar] [CrossRef] [Green Version]
- Waisbren, S.E.; Rones, M.; Read, C.Y.; Marsden, D.; Levy, H.L. Brief report: Predictors of parenting stress among parents of children with biochemical genetic disorders. J. Pediatr. Psychol. 2004, 29, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gozalbo, M.E.; Bosch, A.M.; Burlina, A.; Berry, G.T.; Treacy, E.P. The galactosemia network (GalNet). J. Inherit. Metab. Dis. 2017, 40, 169–170. [Google Scholar] [CrossRef]
- Pasquali, M.; Yu, C.; Coffee, B. Laboratory diagnosis of galactosemia: A technical standard and guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Calderon, F.R.; Phansalkar, A.R.; Crockett, D.K.; Miller, M.; Mao, R. Mutation database for the galactose-1-phosphate uridyltransferase (GALT) gene. Hum. Mutat. 2007, 28, 939–943. [Google Scholar] [CrossRef]
- Coelho, A.I.; Trabuco, M.; Ramos, R.; Silva, M.J.; Tavares de Almeida, I.; Leandro, P.; Rivera, I.; Vicente, J.B. Functional and structural impact of the most prevalent missense mutations in classic galactosemia. Mol. Genet. Genom. Med. 2014, 2, 484–496. [Google Scholar] [CrossRef]
- Coelho, A.I.; Bierau, J.; Lindhout, M.; Achten, J.; Kramer, B.W.; Rubio-Gozalbo, M.E. Classic Galactosemia: Study on the Late Prenatal Development of GALT Specific Activity in a Sheep Model. Anat. Rec. (Hoboken) 2017, 300, 1570–1575. [Google Scholar] [CrossRef] [Green Version]
- Daenzer, J.M.; Jumbo-Lucioni, P.P.; Hopson, M.L.; Garza, K.R.; Ryan, E.L.; Fridovich-Keil, J.L. Acute and long-term outcomes in a Drosophila melanogaster model of classic galactosemia occur independently of galactose-1-phosphate accumulation. Dis. Model. Mech. 2016, 9, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- McCorvie, T.J.; Timson, D.J. Chapter 11—Galactosemia: Opportunities for novel therapies. In Protein Homeostasis Diseases; Pey, A.L., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 221–245. [Google Scholar] [CrossRef]
- Wehrli, S.L.; Berry, G.T.; Palmieri, M.; Mazur, A.; Elsas, L., III; Segal, S. Urinary galactonate in patients with galactosemia: Quantitation by nuclear magnetic resonance spectroscopy. Pediatr. Res. 1997, 42, 855–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, C.M.; De-Souza, E.A.; De-Queiroz, A.; Pimentel, F.S.A.; Silva, G.F.S.; Gomes, F.M.; Montero-Lomeli, M.; Masuda, C.A. The galactose-induced decrease in phosphate levels leads to toxicity in yeast models of galactosemia. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Kushner, R.F.; Ryan, E.L.; Sefton, J.M.; Sanders, R.D.; Lucioni, P.J.; Moberg, K.H.; Fridovich-Keil, J.L. A Drosophila melanogaster model of classic galactosemia. Dis. Model. Mech. 2010, 3, 618–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.; Langley, S.D.; Khwaja, F.W.; Schmitt, E.W.; Elsas, L.J. GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology 2003, 13, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ning, C.; Reynolds, R.; Chen, J.; Yager, C.; Berry, G.T.; Leslie, N.; Segal, S. Galactose metabolism in mice with galactose-1-phosphate uridyltransferase deficiency: Sucklings and 7-week-old animals fed a high-galactose diet. Mol. Genet. Metab. 2001, 72, 306–315. [Google Scholar] [CrossRef]
- Ross, K.L.; Davis, C.N.; Fridovich-Keil, J.L. Differential roles of the Leloir pathway enzymes and metabolites in defining galactose sensitivity in yeast. Mol. Genet. Metab. 2004, 83, 103–116. [Google Scholar] [CrossRef]
- Ryan, E.L.; DuBoff, B.; Feany, M.B.; Fridovich-Keil, J.L. Mediators of a long-term movement abnormality in a Drosophila melanogaster model of classic galactosemia. Dis. Model. Mech. 2012, 5, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Siddiqi, A.; Witt, B.; Yuzyuk, T.; Johnson, B.; Fraser, N.; Chen, W.; Rascon, R.; Yin, X.; Goli, H.; et al. Subfertility and growth restriction in a new galactose-1 phosphate uridylyltransferase (GALT)—Deficient mouse model. Eur. J. Hum. Genet. 2014, 22, 1172–1179. [Google Scholar] [CrossRef] [Green Version]
- Yager, C.; Ning, C.; Reynolds, R.; Leslie, N.; Segal, S. Galactitol and galactonate accumulation in heart and skeletal muscle of mice with deficiency of galactose-1-phosphate uridyltransferase. Mol. Genet. Metab. 2004, 81, 105–111. [Google Scholar] [CrossRef]
- Haskovic, M.; Coelho, A.I.; Lindhout, M.; Zijlstra, F.; Veizaj, R.; Vos, R.; Vanoevelen, J.M.; Bierau, J.; Lefeber, D.J.; Rubio-Gozalbo, M.E. Nucleotide sugar profiles throughout development in wildtype and galt knockout zebrafish. J. Inherit. Metab. Dis. 2020, 43, 994–1001. [Google Scholar] [CrossRef]
- Coss, K.P.; Treacy, E.P.; Cotter, E.J.; Knerr, I.; Murray, D.W.; Shin, Y.S.; Doran, P.P. Systemic gene dysregulation in classical Galactosaemia: Is there a central mechanism? Mol. Genet. Metab. 2014, 113, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Dobbie, J.A.; Holton, J.B.; Clamp, J.R. Defective galactosylation of proteins in cultured skin fibroblasts from galactosaemic patients. Ann. Clin. Biochem. 1990, 27 Pt 3, 274–275. [Google Scholar] [CrossRef]
- Ornstein, K.S.; McGuire, E.J.; Berry, G.T.; Roth, S.; Segal, S. Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose-1-phosphate uridyltransferase deficiency. Pediatr. Res. 1992, 31, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petry, K.; Greinix, H.T.; Nudelman, E.; Eisen, H.; Hakomori, S.; Levy, H.L.; Reichardt, J.K. Characterization of a novel biochemical abnormality in galactosemia: Deficiency of glycolipids containing galactose or N-acetylgalactosamine and accumulation of precursors in brain and lymphocytes. Biochem. Med. Metab. Biol. 1991, 46, 93–104. [Google Scholar] [CrossRef]
- Staubach, S.; Müller, S.; Pekmez, M.; Hanisch, F.G. Classical Galactosemia: Insight into Molecular Pathomechanisms by Differential Membrane Proteomics of Fibroblasts under Galactose Stress. J. Proteome Res. 2017, 16, 516–527. [Google Scholar] [CrossRef]
- Van Erven, B. Classic galactosemia: A zebrafish model and new clinical insights. Ph.D Thesis, Maastricht University, Maastricht, The Netherlands, March 2017. [Google Scholar]
- Balakrishnan, B.; Chen, W.; Tang, M.; Huang, X.; Cakici, D.D.; Siddiqi, A.; Berry, G.; Lai, K. Galactose-1 phosphate uridylyltransferase (GalT) gene: A novel positive regulator of the PI3K/Akt signaling pathway in mouse fibroblasts. Biochem. Biophys. Res. Commun. 2016, 470, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, B.; Nicholas, C.; Siddiqi, A.; Chen, W.; Bales, E.; Feng, M.; Johnson, J.; Lai, K. Reversal of aberrant PI3K/Akt signaling by Salubrinal in a GalT-deficient mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3286–3293. [Google Scholar] [CrossRef]
- De-Souza, E.A.; Pimentel, F.S.A.; Machado, C.M.; Martins, L.S.; da-Silva, W.S.; Montero-Lomelí, M.; Masuda, C.A. The unfolded protein response has a protective role in yeast models of classic galactosemia. Dis. Models Mech. 2014, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Slepak, T.; Tang, M.; Addo, F.; Lai, K. Intracellular galactose-1-phosphate accumulation leads to environmental stress response in yeast model. Mol. Genet. Metab. 2005, 86, 360–371. [Google Scholar] [CrossRef]
- Slepak, T.I.; Tang, M.; Slepak, V.Z.; Lai, K. Involvement of endoplasmic reticulum stress in a novel Classic Galactosemia model. Mol. Genet. Metab. 2007, 92, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Jumbo-Lucioni, P.P.; Hopson, M.L.; Hang, D.; Liang, Y.; Jones, D.P.; Fridovich-Keil, J.L. Oxidative stress contributes to outcome severity in a Drosophila melanogaster model of classic galactosemia. Dis. Model. Mech. 2013, 6, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumbo-Lucioni, P.P.; Ryan, E.L.; Hopson, M.L.; Bishop, H.M.; Weitner, T.; Tovmasyan, A.; Spasojevic, I.; Batinic-Haberle, I.; Liang, Y.; Jones, D.P.; et al. Manganese-based superoxide dismutase mimics modify both acute and long-term outcome severity in a Drosophila melanogaster model of classic galactosemia. Antioxid. Redox Signal. 2014, 20, 2361–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, S.A.; Daenzer, J.M.I.; MacWilliams, J.A.; Head, S.T.; Williams, M.B.; Geurts, A.M.; Schroeder, J.P.; Weinshenker, D.; Fridovich-Keil, J.L. A galactose-1-phosphate uridylyltransferase-null rat model of classic galactosemia mimics relevant patient outcomes and reveals tissue-specific and longitudinal differences in galactose metabolism. J. Inherit. Metab. Dis. 2020, 43, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Vanoevelen, J.M.; van Erven, B.; Bierau, J.; Huang, X.; Berry, G.T.; Vos, R.; Coelho, A.I.; Rubio-Gozalbo, M.E. Impaired fertility and motor function in a zebrafish model for classic galactosemia. J. Inherit. Metab. Dis. 2018, 41, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, G.T. Classic Galactosemia and Clinical Variant Galactosemia; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; GeneReviews®: Seattle, WA, USA, 1993. [Google Scholar]
- Walter, J.; Fridovich-Keil, J. Galactosemia. In The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID); Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K., Mitchell, G., Eds.; McGraw-Hill: New York, NY, USA, 2014. [Google Scholar]
- Cring, M.R.; Sheffield, V.C. Gene therapy and gene correction: Targets, progress, and challenges for treating human diseases. Gene 2020. [Google Scholar] [CrossRef] [PubMed]
- Rutten, M.G.S.; Rots, M.G.; Oosterveer, M.H. Exploiting epigenetics for the treatment of inborn errors of metabolism. J. Inherit. Metab. Dis. 2020, 43, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneller, J.L.; Lee, C.M.; Bao, G.; Venditti, C.P. Genome editing for inborn errors of metabolism: Advancing towards the clinic. BMC Med. 2017, 15, 43. [Google Scholar] [CrossRef] [Green Version]
- Chandler, R.J.; Venditti, C.P. Gene Therapy for Metabolic Diseases. Transl Sci. Rare Dis. 2016, 1, 73–89. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, B.S.; Gurung, S.; Perocheau, D.; Counsell, J.; Baruteau, J. Gene Therapy for Inherited Metabolic Diseases. J. Mother Child. 2020. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Daenzer, J.M.I.; Fridovich-Keil, J.L. A pilot study of neonatal GALT gene replacement using AAV9 dramatically lowers galactose metabolites in blood, liver, and brain and minimizes cataracts in GALT-null rat pups. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Loiler, S.A. Gene therapy for the treatment of galactosemia. U.S. Patent WO2020047472A1, 30 August 2019. [Google Scholar]
- Brophy, M.L.; Chen, T.-W.; Le, K.; Tabet, R.; Ahn, Y.; Murphy, J.E.; Bell, R.D. AAV-Mediated Gene Therapy Rescues GALT Activity and Reduces ER Stress in Classic Galactosemia. Mol. Ther. 2020, 28, 303. [Google Scholar]
- Pan, X.; Sands, S.A.; Yue, Y.; Zhang, K.; LeVine, S.M.; Duan, D. An Engineered Galactosylceramidase Construct Improves AAV Gene Therapy for Krabbe Disease in Twitcher Mice. Hum. Gene 2019, 30, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Molecular 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Martini, P.G.V.; Guey, L.T. A New Era for Rare Genetic Diseases: Messenger RNA Therapy. Hum. Gene 2019, 30, 1180–1189. [Google Scholar] [CrossRef]
- Timson, D.J. Therapies for galactosemia: A patent landscape. Pharm. Pat. Anal. 2020, 9, 45–51. [Google Scholar] [CrossRef]
- Zifu, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Portela Catani, J.; Sanders, N. mRNA therapeutics deliver a hopeful message. Nano Today 2018. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Molecular 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, F.; Guild, B.; Karve, S.; Smith, L.; Love, K.; Dorkin, J.R.; Kauffman, K.J.; Zhang, J.; Yahalom, B.; Anderson, D.G.; et al. Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 2016, 23, 699. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid 2018, 28, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Novakowski, S.; Jiang, K.; Prakash, G.; Kastrup, C. Delivery of mRNA to platelets using lipid nanoparticles. Sci. Rep. 2019, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hou, X.; Vick, O.G.; Dong, Y. RNA delivery biomaterials for the treatment of genetic and rare diseases. Biomaterials 2019, 217, 119291. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, B.; An, D.; Nguyen, V.; DeAntonis, C.; Martini, P.G.V.; Lai, K. Novel mRNA-Based Therapy Reduces Toxic Galactose Metabolites and Overcomes Galactose Sensitivity in a Mouse Model of Classic Galactosemia. Molecular 2020, 28, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Truong, B.; Allegri, G.; Liu, X.B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Haberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef]
- Berraondo, P.; Martini, P.G.V.; Avila, M.A.; Fontanellas, A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 2019, 68, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S.; et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.S.; Theisen, M.; Hong, S.J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2018, 24, 2520. [Google Scholar] [CrossRef]
- Jiang, L.; Berraondo, P.; Jerico, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaria, E.; Alegre, M.; et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency. Molecular 2018, 26, 801–813. [Google Scholar] [CrossRef]
- Roseman, D.S.; Khan, T.; Rajas, F.; Jun, L.S.; Asrani, K.H.; Isaacs, C.; Farelli, J.D.; Subramanian, R.R. G6PC mRNA Therapy Positively Regulates Fasting Blood Glucose and Decreases Liver Abnormalities in a Mouse Model of Glycogen Storage Disease 1a. Molecular 2018, 26, 814–821. [Google Scholar] [CrossRef]
- Haskovic, M.; Delnoy, B.; Bierau, J.; Lindhout, M.; Zimmermann, L.J.; Vanoevelen, J.M.; Coelho, A.I.; Rubio-Gozalbo, M.E. The promise of mRNA therapy as a treatment for classic galactosemia in Classic galactosemia: Natural histroy and new treatment approaches. Ph.D Thesis, Maastricht University, Maastricht, The Netherlands, October 2020; p. 17. [Google Scholar]
- Fan, J.Q. A counterintuitive approach to treat enzyme deficiencies: Use of enzyme inhibitors for restoring mutant enzyme activity. Biol. Chem. 2008, 389, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative strategies to treat protein misfolding in inborn errors of metabolism: Pharmacological chaperones and proteostasis regulators. J. Inherit. Metab. Dis. 2014, 37, 505–523. [Google Scholar] [CrossRef] [PubMed]
- Ulloa-Aguirre, A.; Janovick, J.A.; Brothers, S.P.; Conn, P.M. Pharmacologic rescue of conformationally-defective proteins: Implications for the treatment of human disease. Traffic 2004, 5, 821–837. [Google Scholar] [CrossRef] [PubMed]
- McCorvie, T.J.; Gleason, T.J.; Fridovich-Keil, J.L.; Timson, D.J. Misfolding of galactose 1-phosphate uridylyltransferase can result in type I galactosemia. Biochim. Biophys. Acta 2013, 1832, 1279–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.; Facchiano, A.; Rachamadugu, R.; Calderon, F.; Mao, R.; Milanesi, L.; Marabotti, A.; Lai, K. Correlation assessment among clinical phenotypes, expression analysis and molecular modeling of 14 novel variations in the human galactose-1-phosphate uridylyltransferase gene. Hum. Mutat. 2012, 33, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Sarkar, S.; Paul, S.S.; Roy, S.; Chattopadhyay, K. A small molecule chemical chaperone optimizes its unfolded state contraction and denaturant like properties. Sci. Rep. 2013, 3, 3525. [Google Scholar] [CrossRef]
- Coelho, A.I.; Trabuco, M.; Silva, M.J.; de Almeida, I.T.; Leandro, P.; Rivera, I.; Vicente, J.B. Arginine Functionally Improves Clinically Relevant Human Galactose-1-Phosphate Uridylyltransferase (GALT) Variants Expressed in a Prokaryotic Model. JIMD Rep. 2015, 23, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Haskovic, M.; Derks, B.; van der Ploeg, L.; Trommelen, J.; Nyakayiru, J.; van Loon, L.J.C.; Mackinnon, S.; Yue, W.W.; Peake, R.W.A.; Zha, L.; et al. Arginine does not rescue p.Q188R mutation deleterious effect in classic galactosemia. Orphanet J. Rare Dis. 2018, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Timson, D.J. Purple sweet potato colour—A potential therapy for galactosemia? Int. J. Food Sci. Nutr. 2014, 65, 391–393. [Google Scholar] [CrossRef]
- Bosch, A.M.; Bakker, H.D.; van Gennip, A.H.; van Kempen, J.V.; Wanders, R.J.; Wijburg, F.A. Clinical features of galactokinase deficiency: A review of the literature. J. Inherit. Metab. Dis. 2002, 25, 629–634. [Google Scholar] [CrossRef]
- Hennermann, J.B.; Schadewaldt, P.; Vetter, B.; Shin, Y.S.; Monch, E.; Klein, J. Features and outcome of galactokinase deficiency in children diagnosed by newborn screening. J. Inherit. Metab. Dis. 2011, 34, 399–407. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Derks, B.; Das, A.M.; Meyer, U.; Moslinger, D.; Couce, M.L.; Empain, A.; Ficicioglu, C.; Julia Palacios, N.; De Los Santos De Pelegrin, M.M.; et al. Galactokinase deficiency: Lessons from the GalNet registry. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Odejinmi, S.; Rascon, R.; Tang, M.; Vankayalapati, H.; Lai, K. Structure-activity analysis and cell-based optimization of human galactokinase inhibitors. ACS Med. Chem. Lett. 2011, 2, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Wierenga, K.; Elsas, L.J.; Lai, K. Molecular and biochemical characterization of human galactokinase and its small molecule inhibitors. Chem. Biol. Interact. 2010, 188, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Zhang, Y.Q.; Lee, O.W.; Liu, L.; Tang, M.; Lai, K.; Boxer, M.B.; Hall, M.D.; Shen, M. Discovery of novel inhibitors of human galactokinase by virtual screening. J. Comput Aided Mol. Des. 2019, 33, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tang, M.; Walsh, M.J.; Brimacombe, K.R.; Pragani, R.; Tanega, C.; Rohde, J.M.; Baker, H.L.; Fernandez, E.; Blackman, B.; et al. Structure activity relationships of human galactokinase inhibitors. Bioorg Med. Chem. Lett. 2015, 25, 721–727. [Google Scholar] [CrossRef]
- McAuley, M.; Mesa-Torres, N.; McFall, A.; Morris, S.; Huang, M.; Pey, A.L.; Timson, D.J. Improving the Activity and Stability of Human Galactokinase for Therapeutic and Biotechnological Applications. ChemBioChem 2018, 19, 1088–1095. [Google Scholar] [CrossRef]
- Wierenga, K.J.; Lai, K.; Buchwald, P.; Tang, M. High-throughput screening for human galactokinase inhibitors. J. Biomol. Screen 2008, 13, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.; Boxer, M.B.; Marabotti, A. GALK inhibitors for classic galactosemia. Future Med. Chem. 2014, 6, 1003–1015. [Google Scholar] [CrossRef]
- De Jongh, W.A.; Bro, C.; Ostergaard, S.; Regenberg, B.; Olsson, L.; Nielsen, J. The roles of galactitol, galactose-1-phosphate, and phosphoglucomutase in galactose-induced toxicity in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2008, 101, 317–326. [Google Scholar] [CrossRef]
- Lai, K.; Elsas, L.J.; Wierenga, K.J. Galactose toxicity in animals. IUBMB Life 2009, 61, 1063–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pintor, J. Sugars, the crystalline lens and the development of cataracts. Biochem. Pharm. 2012, 1, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Ai, Y.; Zheng, Z.; O’Brien-Jenkins, A.; Bernard, D.J.; Wynshaw-Boris, T.; Ning, C.; Reynolds, R.; Segal, S.; Huang, K.; Stambolian, D. A mouse model of galactose-induced cataracts. Hum. Mol. Genet. 2000, 9, 1821–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamijo, M.; Basso, M.; Cherian, P.V.; Hohman, T.C.; Sima, A.A. Galactosemia produces ARI-preventable nodal changes similar to those of diabetic neuropathy. Diabetes Res. Clin. Pr. 1994, 25, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Berry, G.T.; Hunter, J.V.; Wang, Z.; Dreha, S.; Mazur, A.; Brooks, D.G.; Ning, C.; Zimmerman, R.A.; Segal, S. In vivo evidence of brain galactitol accumulation in an infant with galactosemia and encephalopathy. J. Pediatr. 2001, 138, 260–262. [Google Scholar] [CrossRef]
- Huttenlocher, P.R.; Hillman, R.E.; Hsia, Y.E. Pseudotumor cerebri in galactosemia. J. Pediatr. 1970, 76, 902–905. [Google Scholar] [CrossRef]
- Lou, M.F.; Dickerson, J.E., Jr.; Chandler, M.L.; Brazzell, R.K.; York, B.M., Jr. The prevention of biochemical changes in lens, retina, and nerve of galactosemic dogs by the aldose reductase inhibitor AL01576. J. Ocul. Pharm. 1989, 5, 233–240. [Google Scholar] [CrossRef]
- Obrosova, I.; Faller, A.; Burgan, J.; Ostrow, E.; Williamson, J.R. Glycolytic pathway, redox state of NAD(P)-couples and energy metabolism in lens in galactose-fed rats: Effect of an aldose reductase inhibitor. Curr. Eye Res. 1997, 16, 34–43. [Google Scholar] [CrossRef]
- Mulhern, M.L.; Madson, C.J.; Danford, A.; Ikesugi, K.; Kador, P.F.; Shinohara, T. The unfolded protein response in lens epithelial cells from galactosemic rat lenses. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3951–3959. [Google Scholar] [CrossRef] [Green Version]
- Mizisin, A.P.; Myers, R.R.; Powell, H.C. Endoneurial sodium accumulation in galactosemic rat nerves. Muscle Nerve 1986, 9, 440–444. [Google Scholar] [CrossRef]
- Mizisin, A.P.; Powell, H.C. Schwann cell injury is attenuated by aldose reductase inhibition in galactose intoxication. J. Neuropathol. Exp. Neurol. 1993, 52, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Mizisin, A.P.; Powell, H.C.; Myers, R.R. Edema and increased endoneurial sodium in galactose neuropathy. Reversal with an aldose reductase inhibitor. J. Neurol. Sci. 1986, 74, 35–43. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT04117711, Safety and Pharmacokinetics of AT-007 in Healthy Subjects and in Adult Subjects With Classic Galactosemia; 2019 Oct 07. Available online: https://clinicaltrials.gov/ct2/show/NCT04117711 (accessed on 27 January 2021).
- Balakrishnan, B.; Siddiqi, A.; Mella, J.; Lupo, A.; Li, E.; Hollien, J.; Johnson, J.; Lai, K. Salubrinal enhances eIF2α phosphorylation and improves fertility in a mouse model of Classic Galactosemia. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165516. [Google Scholar] [CrossRef] [PubMed]
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grunewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. International clinical guideline for the management of classical galactosemia: Diagnosis, treatment, and follow-up. J. Inherit. Metab. Dis. 2017, 40, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Pollitt, R.J.; Green, A.; McCabe, C.J.; Booth, A.; Cooper, N.J.; Leonard, J.V.; Nicholl, J.; Nicholson, P.; Tunaley, J.R.; Virdi, N.K. Neonatal screening for inborn errors of metabolism: Cost, yield and outcome. Health Technol. Assess. 1997, 1, i–iv. [Google Scholar]
- Schweitzer-Krantz, S. Early diagnosis of inherited metabolic disorders towards improving outcome: The controversial issue of galactosaemia. Eur. J. Pediatr. 2003, 162 (Suppl. S1), S50–S53. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Friedman, S.; Moore, A.M.; Platt, B.A.; Feigenbaum, A.S. Selective screening for neonatal galactosemia: An alternative approach. Acta Paediatr. 2001, 90, 948–949. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Read, C.Y.; Ampola, M.; Brewster, T.G.; Demmer, L.; Greenstein, R.; Ingham, C.L.; Korson, M.; Msall, M.; Pueschel, S.; et al. Newborn screening compared to clinical identification of biochemical genetic disorders. J. Inherit. Metab. Dis. 2002, 25, 599–600. [Google Scholar] [CrossRef]
- Walter, J.H. Arguments for early screening: A clinician’s perspective. Eur. J. Pediatr. 2003, 162 (Suppl. S1), S2–S4. [Google Scholar] [CrossRef]
- Abidin, Z.; Treacy, E.P. Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. Int. J. Mol. Sci. 2019, 20, 5236. [Google Scholar] [CrossRef] [Green Version]
- Gubbels, C.S.; Land, J.A.; Rubio-Gozalbo, M.E. Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obs. Gynecol Surv. 2008, 63, 334–343. [Google Scholar] [CrossRef]
- Kaufman, F.; Kogut, M.D.; Donnell, G.N.; Koch, R.; Goebelsmann, U. Ovarian failure in galactosæmia. Lancet 1979, 314, 737–738. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Gubbels, C.S.; Bakker, J.A.; Menheere, P.P.; Wodzig, W.K.; Land, J.A. Gonadal function in male and female patients with classic galactosemia. Hum. Reprod. Update 2010, 16, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waggoner, D.D.; Buist, N.R.; Donnell, G.N. Long-term prognosis in galactosaemia: Results of a survey of 350 cases. J. Inherit. Metab. Dis. 1990, 13, 802–818. [Google Scholar] [CrossRef] [PubMed]
- Mamsen, L.S.; Kelsey, T.W.; Ernst, E.; Macklon, K.T.; Lund, A.M.; Andersen, C.Y. Cryopreservation of ovarian tissue may be considered in young girls with galactosemia. J. Assist. Reprod Genet. 2018, 35, 1209–1217. [Google Scholar] [CrossRef] [Green Version]
- Sozen, B.; Ozekinci, M.; Erman, M.; Gunduz, T.; Demir, N.; Akouri, R. Dehydroepiandrosterone supplementation attenuates ovarian ageing in a galactose-induced primary ovarian insufficiency rat model. J. Assist. Reprod Genet. 2019, 36, 2181–2189. [Google Scholar] [CrossRef]
- Thakur, M.; Feldman, G.; Puscheck, E.E. Primary ovarian insufficiency in classic galactosemia: Current understanding and future research opportunities. J. Assist. Reprod Genet. 2018, 35, 3–16. [Google Scholar] [CrossRef]
- Gubbels, C.S.; Land, J.A.; Evers, J.L.; Bierau, J.; Menheere, P.P.; Robben, S.G.; Rubio-Gozalbo, M.E. Primary ovarian insufficiency in classic galactosemia: Role of FSH dysfunction and timing of the lesion. J. Inherit. Metab. Dis. 2013, 36, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Sanders, R.D.; Spencer, J.B.; Epstein, M.P.; Pollak, S.V.; Vardhana, P.A.; Lustbader, J.W.; Fridovich-Keil, J.L. Biomarkers of ovarian function in girls and women with classic galactosemia. Fertil. Steril. 2009, 92, 344–351. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.B.; Badik, J.R.; Ryan, E.L.; Gleason, T.J.; Broadaway, K.A.; Epstein, M.P.; Fridovich-Keil, J.L. Modifiers of ovarian function in girls and women with classic galactosemia. J. Clin. Endocrinol. Metab. 2013, 98, E1257–E1265. [Google Scholar] [CrossRef] [Green Version]
- Van Erven, B.; Berry, G.T.; Cassiman, D.; Connolly, G.; Forga, M.; Gautschi, M.; Gubbels, C.S.; Hollak, C.E.M.; Janssen, M.C.; Knerr, I.; et al. Fertility in adult women with classic galactosemia and primary ovarian insufficiency. Fertil. Steril. 2017, 108, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Van Erven, B.; Gubbels, C.S.; van Golde, R.J.; Dunselman, G.A.; Derhaag, J.G.; de Wert, G.; Geraedts, J.P.; Bosch, A.M.; Treacy, E.P.; Welt, C.K.; et al. Fertility preservation in female classic galactosemia patients. Orphanet J. Rare Dis. 2013, 8, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamzatova, Z.; Komlichenko, E.; Kostareva, A.; Galagudza, M.; Ulrikh, E.; Zubareva, T.; Sheveleva, T.; Nezhentseva, E.; Kalinina, E. Autotransplantation of cryopreserved ovarian tissue—Effective method of fertility preservation in cancer patients. Gynecol. Endocrinol. 2014, 30 (Suppl. S1), 43–47. [Google Scholar] [CrossRef] [PubMed]
- Rivas Leonel, E.C.; Lucci, C.M.; Amorim, C.A. Cryopreservation of Human Ovarian Tissue: A Review. Transfus. Med. Hemother. 2019, 46, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, M.W.; Dittrich, R.; Lotz, L.; van der Ven, K.; van der Ven, H.H.; Liebenthron, J.; Korell, M.; Frambach, T.; Sutterlin, M.; Schwab, R.; et al. Fertility protection: Complications of surgery and results of removal and transplantation of ovarian tissue. Reprod. Biomed. Online 2018, 36, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Haskovic, M.; Poot, W.J.; van Golde, R.J.T.; Benneheij, S.H.; Oussoren, E.; de Wert, G.; Krumeich, A.; Rubio-Gozalbo, M.E. Intrafamilial oocyte donation in classic galactosemia: Ethical and societal aspects. J. Inherit. Metab. Dis. 2018, 41, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Timmers, I.; Jansma, B.M.; Rubio-Gozalbo, M.E. From mind to mouth: Event related potentials of sentence production in classic galactosemia. PLoS ONE 2012, 7, e52826. [Google Scholar] [CrossRef] [Green Version]
- Timmers, I.; van den Hurk, J.; Hofman, P.A.; Zimmermann, L.J.; Uludag, K.; Jansma, B.M.; Rubio-Gozalbo, M.E. Affected functional networks associated with sentence production in classic galactosemia. Brain Res. 2015, 1616, 166–176. [Google Scholar] [CrossRef]
- Timmers, I.; van der Korput, L.D.; Jansma, B.M.; Rubio-Gozalbo, M.E. Grey matter density decreases as well as increases in patients with classic galactosemia: A voxel-based morphometry study. Brain Res. 2016, 1648, 339–344. [Google Scholar] [CrossRef]
- Van Erven, B.; Jansma, B.M.; Rubio-Gozalbo, M.E.; Timmers, I. Exploration of the Brain in Rest: Resting-State Functional MRI Abnormalities in Patients with Classic Galactosemia. Sci. Rep. 2017, 7, 9095. [Google Scholar] [CrossRef] [Green Version]
- Welsink-Karssies, M.M.; Oostrom, K.J.; Hermans, M.E.; Hollak, C.E.M.; Janssen, M.C.H.; Langendonk, J.G.; Oussoren, E.; Rubio Gozalbo, M.E.; de Vries, M.; Geurtsen, G.J.; et al. Classical galactosemia: Neuropsychological and psychosocial functioning beyond intellectual abilities. Orphanet J. Rare Dis. 2020, 15, 42. [Google Scholar] [CrossRef] [Green Version]
- Welsink-Karssies, M.M.; Schrantee, A.; Caan, M.W.A.; Hollak, C.E.M.; Janssen, M.C.H.; Oussoren, E.; de Vries, M.C.; Roosendaal, S.D.; Engelen, M.; Bosch, A.M. Gray and white matter are both affected in classical galactosemia: An explorative study on the association between neuroimaging and clinical outcome. Mol. Genet. Metab. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hermans, M.E.; Welsink-Karssies, M.M.; Bosch, A.M.; Oostrom, K.J.; Geurtsen, G.J. Cognitive functioning in patients with classical galactosemia: A systematic review. Orphanet J. Rare Dis. 2019, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Del Felice, A.; Castiglia, L.; Formaggio, E.; Cattelan, M.; Scarpa, B.; Manganotti, P.; Tenconi, E.; Masiero, S. Personalized transcranial alternating current stimulation (tACS) and physical therapy to treat motor and cognitive symptoms in Parkinson’s disease: A randomized cross-over trial. Neuroimage Clin. 2019, 22, 101768. [Google Scholar] [CrossRef] [PubMed]
- Elyamany, O.; Leicht, G.; Herrmann, C.S.; Mulert, C. Transcranial alternating current stimulation (tACS): From basic mechanisms towards first applications in psychiatry. Eur. Arch. Psychiatry Clin. Neurosci. 2020. [Google Scholar] [CrossRef]
- Riddle, J.; Rubinow, D.R.; Frohlich, F. A case study of weekly tACS for the treatment of major depressive disorder. Brain Stimul. 2020, 13, 576–577. [Google Scholar] [CrossRef] [Green Version]
- Rufener, K.S.; Krauel, K.; Meyer, M.; Heinze, H.J.; Zaehle, T. Transcranial electrical stimulation improves phoneme processing in developmental dyslexia. Brain Stimul. 2019, 12, 930–937. [Google Scholar] [CrossRef] [Green Version]
- Tavakoli, A.V.; Yun, K. Transcranial Alternating Current Stimulation (tACS) Mechanisms and Protocols. Front. Cell Neurosci. 2017, 11, 214. [Google Scholar] [CrossRef] [Green Version]
- Peter, B.; Potter, N.; Davis, J.; Donenfeld-Peled, I.; Finestack, L.; Stoel-Gammon, C.; Lien, K.; Bruce, L.; Vose, C.; Eng, L.; et al. Toward a paradigm shift from deficit-based to proactive speech and language treatment: Randomized pilot trial of the Babble Boot Camp in infants with classic galactosemia. F1000Research 2019, 8, 271. [Google Scholar] [CrossRef]
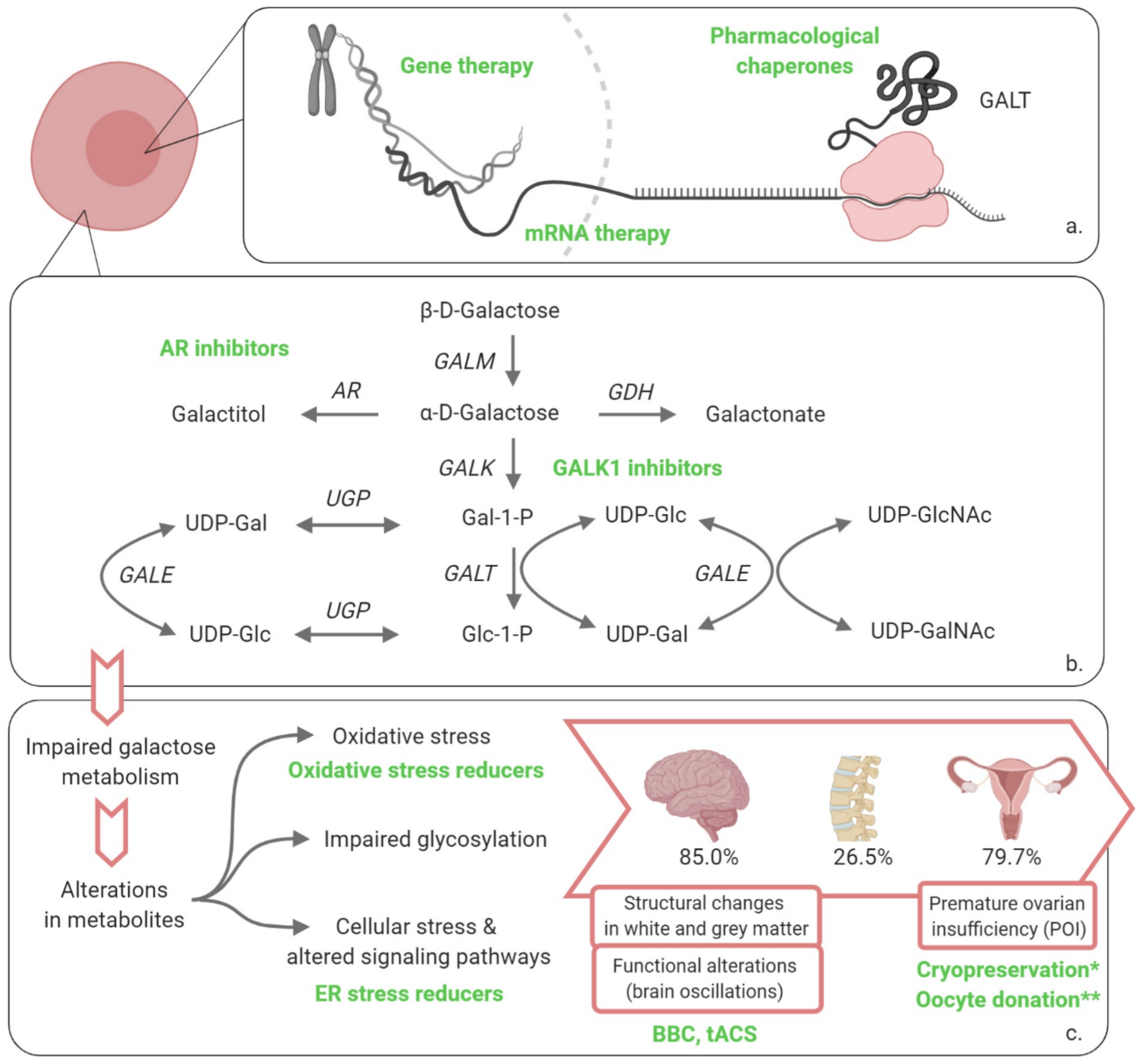
Figure 1.
Overview of disorders of galactose metabolism and novel therapeutic approaches recently investigated or under investigation for classic galactosemia. (a) Transcription and translation leading to the production of enzyme, therapies restoring enzyme activity; (b) Leloir pathway of galactose metabolism and alternative routes of galactose disposal; (c) impaired galactose metabolism leading to long-term organ damage. * cryopreservation: more data have become available in the last 5 years; ** oocyte donation: recommendations for CG regarding this procedure have been formulated by a group of experts in the last 5 years. GALM, galactose mutarotase; GALK1, galactokinase 1; Gal-1-P, galactose-1-phosphate; GALT, galactose 1-phosphate uridylyltransferase; Glc-1-P, glucose-1-phosphate; GALE, UDP-galactose 4′-epimerase; UDP-Gal, uridine diphosphate-galactose; UDP-Glc, uridine diphosphate-glucose; UGP, UDP-glucose pyrophosphorylase; UDP-GlcNAc, UDP-N-acetylglucosamine; UDP-GalNAc, UDP-N-acetylgalactosamine; AR, aldose reductase; GDH, galactose dehydrogenase; ER, endoplasmic reticulum; BBC, Babble Boot Camp; tACS, transcranial alternating current stimulation. Created with BioRender.com.
Figure 1.
Overview of disorders of galactose metabolism and novel therapeutic approaches recently investigated or under investigation for classic galactosemia. (a) Transcription and translation leading to the production of enzyme, therapies restoring enzyme activity; (b) Leloir pathway of galactose metabolism and alternative routes of galactose disposal; (c) impaired galactose metabolism leading to long-term organ damage. * cryopreservation: more data have become available in the last 5 years; ** oocyte donation: recommendations for CG regarding this procedure have been formulated by a group of experts in the last 5 years. GALM, galactose mutarotase; GALK1, galactokinase 1; Gal-1-P, galactose-1-phosphate; GALT, galactose 1-phosphate uridylyltransferase; Glc-1-P, glucose-1-phosphate; GALE, UDP-galactose 4′-epimerase; UDP-Gal, uridine diphosphate-galactose; UDP-Glc, uridine diphosphate-glucose; UGP, UDP-glucose pyrophosphorylase; UDP-GlcNAc, UDP-N-acetylglucosamine; UDP-GalNAc, UDP-N-acetylgalactosamine; AR, aldose reductase; GDH, galactose dehydrogenase; ER, endoplasmic reticulum; BBC, Babble Boot Camp; tACS, transcranial alternating current stimulation. Created with BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Delnoy, B.; Coelho, A.I.; Rubio-Gozalbo, M.E. Current and Future Treatments for Classic Galactosemia. J. Pers. Med. 2021, 11, 75. https://doi.org/10.3390/jpm11020075
AMA Style
Delnoy B, Coelho AI, Rubio-Gozalbo ME. Current and Future Treatments for Classic Galactosemia. Journal of Personalized Medicine. 2021; 11(2):75. https://doi.org/10.3390/jpm11020075
Chicago/Turabian StyleDelnoy, Britt, Ana I. Coelho, and Maria Estela Rubio-Gozalbo. 2021. "Current and Future Treatments for Classic Galactosemia" Journal of Personalized Medicine 11, no. 2: 75. https://doi.org/10.3390/jpm11020075
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.