
Diagnostic Challenges in Rare Causes of Arrhythmogenic Cardiomyopathy—The Role of Cardiac MRI

 , , , and
, , , and
Abstract
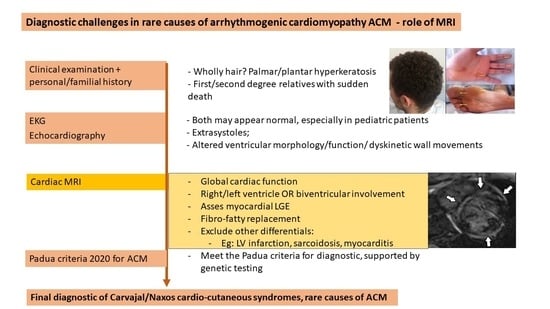
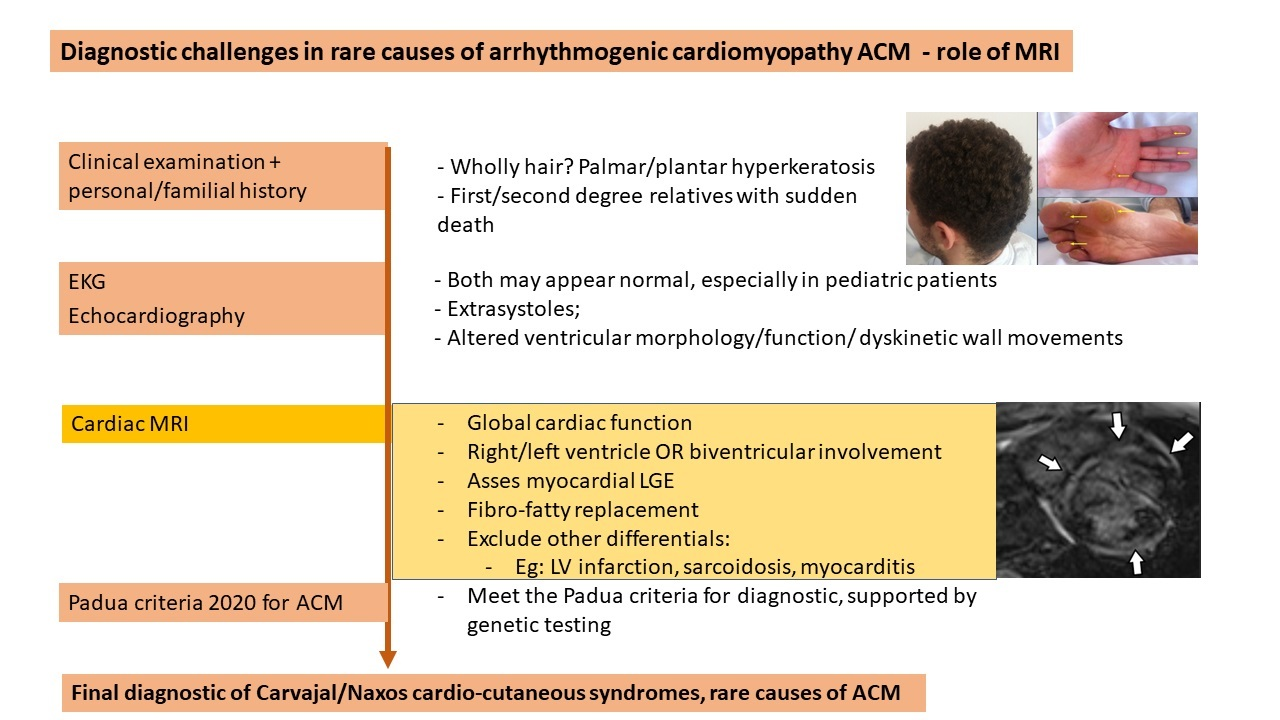
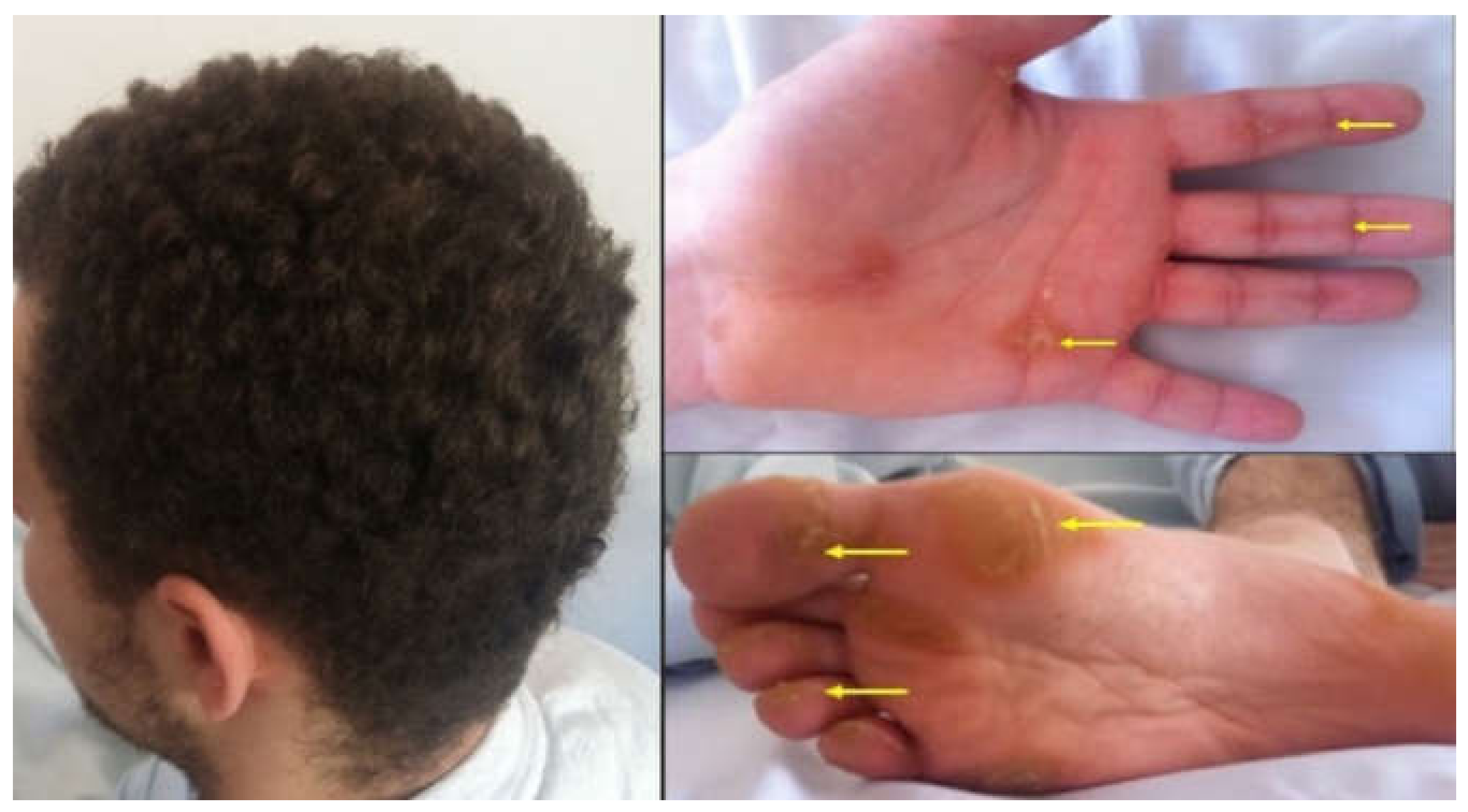
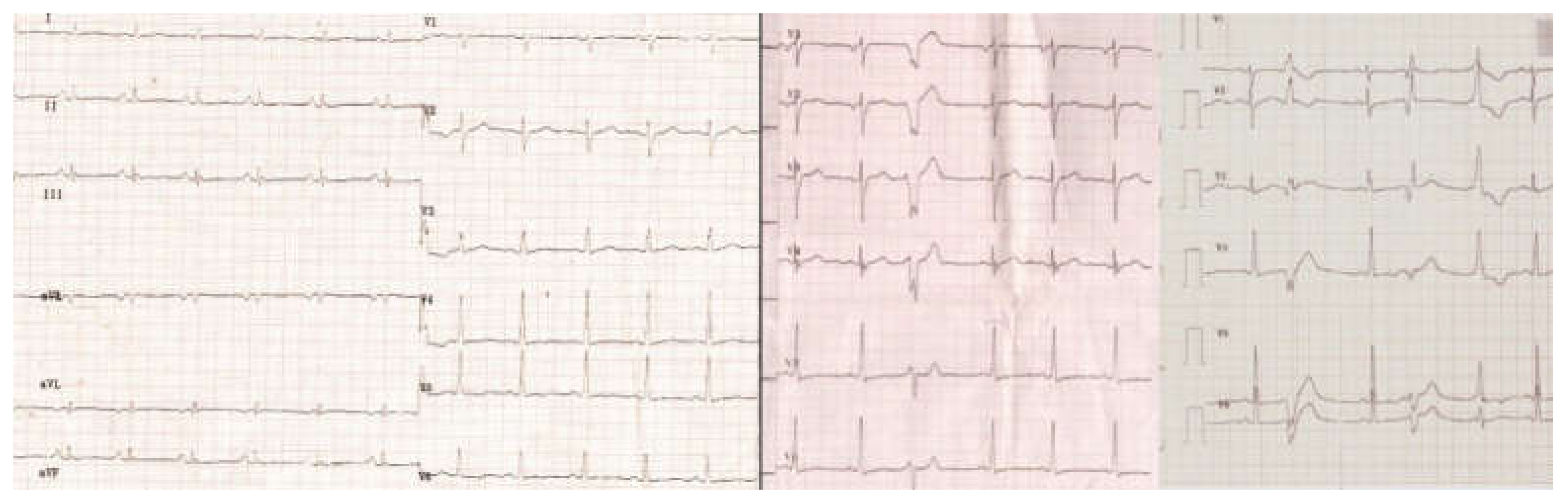
:
1. Introduction
2. Genetics
3. Pathophysiology
4. Imaging—Role of MRI
4.1. MRI Diagnostic Criteria
4.2. Differential Diagnosis—Normal Variants
4.3. Differential Diagnosis—Pathologic Conditions
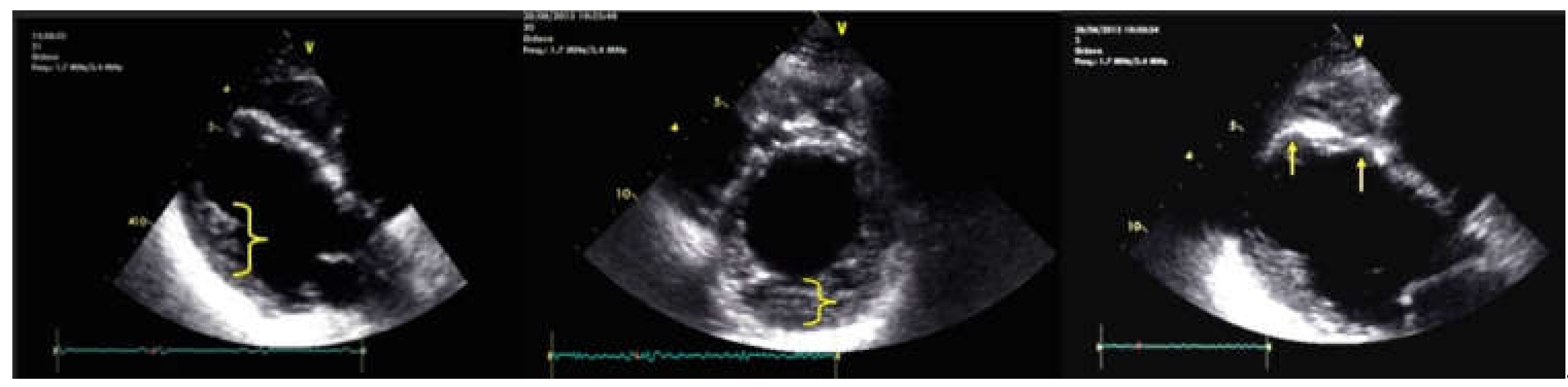
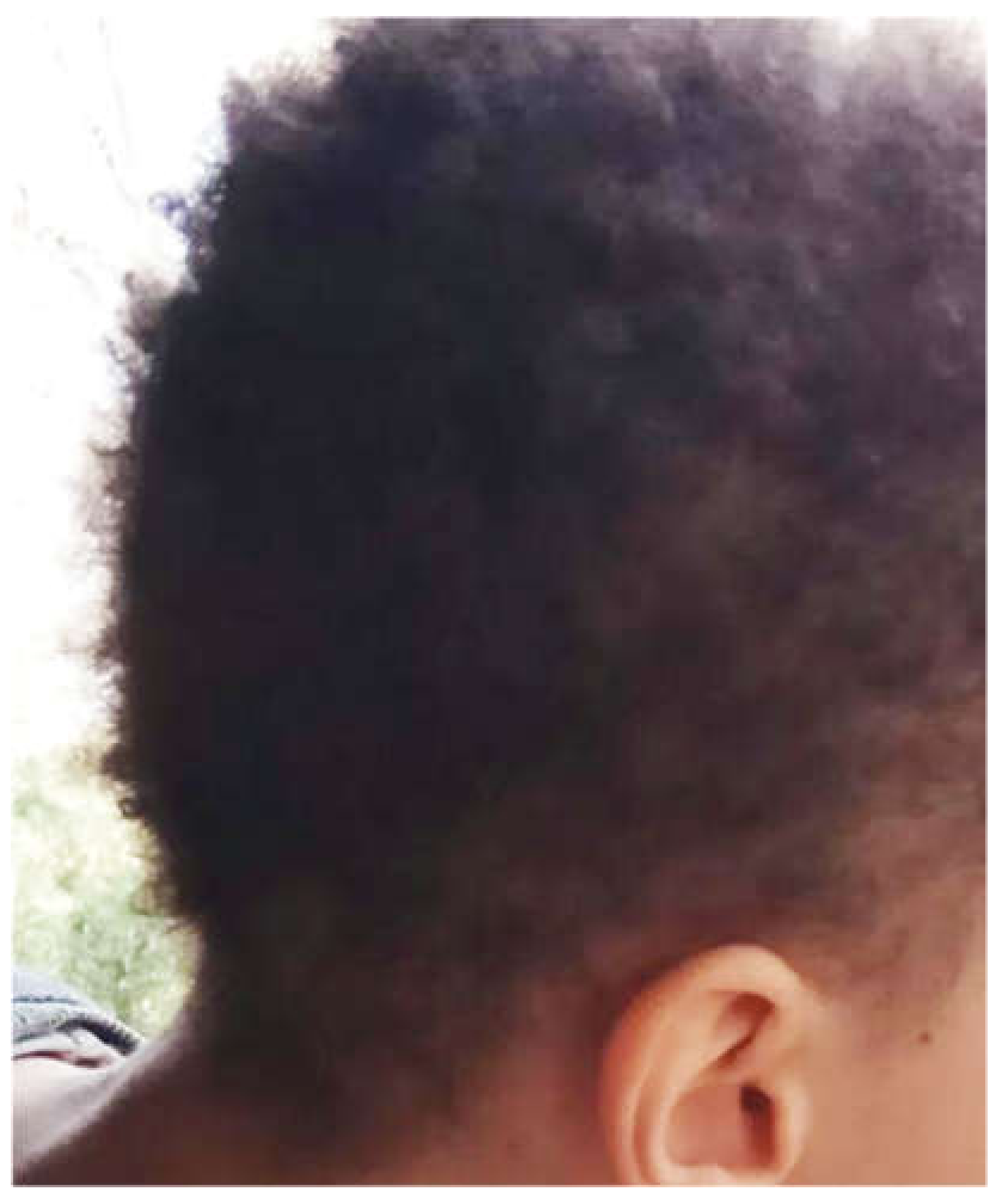
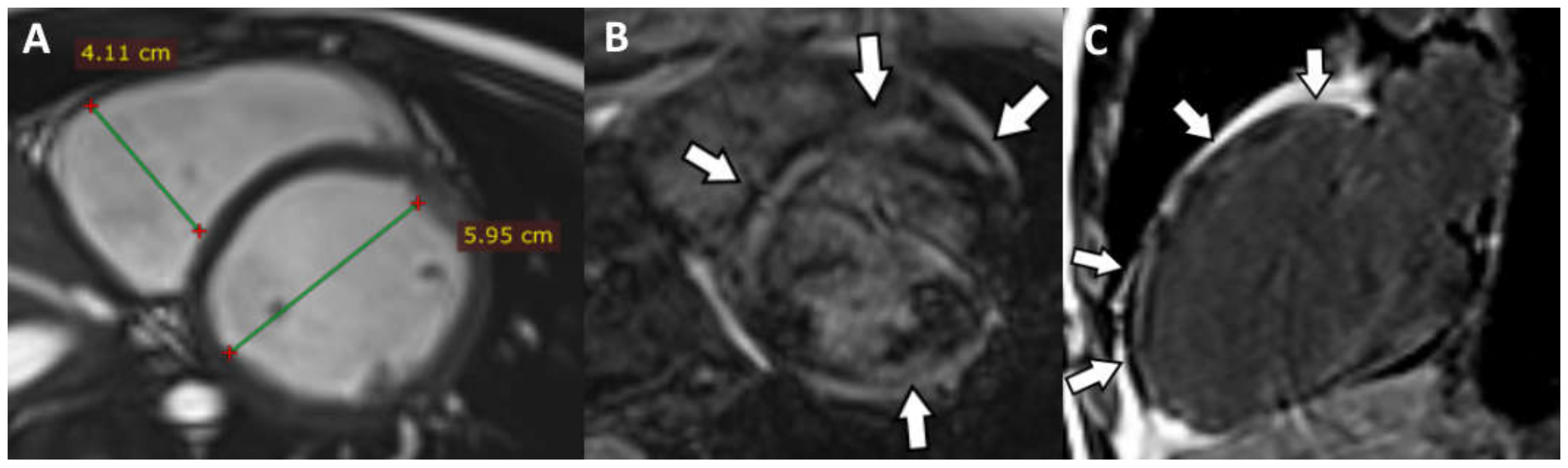
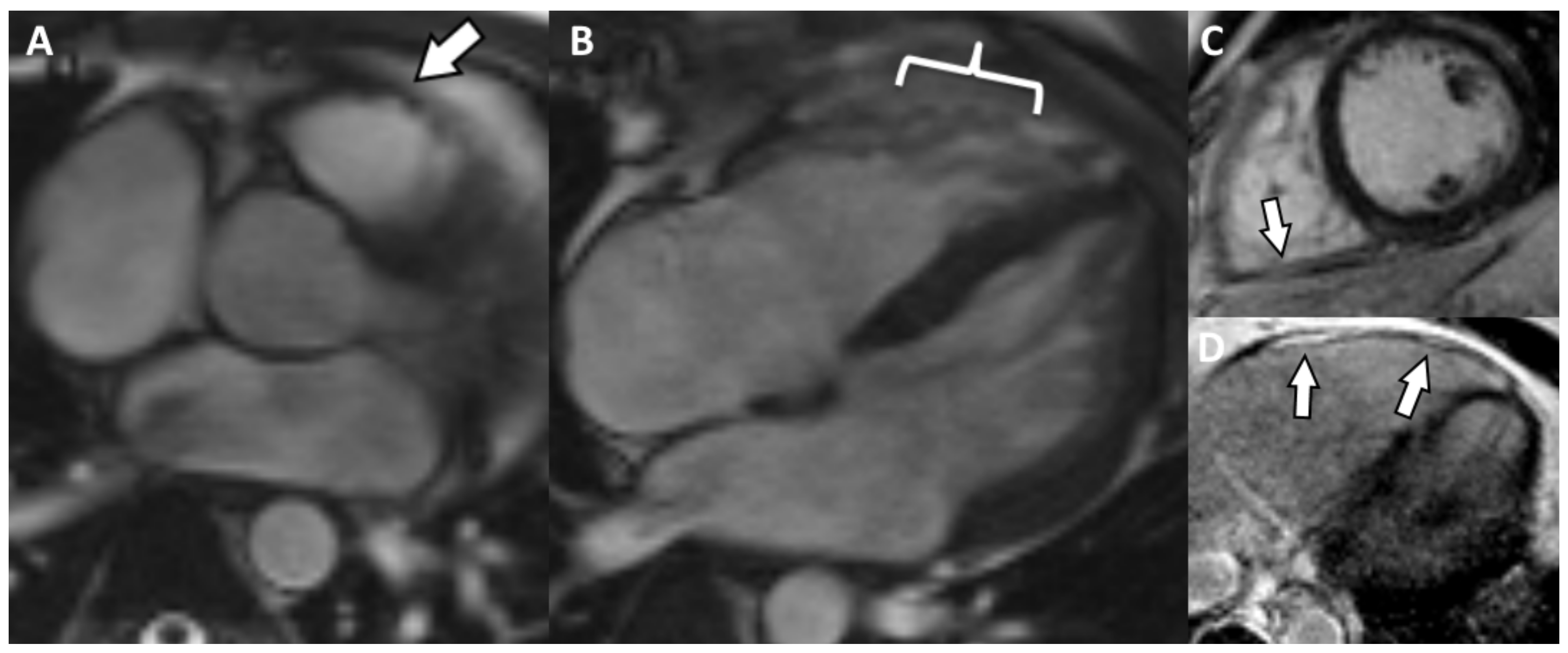
5. Cases Presentation
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pilichou, K.; Thiene, G.; Bauce, B. Arrhythmogenic cardiomyopathy. Orphanet J. Rare Dis. 2016, 11, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, C.; Pilichou, K.; Bauce, B.; Corrado, D.; Thiene, G. Diagnostic Criteria, Genetics, and Molecular Basis of Arrhythmogenic Cardiomyopathy. Heart Fail Clin. 2018, 14, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, E.; Jongbloed, J.D.; Pilichou, K.; Thiene, G.; Basso, C.; Bikker, H.; Charbon, B.; Swertz, M.; van Tintelen, J.P.; van der Zwaag, P.A. The ARVD/C Genetic Variants Database: 2014 Update. Hum. Mutat. 2015, 36, 403–410. [Google Scholar] [CrossRef]
- Bennett, R.G.; Haqqani, H.M.; Berruezo, A.; Della Bella, P.; Marchlinski, F.E.; Hsu, C.-J.; Kumar, S. Arrhythmogenic Cardiomyopathy in 2018–2019: ARVC/ALVC or Both? Heart Lung Circ. 2019, 28, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, A.; Elliott, P.M. Arrhythmogenic Cardiomyopathy: A Disease or Merely a Phenotype? Eur. Cardiol. 2020, 15, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Marra, M.P.; Zorzi, A.; Beffagna, G.; Cipriani, A. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, A.S.; Tandri, H.; Bluemke, D.A. Arrhythmogenic right ventricular cardiomyopathy (ARVC): Cardiovascular magnetic resonance update. J. Cardiovasc. Magn. Reson. 2014, 16, 50. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Agullo-Pascual, E.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Voorn, S.M.; Riele, A.S.J.M.T.; Basso, C.; Calkins, H.; Remme, C.A.; Veen, T.A.B.V. Arrhythmogenic cardiomyopathy: Pathogenesis, pro-arrhythmic remodelling, and novel approaches for risk stratification and therapy. Cardiovasc. Res. 2020, 116, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Marra, M.P.; Rizzo, S.; Bauce, B.; De Lazzari, M.; Pilichou, K. Arrhythmogenic right ventricular cardiomyopathy. Contribution of cardiac magnetic resonance imaging to the diagnosis. Herz 2015, 40, 600–606. [Google Scholar] [CrossRef]
- Marra, M.P.; Leoni, L.; Bauce, B.; Corbetti, F.; Zorzi, A. Imaging study of ventricular scar in arrhythmogenic right ventricular cardiomyopathy: Comparison of 3D standard electroanatomical voltage mapping and contrast-enhanced cardiac magnetic resonance. Circ. Arrhythm. Electrophysiol. 2012, 5, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.; McKenna, W.J. Left-Dominant Arrhythmogenic Cardiomyopathy: An Under-Recognized Clinical Entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te Riele, A.S.; James, C.A.; Philips, B.; Rastegar, N.; Bhonsale, A. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: The triangle of dysplasia displaced. J. Cardiovasc. Electrophysiol. 2013, 24, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, F.; Yoshinaga, K. New Guidelines for Diagnosis of Cardiac Sarcoidosis in Japan. Ann. Nucl. Cardiol. 2017, 3, 42–45. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Saberniak, J.; Leren, I.S.; Haland, T.F.; Beitnes, J.O.; Hopp, E.; Borgquist, R.; Edvardsen, T.; Haugaa, K.H. Comparison of patients with early-phase arrhythmogenic right ventricular cardiomyopathy and right ventricular outflow tract ventricular tachycardia. Eur. Heart J.-Cardiovasc. Imaging 2017, 18, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Walia, J.; Steinberg, C.; Laksman, Z. Brugada syndrome: Updated perspectives. Res. Rep. Clin. Cardiol. 2019, 10, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Bastiaenen, R.; Cox, A.T.; Castelletti, S.; Wijeyeratne, Y.; Colbeck, N.; Pakroo, N.; Ahmed, H.; Bunce, N.; Anderson, L.; Moon, J.; et al. Late gadolinium enhancement in Brugada syndrome: A marker for subtle underlying cardiomyopathy? Heart Rhythm 2017, 14, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, A.; Cipriani, A.; Mattesi, G.; Vio, R.; Bettella, N.; Corrado, D. Arrhythmogenic Cardiomyopathy and Sports Activity. J. Cardiovasc. Transl. Res. 2020, 13, 274–283. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, D.H.; Abbara, S.; Chaithiraphan, V.; Yared, K.; Killeen, R.P. Cardiac MR imaging of nonischemic cardiomyopathies: Imaging protocols and spectra of appearances. Radiology 2012, 262, 403–422. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Bauce, B.; Rampazzo, A.; Basso, C.; Mazzotti, E.; Rigato, I. Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm 2011, 8, 1686–1695. [Google Scholar] [CrossRef]
- Pigors, M.; Schwieger-Briel, A.; Cosgarea, R.; Diaconeasa, A.; Bruckner-Tuderman, L.; Fleck, T.; Has, C. Desmoplakin Mutations with Palmoplantar Keratoderma, Woolly Hair and Cardiomyopathy. Acta Derm. Venereol. 2015, 95, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Cannavale, G.; Francone, M.; Galea, N.; Vullo, F.; Molisso, A.; Carbone, I.; Catalano, C. Fatty Images of the Heart: Spectrum of Normal and Pathological Findings by Computed Tomography and Cardiac Magnetic Resonance Imaging. BioMed Res. Int. 2018, 2018, 5610347. [Google Scholar] [CrossRef]
- Martins, D.; Ovaert, C.; Khraiche, D.; Boddaert, N.; Bonnet, D. Myocardial inflammation detected by cardiac MRI in Arrhythmogenic right ventricular cardiomyopathy: A paediatric case series. Int. J. Cardiol. 2018, 271, 81–86. [Google Scholar] [CrossRef]
- Mavrogeni, S.; Protonotarios, N.; Tsatsopoulou, A.; Papachristou, P.; Sfendouraki, E.; Papadopoulos, G. Naxos disease evolution mimicking acute myocarditis: The role of cardiovascular magnetic resonance imaging. Int. J. Cardiol. 2013, 166, e14–e15. [Google Scholar] [CrossRef]
- Arbustini, E.; Favalli, V.; Narula, N.; Serio, A.; Grasso, M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 949–966. [Google Scholar] [CrossRef]
- Finsterer, J.; Stöllberger, C.; Wollmann, E.; Dertinger, S.; Laccone, F. Autosomal dominant Carvajal plus syndrome due to the novel desmoplakin mutation c.1678A > T (p.Ile560Phe). Mol. Genet. Metab. Rep. 2016, 8, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Bharucha, T.; Lee, K.J.; Daubeney, P.E.; Nugent, A.W.; Turner, C. Sudden death in childhood cardiomyopathy: Results from a long-term national population-based study. J. Am. Coll. Cardiol. 2015, 65, 2302–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predominant RV Involvement | Biventricular Involvement | Predominant LV Involvement |
|---|---|---|
| JUP | TMEM43 | DSP |
| PKP2 | DES | |
| DSG2 | PLN |
| MRI Criteria | Description |
|---|---|
| Major | Focal akinesia/dyskinesia of the RV/asynchronous ventricular contraction and one of the following: RV dilatation (EDV index ≥ 110 mL/m2 men, ≥100 mL/m2 women) RVEF ≤ 40% |
| Minor | Focal akinesia/dyskinesia of the RV/asynchronous ventricular contraction and one of the following: RV dilatation (EDV index ≥ 100 but <110 mL/m2 men, ≥90 but <100 mL/m2 women) RVEF ≤ 45% but >40% |
| MRI Criteria | Description |
|---|---|
| Major | LGE ≥ 1 segment (in two orthogonal views) of the free wall (subepicardial or midmyocardial), IVS or both |
| Minor | Depression of LVEF with or without dilatation of LV, according with data from age, sex, and body surface are nomograms Regional akinesia or hypokinesia of the free wall, IVS or both |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manole, S.; Pintican, R.; Popa, G.; Rancea, R.; Dadarlat-Pop, A.; Vulturar, R.; Palade, E. Diagnostic Challenges in Rare Causes of Arrhythmogenic Cardiomyopathy—The Role of Cardiac MRI. J. Pers. Med. 2022, 12, 187. https://doi.org/10.3390/jpm12020187
Manole S, Pintican R, Popa G, Rancea R, Dadarlat-Pop A, Vulturar R, Palade E. Diagnostic Challenges in Rare Causes of Arrhythmogenic Cardiomyopathy—The Role of Cardiac MRI. Journal of Personalized Medicine. 2022; 12(2):187. https://doi.org/10.3390/jpm12020187
Chicago/Turabian StyleManole, Simona, Roxana Pintican, George Popa, Raluca Rancea, Alexandra Dadarlat-Pop, Romana Vulturar, and Emanuel Palade. 2022. "Diagnostic Challenges in Rare Causes of Arrhythmogenic Cardiomyopathy—The Role of Cardiac MRI" Journal of Personalized Medicine 12, no. 2: 187. https://doi.org/10.3390/jpm12020187