

Overview of Apoptosis, Autophagy, and Inflammatory Processes in Toxoplasma gondii Infected Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
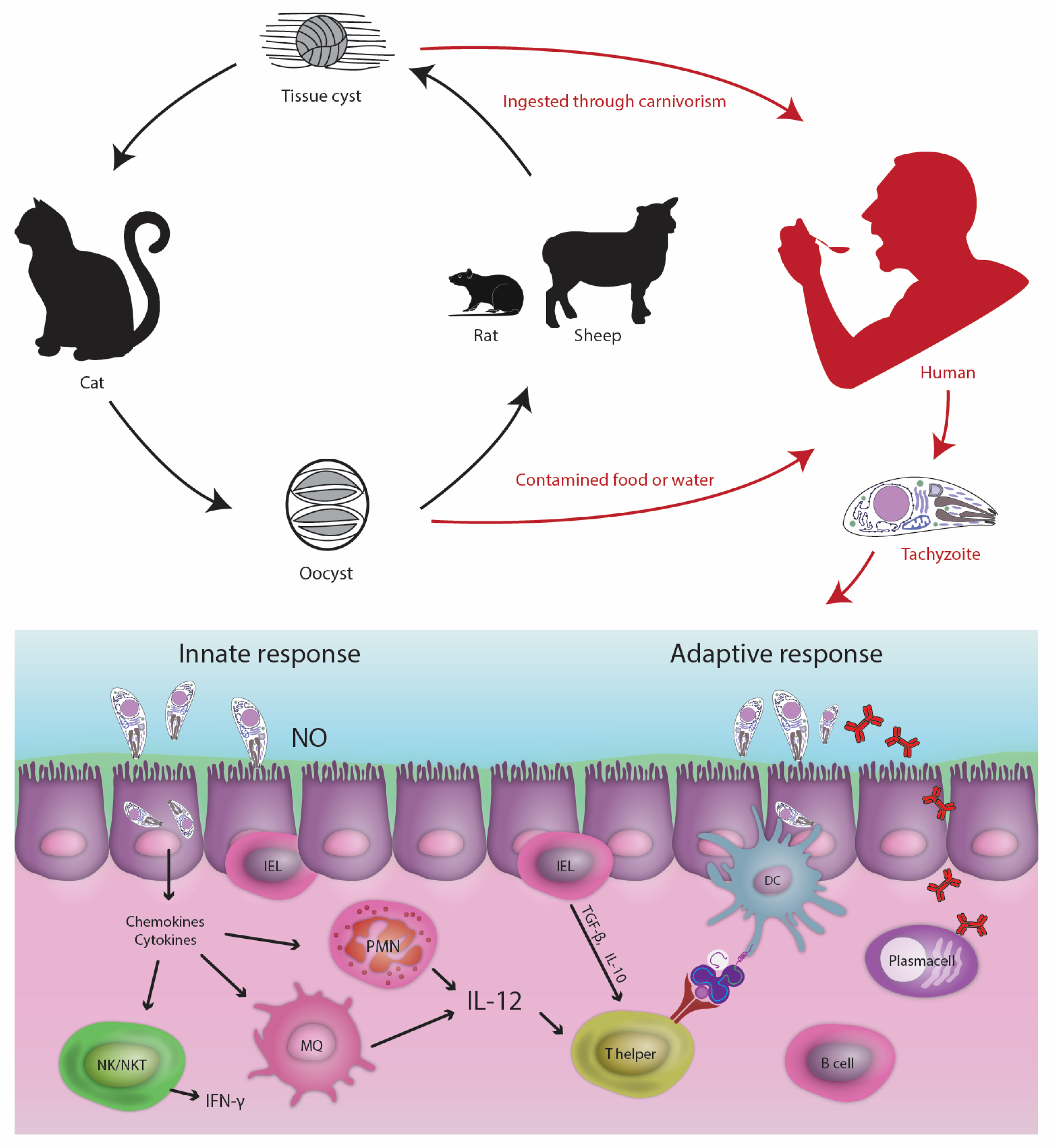
2. T. gondii Infection
3. Interaction of T. gondii with Immune Cell Signaling
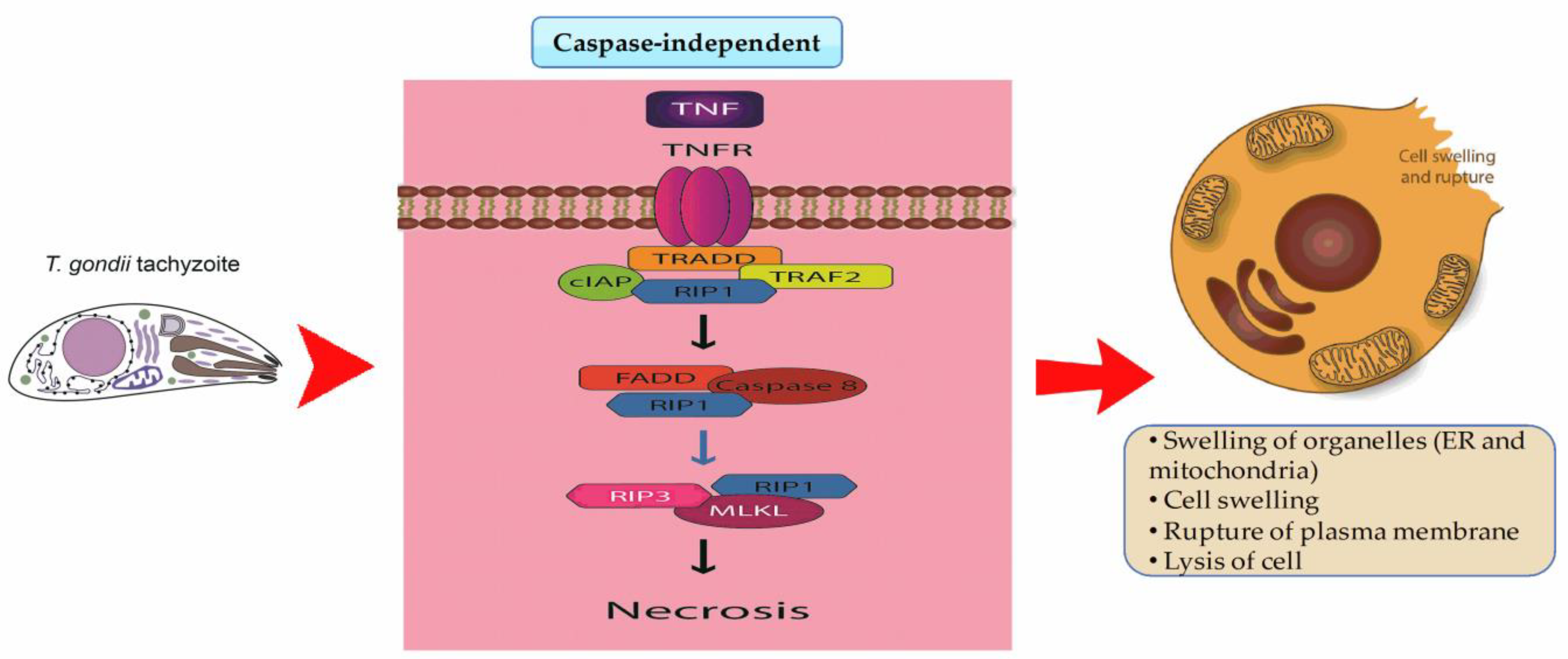
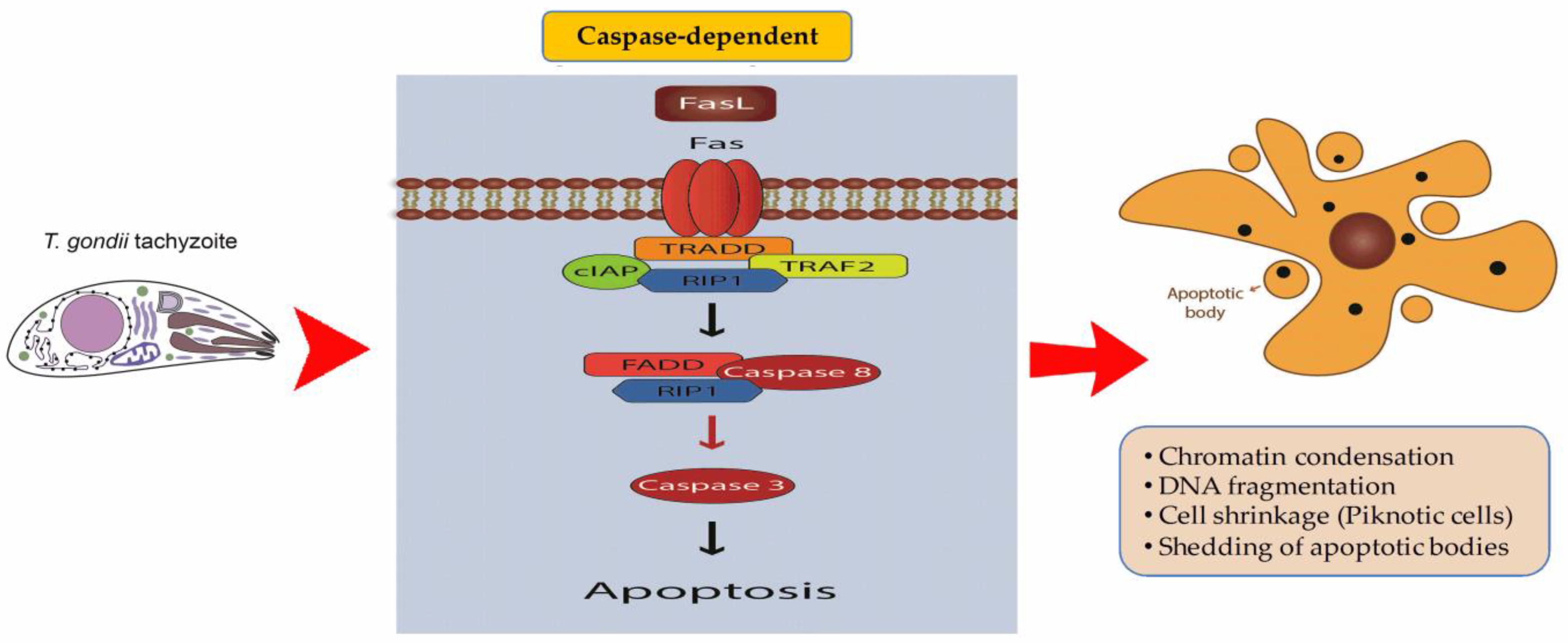
4. Regulated Cell Death (RCD) and T. gondii Infection
Inhibition of Apoptosis by T. gondii
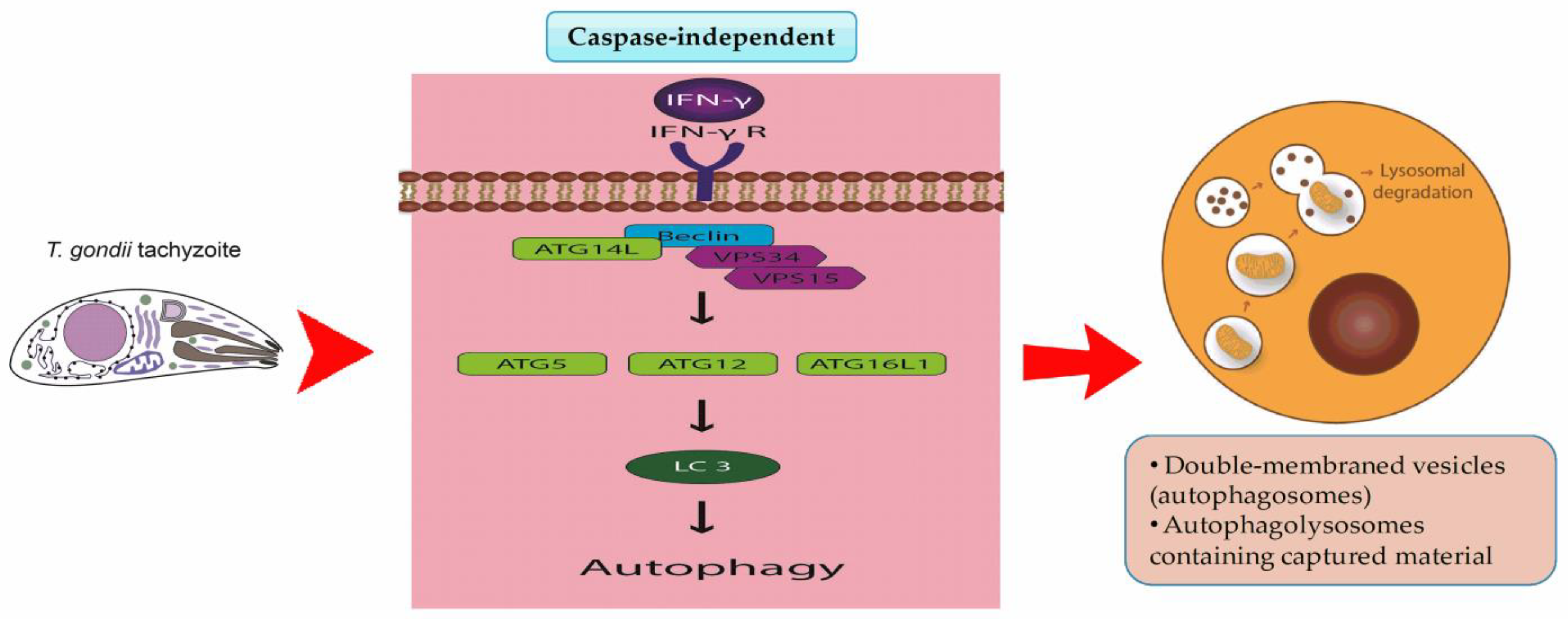
5. Autophagy in T. gondii Infection
5.1. Host Cell Autophagy Pathways Targeting T. gondii
5.2. Mechanisms of Autophagy Control in T. gondiiInfection
6. The Role of RCD in Controlling T. gondii Infection
7. Inflammation-Associated Factors in Toxoplasmosis
8. Effect of T. gondii on Autoimmune Diseases
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PLGA | Poly lactic-co-glycolic acid |
| PEG-poly | Polyethylene glycol |
| Caspase-3 | Cysteine aspartic acid-protease 3 |
| DD | Death domain |
| TNFR | Tumor necrosis factor |
| PCD | Programmed cell death |
| HLA | Human lymphocyte antigen |
| IgG | Immunoglobulin G |
| CDCA | Chenodeoxycholic acid |
| DSS | Dextran sulfate sodium |
| IDO | Indoleamine oxidase |
| IRG | Immunity-related GTPase |
| GBP | Guanylate binding protein |
| iNOS | Inducible nitric oxide synthase |
| NO | Nitric oxide |
| ROS | Reactive oxygen species |
| UB | Ubiquitin |
| PVM | Parasitophorous vacuole membrane |
| GRA | Dense granule |
| NDP | Nuclear Domain Protein |
| HFF | Human foreskin fibroblasts |
| BMDC | Bone marrow dendritic cell |
| IVN | Intravascular network |
| HSPGS | Heparan sulfate proteoglycans |
References
- Kochanowsky, J.A.; Koshy, A.A. Toxoplasma gondii. Curr. Biol. 2018, 28, R770–R771. [Google Scholar] [CrossRef] [PubMed]
- Lambert, H.; Barragan, A. Modelling parasite dissemination: Host cell subversion and immune evasion by Toxoplasma gondii. Cell. Microbiol. 2010, 12, 292–300. [Google Scholar] [CrossRef]
- Attias, M.; Teixeira, D.E.; Benchimol, M.; Vommaro, R.C.; Crepaldi, P.H.; De Souza, W. The life-cycle of Toxoplasma gondii reviewed using animations. Parasites Vectors 2020, 13, 1–13. [Google Scholar] [CrossRef]
- Dubey, J. A review of toxoplasmosis in wild birds. Vet. Parasitol. 2002, 106, 121–153. [Google Scholar] [CrossRef] [PubMed]
- Hatam-Nahavandi, K.; Calero-Bernal, R.; Rahimi, M.; Pagheh, A.; Mehdi, Z.; Dezhkam, A. Toxoplasma gondii infection in domestic and wild felids as public health concerns: A systematic review and meta-analysis. Sci Rep. 2021, 11, 9590. [Google Scholar] [CrossRef]
- Azadi, Y.; Ahmadpour, E.; Ahmadi, A. Targeting Strategies in Therapeutic Applications of Toxoplasmosis: Recent Advances in Liposomal Vaccine Delivery Systems. Curr. Drug Targets 2020, 21, 541–558. [Google Scholar] [CrossRef]
- Eaton, M.S.; Weiss, L.M.; Kim, K. Cyclic nucleotide kinases and tachyzoite–bradyzoite transition in Toxoplasma gondii. Int. J. Parasitol. 2006, 36, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.A.; Guevara, R.B.; Rommereim, L.M.; Falla, A.; Bellini, V.; Petre, G.; Rak, C.; Cantillana, V.; Dubremetz, J.-F.; Cesbron-Delauw, M.-F. Toxoplasma gondii parasitophorous vacuole membrane-associated dense granule proteins orchestrate chronic infection and GRA12 underpins resistance to host gamma interferon. MBio 2019, 10, e00589-19. [Google Scholar] [CrossRef]
- Blader, I.J.; Coleman, B.I.; Chen, C.-T.; Gubbels, M.-J. Lytic cycle of Toxoplasma gondii: 15 years later. Annu. Rev. Microbiol. 2015, 69, 463–485. [Google Scholar] [CrossRef]
- Denkers, E.Y.; Gazzinelli, R.T. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 1998, 11, 569–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahiaoui, B.; Dzierszinski, F.; Bernigaud, A.; Slomianny, C.; Camus, D.; Tomavo, S. Isolation and characterization of a subtractive library enriched for developmentally regulated transcripts expressed during encystation of Toxoplasma gondii. Mol. Biochem. Parasitol. 1999, 99, 223–235. [Google Scholar] [CrossRef]
- da Silva, R.C.; Langoni, H. Toxoplasma gondii: Host–parasite interaction and behavior manipulation. Parasitol. Res. 2009, 105, 893–898. [Google Scholar] [CrossRef]
- Boothroyd, J.C.; Grigg, M.E. Population biology of Toxoplasma gondii and its relevance to human infection: Do different strains cause different disease? Curr. Opin. Microbiol. 2002, 5, 438–442. [Google Scholar] [CrossRef]
- Mammari, N.; Halabi, M.A.; Yaacoub, S.; Chlala, H.; Dardé, M.-L.; Courtioux, B. Toxoplasma gondii modulates the host cell responses: An overview of apoptosis pathways. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- de Abreu Cabral, G.R.; Wang, Z.T.; Sibley, L.; DaMatta, R.A. Inhibition of nitric oxide production in activated macrophages caused by Toxoplasma gondii infection occurs by distinct mechanisms in different mouse macrophage cell lines. Front. Microbiol. 2018, 9. [Google Scholar]
- Bliss, S.K.; Gavrilescu, L.C.; Alcaraz, A.; Denkers, E.Y. Neutrophil depletion during Toxoplasma gondii infection leads to impaired immunity and lethal systemic pathology. Infect. Immun. 2001, 69, 4898–4905. [Google Scholar] [CrossRef]
- Lüder, C.; Gross, U. Apoptosis and its modulation during infection with Toxoplasma gondii: Molecular mechanisms and role in pathogenesis. Role Apoptosis Infect. 2005, 219–237. [Google Scholar]
- Wang, L.; Chen, H.; Liu, D.; Huo, X.; Gao, J.; Song, X.; Xu, X.; Huang, K.; Liu, W.; Wang, Y. Genotypes and mouse virulence of Toxoplasma gondii isolates from animals and humans in China. PLoS ONE 2013, 8, e53483. [Google Scholar] [CrossRef] [PubMed]
- Flegr, J.; Prandota, J.; Sovičková, M.; Israili, Z.H. Toxoplasmosis–a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS ONE 2014, 9, e90203. [Google Scholar]
- Hunter, C.A.; Sibley, L.D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 2012, 10, 766. [Google Scholar] [CrossRef]
- Khan, A.; Fux, B.; Su, C.; Dubey, J.; Dardé, M.-L.; Ajioka, J.; Rosenthal, B.; Sibley, L. Recent transcontinental sweep of Toxoplasma gondii driven by a single monomorphic chromosome. Proc. Natl. Acad. Sci. USA 2007, 104, 14872–14877. [Google Scholar] [CrossRef] [PubMed]
- Mercier, A.; Devillard, S.; Ngoubangoye, B.; Bonnabau, H.; Bañuls, A.-L.; Durand, P.; Salle, B.; Ajzenberg, D.; Dardé, M.-L. Additional haplogroups of Toxoplasma gondii out of Africa: Population structure and mouse-virulence of strains from Gabon. PLoS Negl. Trop. Dis. 2010, 4, e876. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yolken, R.H. Strain hypothesis of Toxoplasma gondii infection on the outcome of human diseases. Acta Physiol. 2015, 213, 828–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, C. Schizophrenia susceptibility genes directly implicated in the life cycles of pathogens: Cytomegalovirus, influenza, herpes simplex, rubella, and Toxoplasma gondii. Schizophr. Bull. 2008, 35, 1163–1182. [Google Scholar] [CrossRef]
- Dubey, J.P.; Beattie, C. Toxoplasmosis of Animals and Man; CRC Press, Inc: Boca Raton, FL, USA, 1988. [Google Scholar]
- Jacobs, L. Propagation, morphology, and biology of Toxoplasma. Ann. New York Acad. Sci. 1956, 64, 154–179. [Google Scholar] [CrossRef]
- Sroka, J. Seroepidemiology of toxoplasmosis in the Lubin region. Ann. Agric. Environ. Med. 2001, 8, 25–32. [Google Scholar]
- Petersen, E.; Vesco, G.; Villari, S.; Buffolano, W. What do we know about risk factors for infection in humans with Toxoplasma gondii and how can we prevent infections? Zoonoses Public Health 2010, 57, 8–17. [Google Scholar] [CrossRef]
- Kolbekova, P.; Kourbatova, E.; Novotna, M.; Kodym, P.; Flegr, J. New and old risk-factors for Toxoplasma gondii infection: Prospective cross-sectional study among military personnel in the Czech Republic. Clin. Microbiol. Infect. 2007, 13, 1012–1017. [Google Scholar] [CrossRef]
- Dubey, J.; Murata, F.; Cerqueira-Cézar, C.; Kwok, O. Epidemiologic and public health significance of Toxoplasma gondii infections in venison: 2009–2020. J. Parasitol. 2021, 107, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Ahmadpour, E.; Rahimi, M.T.; Ghojoghi, A.; Rezaei, F.; Hatam-Nahavandi, K.; Oliveira, S.M.; de Lourdes Pereira, M.; Majidiani, H.; Siyadatpanah, A.; Elhamirad, S. Toxoplasma gondii Infection in Marine Animal Species, as a Potential Source of Food Contamination: A Systematic Review and Meta-Analysis. Acta Parasitol. 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D. Identification of faecal transmission of Toxoplasma gondii: Small science, large characters. Int. J. Parasitol. 2009, 39, 871–875. [Google Scholar] [CrossRef]
- Hafid, J.; Bellete, B.; Flori, P.; Sawadogo, P.; Boyer, Y.; Raberin, H.; Tran Manh Sung, R. Materno-foetal transmission of murine toxoplasmosis after oral infection. Am J Immunol 2005, 1, 1–5. [Google Scholar] [CrossRef]
- Yazar, S.; Eser, B.; Yay, M. Prevalence of anti-toxoplasma Gondii antibodies in Turkish blood donors. Ethiop. Med J. 2006, 44, 257–261. [Google Scholar] [PubMed]
- Fischer, S.A. Infections complicating solid organ transplantation. Surg. Clin. 2006, 86, 1127–1145. [Google Scholar] [CrossRef] [PubMed]
- Fricker-Hidalgo, H.; Bulabois, C.-E.; Brenier-Pinchart, M.-P.; Hamidfar, R.; Garban, F.; Brion, J.-P.; Timsit, J.-F.; Cahn, J.-Y.; Pelloux, H. Diagnosis of toxoplasmosis after allogeneic stem cell transplantation: Results of DNA detection and serological techniques. Clin. Infect. Dis. 2009, 48, e9–e15. [Google Scholar] [CrossRef]
- Edvinsson, B.; Lundquist, J.; Ljungman, P.; RingdÉn, O.; EvengÅrd, B. A prospective study of diagnosis of Toxoplasma gondii infection after bone marrow transplantation. Apmis 2008, 116, 345–351. [Google Scholar] [CrossRef]
- Laibe, S.; Ranque, S.; Curtillet, C.; Faraut, F.; Dumon, H.; Franck, J. Timely diagnosis of disseminated toxoplasmosis by sputum examination. J. Clin. Microbiol. 2006, 44, 646–648. [Google Scholar] [CrossRef]
- Arantes, T.P.; Lopes, W.D.Z.; Ferreira, R.M.; Pieroni, J.S.P.; Pinto, V.M.; Sakamoto, C.A.; da Costa, A.J. Toxoplasma gondii: Evidence for the transmission by semen in dogs. Exp. Parasitol. 2009, 123, 190–194. [Google Scholar] [CrossRef]
- Safarpour, H.; Cevik, M.; Zarean, M.; Barac, A.; Hatam-Nahavandi, K.; Rahimi, M.T.; Baghi, H.B.; Koshki, T.J.; Pagheh, A.S.; Shahrivar, F. Global status of Toxoplasma gondii infection and associated risk factors in people living with HIV. Aids 2020, 34, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Elsheikha, H. Congenital toxoplasmosis: Priorities for further health promotion action. Public Health 2008, 122, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Lambert, H.; Hitziger, N.; Dellacasa, I.; Svensson, M.; Barragan, A. Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination. Cell. Microbiol. 2006, 8, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Persson, C.M.; Lambert, H.; Vutova, P.P.; Dellacasa-Lindberg, I.; Nederby, J.; Yagita, H.; Ljunggren, H.-G.; Grandien, A.; Barragan, A.; Chambers, B.J. Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect. Immun. 2009, 77, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Sultana, M.A.; Du, A.; Carow, B.; Angbjär, C.M.; Weidner, J.M.; Kanatani, S.; Fuks, J.M.; Muliaditan, T.; James, J.; Mansfield, I.O. Downmodulation of effector functions in NK cells upon Toxoplasma gondii infection. Infect. Immun. 2017, 85, e00069-17. [Google Scholar] [CrossRef]
- Sasai, M.; Pradipta, A.; Yamamoto, M. Host immune responses to Toxoplasma gondii. Int. Immunol. 2018, 30, 113–119. [Google Scholar] [CrossRef]
- Black, M.W.; Boothroyd, J.C. Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev. 2000, 64, 607–623. [Google Scholar] [CrossRef]
- Ihara, F.; Nishikawa, Y. Toxoplasma gondii manipulates host cell signaling pathways via its secreted effector molecules. Parasitol. Int. 2021, 83, 102368. [Google Scholar] [CrossRef] [PubMed]
- Sibley, L. Intracellular parasite invasion strategies. Science 2004, 304, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Xue, Y.; Quake, S.R.; Boothroyd, J.C. Differential impacts on host transcription by ROP and GRA effectors from the intracellular parasite Toxoplasma gondii. MBio 2020, 11, e00182-20. [Google Scholar] [CrossRef]
- Lopez, J.; Bittame, A.; Massera, C.; Vasseur, V.; Effantin, G.; Valat, A.; Buaillon, C.; Allart, S.; Fox, B.A.; Rommereim, L.M. Intravacuolar membranes regulate CD8 T cell recognition of membrane-bound Toxoplasma gondii protective antigen. Cell Rep. 2015, 13, 2273–2286. [Google Scholar] [CrossRef]
- Butcher, B.A.; Fox, B.A.; Rommereim, L.M.; Kim, S.G.; Maurer, K.J.; Yarovinsky, F.; De’Broski, R.H.; Bzik, D.J.; Denkers, E.Y. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 2011, 7, e1002236. [Google Scholar] [CrossRef]
- Ong, Y.-C.; Reese, M.L.; Boothroyd, J.C. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 2010, 285, 28731–28740. [Google Scholar] [CrossRef]
- Martí i Líndez, A.-A.; Reith, W. Arginine-dependent immune responses. Cell. Mol. Life Sci. 2021, 78, 5303–5324. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Christian, D.A.; Kochanowsky, J.A.; Phan, A.T.; Clark, J.T.; Wang, S.; Berry, C.; Oh, J.; Chen, X.; Roos, D.S. The Toxoplasma gondii virulence factor ROP16 acts in cis and trans, and suppresses T cell responses. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.D.; Wang, Y.; Wojno, E.D.T.; Shastri, A.J.; Hu, K.; Cornel, L.; Boedec, E.; Ong, Y.-C.; Chien, Y.-h.; Hunter, C.A. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 2011, 9, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Standley, D.M.; Takashima, S.; Saiga, H.; Okuyama, M.; Kayama, H.; Kubo, E.; Ito, H.; Takaura, M.; Matsuda, T. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 2009, 206, 2747–2760. [Google Scholar] [CrossRef]
- Butcher, B.A.; Kim, L.; Panopoulos, A.D.; Watowich, S.S.; Murray, P.J.; Denkers, E.Y. Cutting edge: IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-α in host macrophages. J. Immunol. 2005, 174, 3148–3152. [Google Scholar] [CrossRef]
- Saeij, J.; Coller, S.; Boyle, J.; Jerome, M.; White, M.; Boothroyd, J. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 2007, 445, 324. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.D.; Hu, K.; Whitmarsh, R.J.; Hassan, M.A.; Julien, L.; Lu, D.; Chen, L.; Hunter, C.A.; Saeij, J.P. Toxoplasma rhoptry kinase ROP16 promotes host resistance to oral infection and intestinal inflammation only in the context of the dense granule protein GRA15. Infect. Immun. 2013, 81, 2156–2167. [Google Scholar] [CrossRef]
- Schneider, A.G.; Abdallah, D.S.A.; Butcher, B.A.; Denkers, E.Y. Toxoplasma gondii triggers phosphorylation and nuclear translocation of dendritic cell STAT1 while simultaneously blocking IFNγ-induced STAT1 transcriptional activity. PLoS ONE 2013, 8, e60215. [Google Scholar] [CrossRef]
- Lang, C.; Hildebrandt, A.; Brand, F.; Opitz, L.; Dihazi, H.; Lüder, C.G. Impaired chromatin remodelling at STAT1-regulated promoters leads to global unresponsiveness of Toxoplasma gondii-infected macrophages to IFN-γ. PLoS Pathog. 2012, 8, e1002483. [Google Scholar] [CrossRef]
- Sturge, C.R.; Yarovinsky, F. Complex immune cell interplay in the IFN-γ response during Toxoplasma gondii infection. Infect. Immun. 2014, IAI, 01714–01722. [Google Scholar]
- Brasil, T.R.; Freire-de-Lima, C.G.; Morrot, A.; Vetö Arnholdt, A.C. Host-Toxoplasma gondii Coadaptation Leads to Fine Tuning of the immune Response. Front. Immunol. 2017, 8, 1080. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K.; Fouts, A.E.; Boothroyd, J.C. Toxoplasma gondii dysregulates IFN-γ-inducible gene expression in human fibroblasts: Insights from a genome-wide transcriptional profiling. J. Immunol. 2007, 178, 5154–5165. [Google Scholar] [CrossRef] [PubMed]
- Rosowski, E.E.; Nguyen, Q.P.; Camejo, A.; Spooner, E.; Saeij, J.P. Toxoplasma gondii inhibits gamma interferon (IFN-γ)-and IFN-β-induced host cell STAT1 transcriptional activity by increasing the association of STAT1 with DNA. Infect. Immun. 2014, 82, 706–719. [Google Scholar] [CrossRef] [Green Version]
- Dupont, C.D.; Christian, D.A.; Hunter, C.A. Immune response and immunopathology during toxoplasmosis. In Proceedings of Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022; pp. 793–813. [Google Scholar]
- Machado, F.S.; Aliberti, J. Impact of lipoxin-mediated regulation on immune response to infectious disease. Immunol. Res. 2006, 35, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S.; Sasai, M.; Ohshima, J.; Lee, Y.; Bando, H.; Takeda, K.; Yamamoto, M. Selective and strain-specific NFAT4 activation by the Toxoplasma gondii polymorphic dense granule protein GRA6. J. Exp. Med. 2014, 211, 2013–2032. [Google Scholar] [CrossRef]
- Morgado, P.; Sudarshana, D.M.; Gov, L.; Harker, K.S.; Lam, T.; Casali, P.; Boyle, J.P.; Lodoen, M.B. Type II Toxoplasma gondii induction of CD40 on infected macrophages enhances IL-12 responses. Infect. Immun. 2014, IAI, 01614–01615. [Google Scholar]
- Laliberte, J.; Carruthers, V.B. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell. Mol. Life Sci. 2008, 65, 1900–1915. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Arranz-Solís, D.; Saeij, J.P. Influence of the host and parasite strain on the immune response during Toxoplasma infection. Front. Cell. Infect. Microbiol. 2020, 10, 580425. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Raouf-Rahmati, A.; Ansar, A.-R.; Rezaee, S.A.; Hosseini, S.M.; Garweg, J.G.; Ghezeldasht, S.A.; Vaghei, S.; Zarean, M.; Shamsian, S.A.; Moghaddas, E. Local and systemic gene expression levels of IL-10, IL-17 and TGF-β in active ocular toxoplasmosis in humans. Cytokine 2021, 146, 155643. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.A.; Zaki, M.S.A.; Al-Shraim, M.; Eleawa, S.M.; El-Kott, A.F.; Al-Hashem, F.H.; Eldeen, M.A.; Ibrahim, H.; Aldera, H.; Alkhateeb, M.A. Subacute ghrelin administration inhibits apoptosis and improves ultrastructural abnormalities in remote myocardium post-myocardial infarction. Biomed. Pharmacother. 2018, 101, 920–928. [Google Scholar] [CrossRef]
- Poon, I.K.; Parkes, M.A.; Jiang, L.; Atkin-Smith, G.K.; Tixeira, R.; Gregory, C.D.; Ozkocak, D.C.; Rutter, S.F.; Caruso, S.; Santavanond, J.P. Moving beyond size and phosphatidylserine exposure: Evidence for a diversity of apoptotic cell-derived extracellular vesicles in vitro. J. Extracell. Vesicles 2019, 8, 1608786. [Google Scholar] [CrossRef]
- Naeini, M.B.; Bianconi, V.; Pirro, M.; Sahebkar, A. The role of phosphatidylserine recognition receptors in multiple biological functions. Cell. Mol. Biol. Lett. 2020, 25, 1–17. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, T.A.T.; de Araújo Portes, J.; Damasceno-Sá, J.C.; Caldas, L.A.; de Souza, W.; DaMatta, R.A.; Seabra, S.H. Phosphatidylserine exposure by Toxoplasma gondii is fundamental to balance the immune response granting survival of the parasite and of the host. PLoS ONE 2011, 6, e27867. [Google Scholar] [CrossRef]
- Vestal, D.J.; Jeyaratnam, J.A. The guanylate-binding proteins: Emerging insights into the biochemical properties and functions of this family of large interferon-induced guanosine triphosphatase. J. Interferon Cytokine Res. 2011, 31, 89–97. [Google Scholar] [CrossRef]
- Li, H.; Yao, X.-Q.; Grant, B.J. Comparative structural dynamic analysis of GTPases. PLoS Comput. Biol. 2018, 14, e1006364. [Google Scholar] [CrossRef] [PubMed]
- Heussler, V.T.; Küenzi, P.; Rottenberg, S. Inhibition of Apoptosis by Intracellular Protozoan Parasites; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Sacks, D.; Sher, A. Evasion of innate immunity by parasitic protozoa. Nat. Immunol. 2002, 3, 1041. [Google Scholar] [CrossRef]
- Mordue, D.G.; Monroy, F.; La Regina, M.; Dinarello, C.A.; Sibley, L.D. Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J. Immunol. 2001, 167, 4574–4584. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135. [Google Scholar] [CrossRef]
- Ha, H.C.; Snyder, S.H. Poly (ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef] [PubMed]
- Van Cruchten, S.; Van Den Broeck, W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol. 2002, 31, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279. [Google Scholar] [CrossRef]
- Rock, K.L.; Lai, J.J.; Kono, H. Innate and adaptive immune responses to cell death. Immunol. Rev. 2011, 243, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Dunay, I.R.; Fuchs, A.; Sibley, L.D. Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect. Immun. 2010, 78, 1564–1570. [Google Scholar] [CrossRef]
- Lüder, C.G.; Gross, U.; Lopes, M.F. Intracellular protozoan parasites and apoptosis: Diverse strategies to modulate parasite–host interactions. Trends Parasitol. 2001, 17, 480–486. [Google Scholar] [CrossRef]
- Lamberton, P.; Donnelly, C.; Webster, J. Specificity of the Toxoplasma gondii-altered behaviour to definitive versus non-definitive host predation risk. Parasitology 2008, 135, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Besteiro, S. Toxoplasma control of host apoptosis: The art of not biting too hard the hand that feeds you. Microb. Cell 2015, 2, 178. [Google Scholar] [CrossRef]
- Gaskell, E.A.; Smith, J.E.; Pinney, J.W.; Westhead, D.R.; McConkey, G.A. A unique dual activity amino acid hydroxylase in Toxoplasma gondii. PLoS ONE 2009, 4, e4801. [Google Scholar] [CrossRef]
- el Kouni, M.H. Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol. Ther. 2003, 99, 283–309. [Google Scholar] [CrossRef]
- Kato, K. How does Toxoplama gondii invade host cells? J. Vet. Med Sci. 2018, 18–0344. [Google Scholar] [CrossRef]
- Saeij, J.P.; Boyle, J.P.; Boothroyd, J.C. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 2005, 21, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Payne, T.M.; Molestina, R.E.; Sinai, A.P. Inhibition of caspase activation and a requirement for NF-κB function in the Toxoplasma gondii-mediated blockade of host apoptosis. J. Cell Sci. 2003, 116, 4345–4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Liu, T.; Zhang, A.; Huo, X.; Luo, Q.; Chen, Z.; Yu, L.; Li, Q.; Liu, L.; Lun, Z.-r. Reactive oxygen species-triggered trophoblast apoptosis is initiated by endoplasmic reticulum stress via activation of caspase-12, CHOP, and the JNK pathway in Toxoplasma gondii infection in mice. Infect. Immun. 2012, 80, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Gao, S.; Jiang, L.; Shang, L.; Men, J.; Wang, Z.; Zhai, Y.; Xia, Z.; Hu, R.; Zhang, X. A recombinant pseudorabies virus expressing TgSAG1 protects against challenge with the virulent Toxoplasma gondii RH strain and pseudorabies in BALB/c mice. Microbes Infect. 2008, 10, 1355–1362. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Wang, Y.; Gadahi, J.A.; Xie, Q.; Xu, L.; Yan, R.; Song, X.; Li, X. Toxoplasma gondii excretory/secretory antigens (TgESAs) suppress pro-inflammatory cytokine secretion by inhibiting TLR-induced NF-κB activation in LPS-stimulated murine macrophages. Oncotarget 2017, 8, 88351. [Google Scholar] [CrossRef] [PubMed]
- Goebel, S.; Gross, U.; Lüder, C.G. Inhibition of host cell apoptosis by Toxoplasma gondii is accompanied by reduced activation of the caspase cascade and alterations of poly (ADP-ribose) polymerase expression. J. Cell Sci. 2001, 114, 3495–3505. [Google Scholar] [CrossRef]
- Wan, L.; Gong, L.; Wang, W.; An, R.; Zheng, M.; Jiang, Z.; Tang, Y.; Zhang, Y.; Chen, H.; Yu, L.T. gondii rhoptry protein ROP18 induces apoptosis of neural cells via endoplasmic reticulum stress pathway. Parasites Vectors 2015, 8, 554. [Google Scholar] [CrossRef] [PubMed]
- Molestina, R.E.; Payne, T.M.; Coppens, I.; Sinai, A.P. Activation of NF-κB by Toxoplasma gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated IκB to the parasitophorous vacuole membrane. J. Cell Sci. 2003, 116, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Nash, P.B.; Purner, M.B.; Leon, R.P.; Clarke, P.; Duke, R.C.; Curiel, T.J. Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J. Immunol. 1998, 160, 1824–1830. [Google Scholar] [CrossRef]
- Dotiwala, F.; Mulik, S.; Polidoro, R.B.; Ansara, J.A.; Burleigh, B.A.; Walch, M.; Gazzinelli, R.T.; Lieberman, J. Killer lymphocytes use granulysin, perforin and granzymes to kill intracellular parasites. Nat. Med. 2016, 22, 210. [Google Scholar] [CrossRef]
- Goping, S.; Barry, M.; Liston, P.; Sawchuk, T.; Constantinescu, G.; Michalak, K.M.; Shostak, I.; Roberts, D.L.; Hunter, A.M.; Korneluk, R. Granzyme B-induced apoptosis requires both direct caspase activation and relief of caspase inhibition. Immunity 2003, 18, 355–365. [Google Scholar] [CrossRef]
- Yamada, T.; Tomita, T.; Weiss, L.M.; Orlofsky, A. Toxoplasma gondii inhibits granzyme B-mediated apoptosis by the inhibition of granzyme B function in host cells. Int. J. Parasitol. 2011, 41, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Pang, M.-H.; Ye, X.-H.; Yang, G.; Lin, C. The Toxoplasma gondii ME-49 strain upregulates levels of A20 that inhibit NF-κ B activation and promotes apoptosis in human leukaemia T-cell lines. Parasites Vectors 2018, 11, 305. [Google Scholar] [CrossRef]
- Babaie, F.; Ebrazeh, M.; Hemmatzadeh, M.; Sadat Mohammadi, F.; Gowhari Shabgah, A.; Hajaliloo, M.; Ebrahimi, A.A.; Shirafkan, N.; Azizi, G.; Mohammadi, H.; et al. Association analysis of ERAP1 gene single nucleotide polymorphism in susceptibility to ankylosing spondylitis in Iranian population. Immunol. Lett. 2018. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Ahn, M.-H.; Jun, H.-S.; Jung, J.-W.; Ryu, J.-S.; Min, D.-Y. Toxoplasma gondii inhibits apoptosis in infected cells by caspase inactivation and NF-κB activation. Yonsei Med. J. 2006, 47, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Gavrilescu, L.C.; Denkers, E.Y. Interleukin-12 p40-and Fas ligand-dependent apoptotic pathways involving STAT-1 phosphorylation are triggered during infection with a virulent strain of Toxoplasma gondii. Infect. Immun. 2003, 71, 2577–2583. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Makala, L.; Otsuka, H.; Mikami, T.; Nagasawa, H. Mechanisms of apoptosis in murine fibroblasts by two intracellular protozoan parasites, Toxoplasma gondii and Neospora caninum. Parasite Immunol. 2002, 24, 347–354. [Google Scholar] [CrossRef]
- Karin, M.; Lin, A. NF-κB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Hisaeda, H.; Sakai, T.; Zhang, M.; Maekawa, Y.; Zhang, T.; Nishitani, M.; Ishikawa, H.; Himeno, K. Granule-dependent killing of Toxoplasma gondii by CD8+ T cells. Immunology 2001, 104, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Hisaeda, H.; Sakai, T.; Ishikawa, H.; Maekawa, Y.; Yasutomo, K.; Good, R.A.; Himeno, K. Heat shock protein 65 induced by gammadelta T cells prevents apoptosis of macrophages and contributes to host defense in mice infected with Toxoplasma gondii. J. Immunol. 1997, 159, 2375–2381. [Google Scholar] [CrossRef]
- Saleh, A.; Srinivasula, S.M.; Balkir, L.; Robbins, P.D.; Alnemri, E.S. Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat. Cell Biol. 2000, 2, 476. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.-Q.; Jing, K.-P.; Gao, X.; Li, P.; Huang, R.; Niu, Y.-R.; Yan, S.-Q.; Kong, J.-C.; Yu, C.-Y.; Shi, G. Toxoplasma gondii induces autophagy and apoptosis in human umbilical cord mesenchymal stem cells via downregulation of Mcl− 1. Cell Cycle 2017, 16, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.; Denkers, E.Y. Toxoplasma gondii triggers Gi-dependent PI 3-kinase signaling required for inhibition of host cell apoptosis. J. Cell Sci. 2006, 119, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.-H.; Cha, G.-H.; Zhou, W.; Chu, J.-Q.; Nishikawa, Y.; Lee, Y.-H. Involvement of PI 3 kinase/Akt-dependent Bad phosphorylation in Toxoplasma gondii-mediated inhibition of host cell apoptosis. Exp. Parasitol. 2013, 133, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.W.; Ferrel, A.; Naor, A.; Tenborg, E.; Lorenzi, H.A.; Boothroyd, J.C. Translocation of dense granule effectors across the parasitophorous vacuole membrane in Toxoplasma-infected cells requires the activity of ROP17, a rhoptry protein kinase. MSphere 2019, 4, e00276-19. [Google Scholar] [CrossRef]
- El Hajj, H.; Lebrun, M.; Fourmaux, M.N.; Vial, H.; Dubremetz, J.F. Inverted topology of the Toxoplasma gondii ROP5 rhoptry protein provides new insights into the association of the ROP2 protein family with the parasitophorous vacuole membrane. Cell. Microbiol. 2007, 9, 54–64. [Google Scholar] [CrossRef]
- El Hajj, H.; Lebrun, M.; Arold, S.T.; Vial, H.; Labesse, G.; Dubremetz, J.F. ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathog. 2007, 3, e14. [Google Scholar] [CrossRef]
- Taylor, S.; Barragan, A.; Su, C.; Fux, B.; Fentress, S.; Tang, K.; Beatty, W.; El Hajj, H.; Jerome, M.; Behnke, M. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 2006, 314, 1776–1780. [Google Scholar] [CrossRef]
- Saeij, J.; Boyle, J.; Coller, S.; Taylor, S.; Sibley, L.; Brooke-Powell, E.; Ajioka, J.; Boothroyd, J. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 2006, 314, 1780–1783. [Google Scholar] [CrossRef]
- Kemp, L.E.; Yamamoto, M.; Soldati-Favre, D. Subversion of host cellular functions by the apicomplexan parasites. FEMS Microbiol. Rev. 2013, 37, 607–631. [Google Scholar] [CrossRef] [PubMed]
- Steinfeldt, T.; Könen-Waisman, S.; Tong, L.; Pawlowski, N.; Lamkemeyer, T.; Sibley, L.D.; Hunn, J.P.; Howard, J.C. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 2010, 8, e1000576. [Google Scholar] [CrossRef]
- Fentress, S.J.; Steinfeldt, T.; Howard, J.C.; Sibley, L.D. The arginine-rich N-terminal domain of ROP18 is necessary for vacuole targeting and virulence of Toxoplasma gondii. Cell. Microbiol. 2012, 14, 1921–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.J.; Ahn, H.-J.; Nam, H.-W. Anti-apoptotic effects of SERPIN B3 and B4 via STAT6 activation in macrophages after infection with Toxoplasma gondii. Korean J. Parasitol. 2012, 50, 1. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chen, H.; Mo, X.; Tang, Y.; Xu, X.; Zhang, A.; Lun, Z.; Lu, F.; Wang, Y.; Shen, J. Toxoplasma gondii inhibits apoptosis via a novel STAT3-miR-17–92-Bim pathway in macrophages. Cell. Signal. 2014, 26, 1204–1212. [Google Scholar] [CrossRef]
- Duszenko, M.; Ginger, M.L.; Brennand, A.; Gualdrón-López, M.; Colombo, M.I.; Coombs, G.H.; Coppens, I.; Jayabalasingham, B.; Langsley, G.; Lisboa de Castro, S. Autophagy in protists. Autophagy 2011, 7, 127–158. [Google Scholar] [CrossRef]
- Saeij, J.P.; Frickel, E.-M. Exposing Toxoplasma gondii hiding inside the vacuole: A role for GBPs, autophagy and host cell death. Curr. Opin. Microbiol. 2017, 40, 72–80. [Google Scholar] [CrossRef]
- Choi, J.; Park, S.; Biering, S.B.; Selleck, E.; Liu, C.Y.; Zhang, X.; Fujita, N.; Saitoh, T.; Akira, S.; Yoshimori, T. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity 2014, 40, 924–935. [Google Scholar] [CrossRef]
- Fentress, S.J.; Behnke, M.S.; Dunay, I.R.; Mashayekhi, M.; Rommereim, L.M.; Fox, B.A.; Bzik, D.J.; Taylor, G.A.; Turk, B.E.; Lichti, C.F. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 2010, 8, 484–495. [Google Scholar] [CrossRef]
- Jensen, K.D.; Camejo, A.; Melo, M.B.; Cordeiro, C.; Julien, L.; Grotenbreg, G.M.; Frickel, E.-M.; Ploegh, H.L.; Young, L.; Saeij, J.P. Toxoplasma gondii superinfection and virulence during secondary infection correlate with the exact ROP5/ROP18 allelic combination. MBio 2015, 6, e02280-14. [Google Scholar] [CrossRef] [PubMed]
- Suwanti, L.T.; Mufasirin, M. Increased apoptosis skull of pups born to Toxoplasma gondii-infected mice associated with increased expression of interferon gamma, but not tumor necrosis factor alfa. Afr. J. Infect. Dis. 2018, 12, 68–71. [Google Scholar] [CrossRef]
- Yao, Y.; Liu, M.; Ren, C.; Shen, J.; Ji, Y. Exogenous tumor necrosis factor-alpha could induce egress of Toxoplasma gondii from human foreskin fibroblast cells. Parasite 2017, 24. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Kong, L.; Zhou, L.-J.; Wu, S.-Z.; Yao, L.-J.; He, C.; He, C.Y.; Peng, H.-J. Genome-Wide Bimolecular Fluorescence Complementation-Based Proteomic Analysis of Toxoplasma gondii ROP18’s Human Interactome Shows Its Key Role in Regulation of Cell Immunity and Apoptosis. Front. Immunol. 2018, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subauste, C.S. Autophagy in immunity against Toxoplasma gondii. In Autophagy in Infection and Immunity; Springer: Berlin/Heidelberg, Germany, 2009; pp. 251–265. [Google Scholar]
- Niedelman, W.; Gold, D.A.; Rosowski, E.E.; Sprokholt, J.K.; Lim, D.; Arenas, A.F.; Melo, M.B.; Spooner, E.; Yaffe, M.B.; Saeij, J.P. The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog. 2012, 8, e1002784. [Google Scholar] [CrossRef]
- Winter, S.V.; Niedelman, W.; Jensen, K.D.; Rosowski, E.E.; Julien, L.; Spooner, E.; Caradonna, K.; Burleigh, B.A.; Saeij, J.P.; Ploegh, H.L. Determinants of GBP recruitment to Toxoplasma gondii vacuoles and the parasitic factors that control it. PLoS ONE 2011, 6, e24434. [Google Scholar]
- Etheridge, R.D.; Alaganan, A.; Tang, K.; Lou, H.J.; Turk, B.E.; Sibley, L.D. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe 2014, 15, 537–550. [Google Scholar] [CrossRef]
- Lüder, C.G.; Walter, W.; Beuerle, B.; Maeurer, M.J.; Gross, U. Toxoplasma gondii down-regulates MHC class II gene expression and antigen presentation by murine macrophages via interference with nuclear translocation of STAT1α. Eur. J. Immunol. 2001, 31, 1475–1484. [Google Scholar] [CrossRef]
- Haldar, A.K.; Foltz, C.; Finethy, R.; Piro, A.S.; Feeley, E.M.; Pilla-Moffett, D.M.; Komatsu, M.; Frickel, E.-M.; Coers, J. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc. Natl. Acad. Sci. 2015, 112, E5628–E5637. [Google Scholar] [CrossRef]
- Liu, E.; Van Grol, J.; Subauste, C.S. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-γ production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015, 17, 275–284. [Google Scholar] [CrossRef]
- Adeyemi, O.S.; Murata, Y.; Sugi, T.; Han, Y.; Kato, K. Modulation of host HIF-1α activity and the tryptophan pathway contributes to the anti-Toxoplasma gondii potential of nanoparticles. Biochem. Biophys. Rep. 2017, 11, 84–92. [Google Scholar] [CrossRef]
- Lee, Y.; Sasai, M.; Ma, J.S.; Sakaguchi, N.; Ohshima, J.; Bando, H.; Saitoh, T.; Akira, S.; Yamamoto, M. p62 plays a specific role in interferon-γ-induced presentation of a toxoplasma vacuolar antigen. Cell Rep. 2015, 13, 223–233. [Google Scholar] [CrossRef]
- Clough, B.; Wright, J.D.; Pereira, P.M.; Hirst, E.M.; Johnston, A.C.; Henriques, R.; Frickel, E.-M. K63-linked ubiquitination targets Toxoplasma gondii for endo-lysosomal destruction in IFNγ-stimulated human cells. PLoS Pathog. 2016, 12, e1006027. [Google Scholar] [CrossRef] [PubMed]
- Foltz, C.; Napolitano, A.; Khan, R.; Clough, B.; Hirst, E.M.; Frickel, E.-M. TRIM21 is critical for survival of Toxoplasma gondii infection and localises to GBP-positive parasite vacuoles. Sci. Rep. 2017, 7, 5209. [Google Scholar] [CrossRef] [PubMed]
- Slobodkin, M.R.; Elazar, Z. The Atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013, 55, 51–64. [Google Scholar] [PubMed] [Green Version]
- Andrade, R.M.; Wessendarp, M.; Gubbels, M.-J.; Striepen, B.; Subauste, C.S. CD40 induces macrophage anti–Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Investig. 2006, 116, 2366–2377. [Google Scholar] [CrossRef]
- Zhao, Y.O.; Khaminets, A.; Hunn, J.P.; Howard, J.C. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNγ-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 2009, 5, e1000288. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohusmi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Selleck, E.M.; Orchard, R.C.; Lassen, K.G.; Beatty, W.L.; Xavier, R.J.; Levine, B.; Virgin, H.W.; Sibley, L.D. A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-γ-activated human cells. MBio 2015, 6, e01157-15. [Google Scholar] [CrossRef]
- Haldar, A.K.; Piro, A.S.; Pilla, D.M.; Yamamoto, M.; Coers, J. The E2-like conjugation enzyme Atg3 promotes binding of IRG and Gbp proteins to Chlamydia-and Toxoplasma-containing vacuoles and host resistance. PLoS ONE 2014, 9, e86684. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, J.; Lee, Y.; Sasai, M.; Saitoh, T.; Ma, J.S.; Kamiyama, N.; Matsuura, Y.; Pann-Ghill, S.; Hayashi, M.; Ebisu, S. Role of mouse and human autophagy proteins in IFN-γ–induced cell-autonomous responses against Toxoplasma gondii. J. Immunol. 2014, 1302822. [Google Scholar] [CrossRef]
- Selleck, E.M.; Fentress, S.J.; Beatty, W.L.; Degrandi, D.; Pfeffer, K.; Virgin IV, H.W.; MacMicking, J.D.; Sibley, L.D. Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii. PLoS Pathog. 2013, 9, e1003320. [Google Scholar] [CrossRef] [PubMed]
- Sasai, M.; Sakaguchi, N.; Ma, J.S.; Nakamura, S.; Kawabata, T.; Bando, H.; Lee, Y.; Saitoh, T.; Akira, S.; Iwasaki, A. Essential role for GABARAP autophagy proteins in interferon-inducible GTPase-mediated host defense. Nat. Immunol. 2017, 18, 899. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Bean, C.; Boothroyd, J.C.; Scorrano, L. Mitochondria restrict growth of the intracellular parasite toxoplasma gondii by limiting its uptake of fatty acids. Cell Metab. 2018, 27, 886–897.e884. [Google Scholar] [CrossRef]
- Wacker, R.; Eickel, N.; Schmuckli-Maurer, J.; Annoura, T.; Niklaus, L.; Khan, S.M.; Guan, J.L.; Heussler, V.T. LC3-association with the parasitophorous vacuole membrane of Plasmodium berghei liver stages follows a noncanonical autophagy pathway. Cell. Microbiol. 2017, 19, e12754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.-c.; Devenish, R.J. LC3-associated phagocytosis (LAP): Connections with host autophagy. Cells 2012, 1, 396–408. [Google Scholar] [CrossRef]
- Mehta, P.; Henault, J.; Kolbeck, R.; Sanjuan, M.A. Noncanonical autophagy: One small step for LC3, one giant leap for immunity. Curr. Opin. Immunol. 2014, 26, 69–75. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef]
- Traver, M.K.; Henry, S.C.; Cantillana, V.; Oliver, T.; Hunn, J.P.; Howard, J.C.; Beer, S.; Pfeffer, K.; Coers, J.; Taylor, G.A. Immunity-related GTPase M (IRGM) proteins influence the localization of guanylate-binding protein 2 (GBP2) by modulating macroautophagy. J. Biol. Chem. 2011, 286, 30471–30480. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.C.; Hunn, J.P.; Steinfeldt, T. The IRG protein-based resistance mechanism in mice and its relation to virulence in Toxoplasma gondii. Curr. Opin. Microbiol. 2011, 14, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Muniz-Feliciano, L.; Van Grol, J.; Portillo, J.-A.C.; Liew, L.; Liu, B.; Carlin, C.R.; Carruthers, V.B.; Matthews, S.; Subauste, C.S. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog. 2013, 9, e1003809. [Google Scholar] [CrossRef]
- Portillo, J.-A.C.; Muniz-Feliciano, L.; Corcino, Y.L.; Lee, S.J.; Van Grol, J.; Parsons, S.J.; Schiemman, W.P.; Subauste, C.S. Toxoplasma gondii induces FAK-Src-STAT3 signaling during infection of host cells that prevents parasite targeting by autophagy. PLoS Pathog. 2017, 13, e1006671. [Google Scholar] [CrossRef]
- Carruthers, V.B.; Giddings, O.K.; Sibley, L.D. Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell. Microbiol. 1999, 1, 225–235. [Google Scholar] [CrossRef]
- Carruthers, V.; Boothroyd, J.C. Pulling together: An integrated model of Toxoplasma cell invasion. Curr. Opin. Microbiol. 2007, 10, 83–89. [Google Scholar] [CrossRef]
- Koshy, A.A.; Fouts, A.E.; Lodoen, M.B.; Alkan, O.; Blau, H.M.; Boothroyd, J.C. Toxoplasma secreting Cre recombinase for analysis of host-parasite interactions. Nat. Methods 2010, 7, 307. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhang, F.; Chen, H.; Tang, Y.; Xing, T.; Luo, Q.; Yu, L.; Du, J.; Shen, J.; Zhang, L. Toxoplasma gondii dense granule protein 15 induces apoptosis in choriocarcinoma JEG-3 cells through endoplasmic reticulum stress. Parasites Vectors 2018, 11, 251. [Google Scholar] [CrossRef]
- Koshy, A.A.; Dietrich, H.K.; Christian, D.A.; Melehani, J.H.; Shastri, A.J.; Hunter, C.A.; Boothroyd, J.C. Toxoplasma co-opts host cells it does not invade. PLoS Pathog. 2012, 8, e1002825. [Google Scholar] [CrossRef]
- Menard, K.L.; Haskins, B.E.; Denkers, E.Y. Impact of Toxoplasma gondii infection on host non-coding RNA responses. Front. Cell. Infect. Microbiol. 2019, 9, 132. [Google Scholar] [CrossRef]
- Blader, I.J.; Koshy, A.A. Toxoplasma Development of Its Replicative Niche: In Its Host Cell and Beyond. Eukaryot. Cell 2014, 13, 965–976. [Google Scholar] [CrossRef]
- Lima, T.S.; Lodoen, M.B. Mechanisms of human innate immune evasion by Toxoplasma gondii. Front. Cell. Infect. Microbiol. 2019, 9, 103. [Google Scholar] [CrossRef]
- Kawabe, T.; Matsushima, M.; Hashimoto, N.; Imaizumi, K.; Hasegawa, Y. CD40/CD40 ligand interactions in immune responses and pulmonary immunity. Nagoya J. Med Sci. 2011, 73, 69. [Google Scholar] [PubMed]
- Mahmoudzadeh, S.; Nozad Charoudeh, H.; Marques, C.S.; Bahadory, S.; Ahmadpour, E. The role of IL-12 in stimulating NK cells against Toxoplasma gondii infection: A mini-review. Parasitol. Res. 2021, 120, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Niedelman, W.; Sprokholt, J.K.; Clough, B.; Frickel, E.-M.; Saeij, J.P. Cell death of interferon-gamma stimulated human fibroblasts upon Toxoplasma gondii infection induces early parasite egress and limits parasite replication. Infect. Immun. 2013, IAI, 00413–00416. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.L.; Carlin, J.M.; Pyati, P.; Dai, W.; Pfefferkorn, E.R.; Murphy, M. Antiparasitic and antiproliferative effects of indoleamine 2, 3-dioxygenase enzyme expression in human fibroblasts. Infect. Immun. 1994, 62, 2277–2284. [Google Scholar] [CrossRef]
- Fasshauer, V.; Gross, U.; Bohne, W. The parasitophorous vacuole membrane of Encephalitozoon cuniculi lacks host cell membrane proteins immediately after invasion. Eukaryot. Cell 2005, 4, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.M.; Boulter, N.R.; Fuller, S.J.; Zakrzewski, A.M.; Lees, M.P.; Saunders, B.M.; Wiley, J.S.; Smith, N.C. The role of the P2X7 receptor in infectious diseases. PLoS Pathog. 2011, 7, e1002212. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol. 2017, 12, 103. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 2016, 213, 617–629. [Google Scholar] [CrossRef]
- Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 2011, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Aachoui, Y.; Sagulenko, V.; Miao, E.A.; Stacey, K.J. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 2013, 16, 319–326. [Google Scholar] [CrossRef]
- Ewald, S.E.; Chavarria-Smith, J.; Boothroyd, J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014, 82, 460–468. [Google Scholar] [CrossRef]
- Cirelli, K.M.; Gorfu, G.; Hassan, M.A.; Printz, M.; Crown, D.; Leppla, S.H.; Grigg, M.E.; Saeij, J.P.; Moayeri, M. Inflammasome sensor NLRP1 controls rat macrophage susceptibility to Toxoplasma gondii. PLoS Pathog. 2014, 10, e1003927. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, S.; Spotin, A.; Mahami-Oskouei, M.; Baradaran, B.; Babaie, F.; Azadi, Y.; Alizadeh, P.; Valadan, R.; Barac, A.; Ahmadpour, E. Toxoplasma gondii activates NLRP12 inflammasome pathway in the BALB/c murine model. Acta Trop. 2022, 225, 106202. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.-Q.; Shi, G.; Fan, Y.-M.; Choi, I.-W.; Cha, G.-H.; Zhou, Y.; Lee, Y.-H.; Quan, J.-H. Production of IL-1β and inflammasome with up-regulated expressions of NOD-like receptor related genes in Toxoplasma gondii-infected THP-1 macrophages. Korean J. Parasitol. 2016, 54, 711. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhu, J.; Cao, Y.; Shen, J.; Yu, L. Insight into inflammasome signaling: Implications for Toxoplasma gondii infection. Front. Immunol. 2020, 11, 583193. [Google Scholar] [CrossRef]
- Butcher, B.A.; Greene, R.I.; Henry, S.C.; Annecharico, K.L.; Weinberg, J.B.; Denkers, E.Y.; Sher, A.; Taylor, G.A. p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infect. Immun. 2005, 73, 3278–3286. [Google Scholar] [CrossRef]
- Martens, S.; Parvanova, I.; Zerrahn, J.; Griffiths, G.; Schell, G.; Reichmann, G.; Howard, J.C. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005, 1, e24. [Google Scholar] [CrossRef]
- Andrade, W.A.; do Carmo Souza, M.; Ramos-Martinez, E.; Nagpal, K.; Dutra, M.S.; Melo, M.B.; Bartholomeu, D.C.; Ghosh, S.; Golenbock, D.T.; Gazzinelli, R.T. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 2013, 13, 42–53. [Google Scholar] [CrossRef]
- Murray, P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Witola, W.H.; Mui, E.; Hargrave, A.; Liu, S.; Hypolite, M.; Montpetit, A.; Cavailles, P.; Bisanz, C.; Cesbron-Delauw, M.-F.; Fournié, G.J. NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect. Immun. 2011, 79, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Gov, L.; Karimzadeh, A.; Ueno, N.; Lodoen, M.B. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. MBio 2013, 4, e00255-13. [Google Scholar] [CrossRef]
- Cavaillès, P.; Sergent, V.; Bisanz, C.; Papapietro, O.; Colacios, C.; Mas, M.; Subra, J.-F.; Lagrange, D.; Calise, M.; Appolinaire, S. The rat Toxo1 locus directs toxoplasmosis outcome and controls parasite proliferation and spreading by macrophage-dependent mechanisms. Proc. Natl. Acad. Sci. USA 2006, 103, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Kappler, J.; Kotzin, B.L. Autoimmune disease: Why and where it occurs. Nat. Med. 2001, 7, 899–905. [Google Scholar] [CrossRef]
- Hosseininejad, Z.; Sharif, M.; Sarvi, S.; Amouei, A.; Hosseini, S.A.; Nayeri Chegeni, T.; Anvari, D.; Saberi, R.; Gohardehi, S.; Mizani, A. Toxoplasmosis seroprevalence in rheumatoid arthritis patients: A systematic review and meta-analysis. PLoS Negl. Trop. Dis. 2018, 12, e0006545. [Google Scholar] [CrossRef]
- Jamal, Q.W.; Abdulrahman, T.R.; Al-Marsomy, H.D.; AL-Bassam, W.W. The role of Toxoplasma gondii in Trigger an Autoimmune Disease. Infection 2021, 3, 5. [Google Scholar]
- Prandota, J. Possible critical role of latent chronic Toxoplasma gondii infection in triggering, development and persistence of autoimmune diseases. Int. J. Neurol. Res. 2018, 4, 379–463. [Google Scholar] [CrossRef]
- Melo, M.B.; Jensen, K.D.; Saeij, J.P. Toxoplasma gondii effectors are master regulators of the inflammatory response. Trends Parasitol. 2011, 27, 487–495. [Google Scholar] [CrossRef]
- Mahoney, J.A.; Rosen, A. Apoptosis and autoimmunity. Curr. Opin. Immunol. 2005, 17, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Lu, F.; Xie, C.; Ding, H.; Shen, Y.; Gao, Y.; Lu, S.; Zhuo, X. Toxoplasma gondii infection induces cell apoptosis via multiple pathways revealed by transcriptome analysis. J. Zhejiang Univ.-Sci. 2022, 23, 315–327. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmadpour, E.; Babaie, F.; Kazemi, T.; Mehrani Moghaddam, S.; Moghimi, A.; Hosseinzadeh, R.; Nissapatorn, V.; Pagheh, A.S. Overview of Apoptosis, Autophagy, and Inflammatory Processes in Toxoplasma gondii Infected Cells. Pathogens 2023, 12, 253. https://doi.org/10.3390/pathogens12020253
Ahmadpour E, Babaie F, Kazemi T, Mehrani Moghaddam S, Moghimi A, Hosseinzadeh R, Nissapatorn V, Pagheh AS. Overview of Apoptosis, Autophagy, and Inflammatory Processes in Toxoplasma gondii Infected Cells. Pathogens. 2023; 12(2):253. https://doi.org/10.3390/pathogens12020253
Chicago/Turabian StyleAhmadpour, Ehsan, Farhad Babaie, Tohid Kazemi, Sirous Mehrani Moghaddam, Ata Moghimi, Ramin Hosseinzadeh, Veeranoot Nissapatorn, and Abdol Sattar Pagheh. 2023. "Overview of Apoptosis, Autophagy, and Inflammatory Processes in Toxoplasma gondii Infected Cells" Pathogens 12, no. 2: 253. https://doi.org/10.3390/pathogens12020253