The Diversity, Composition, and Putative Functions of Gill-Associated Bacteria of Bathymodiolin Mussel and Vesicomyid Clam from Haima Cold Seep, South China Sea

Abstract
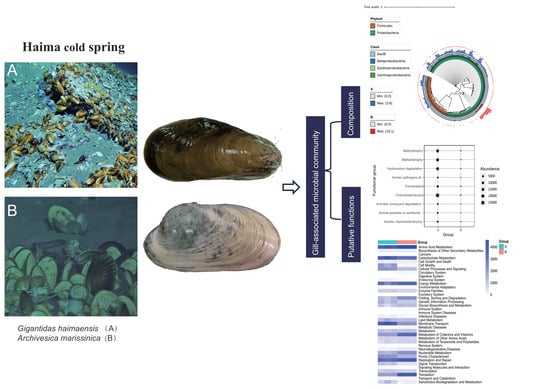
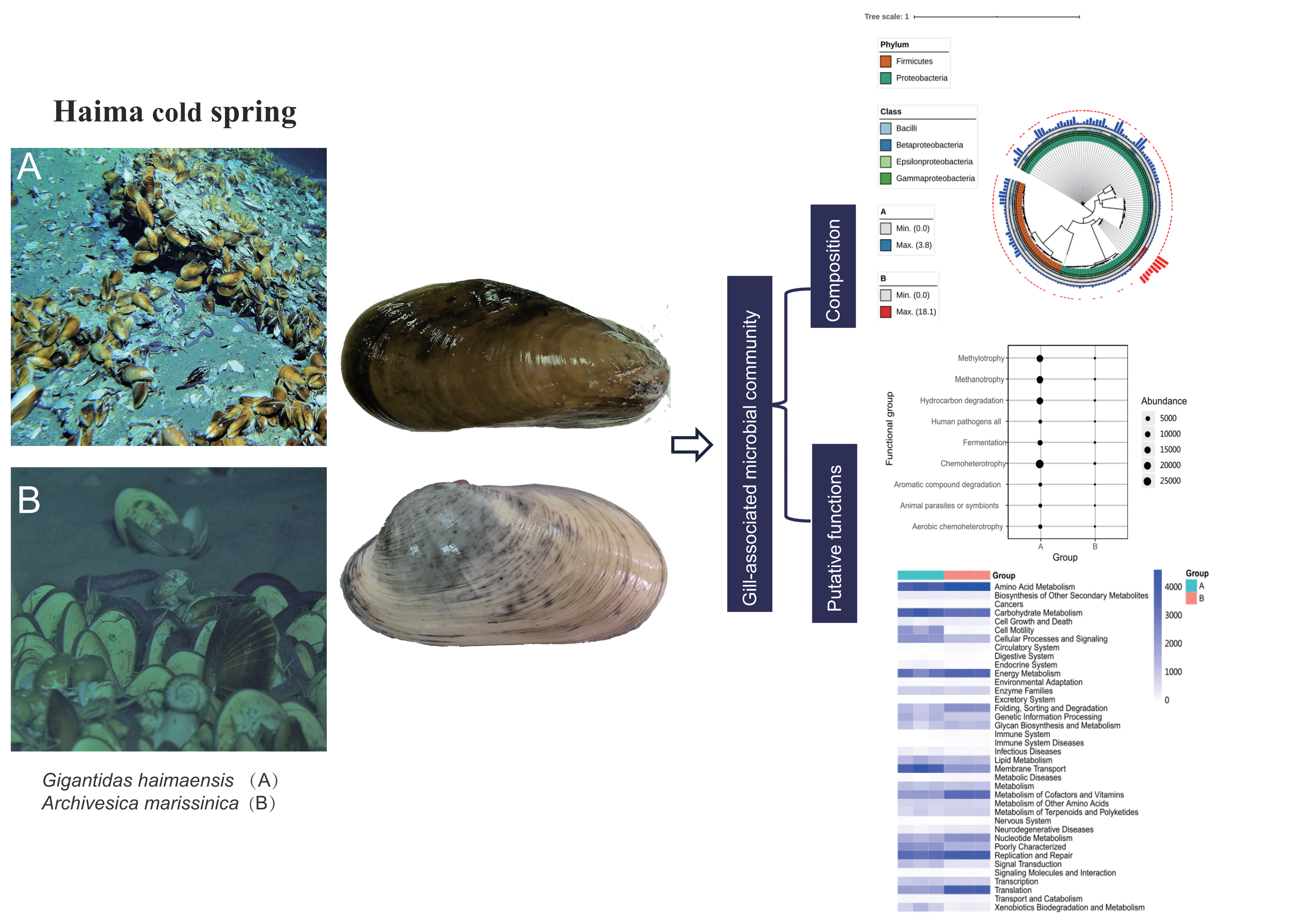
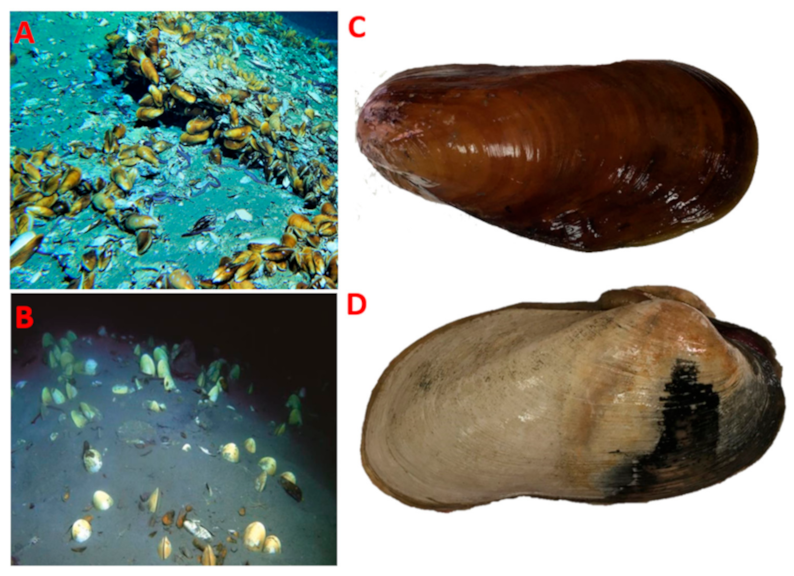
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction, PCR Amplification, and 16S rDNA Amplicon Sequencing
2.3. Sequence Analysis
2.4. Statistical Analysis
2.5. FAPROTAX Analysis
2.6. PICRUSt Analysis
3. Results
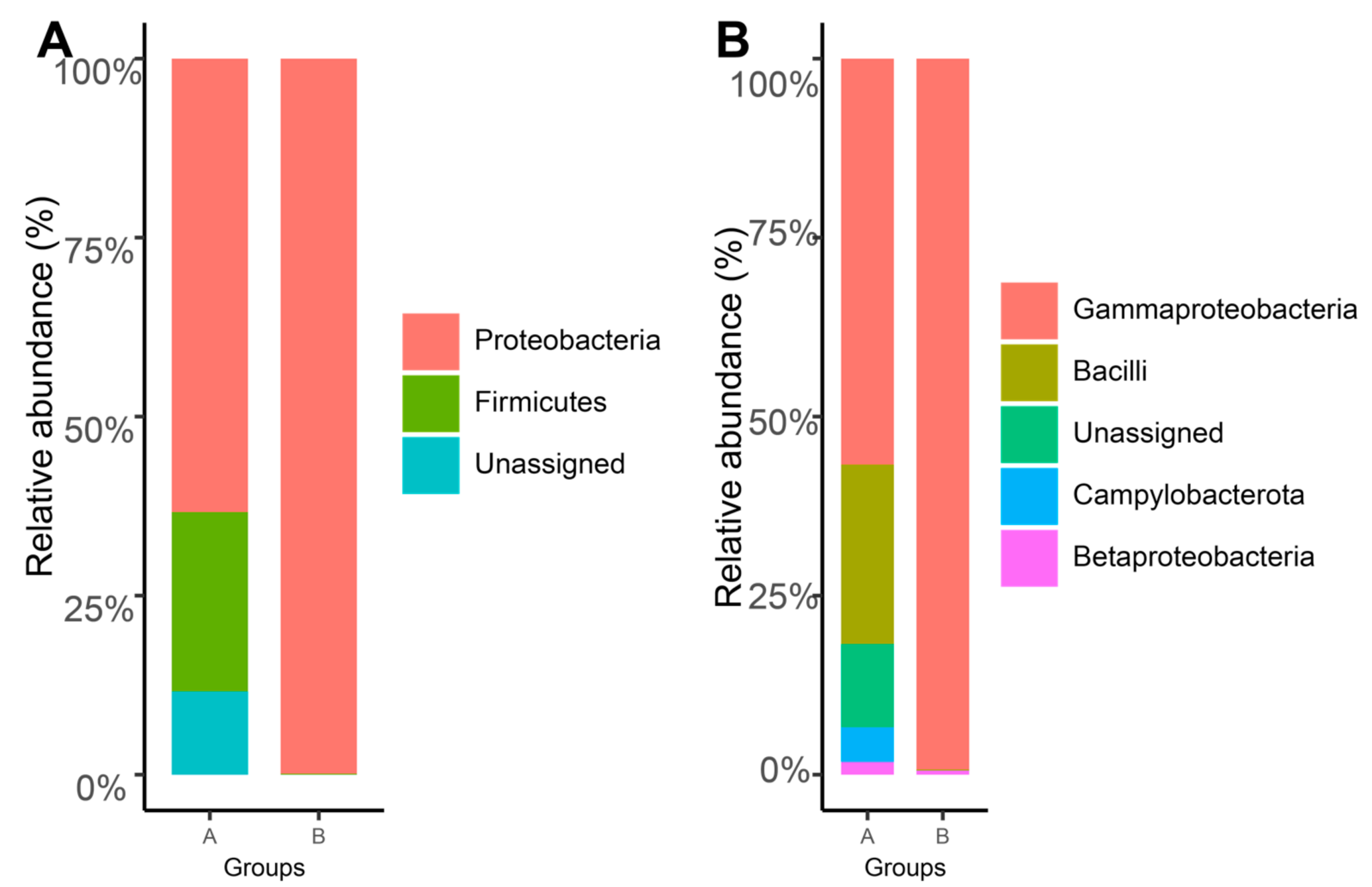
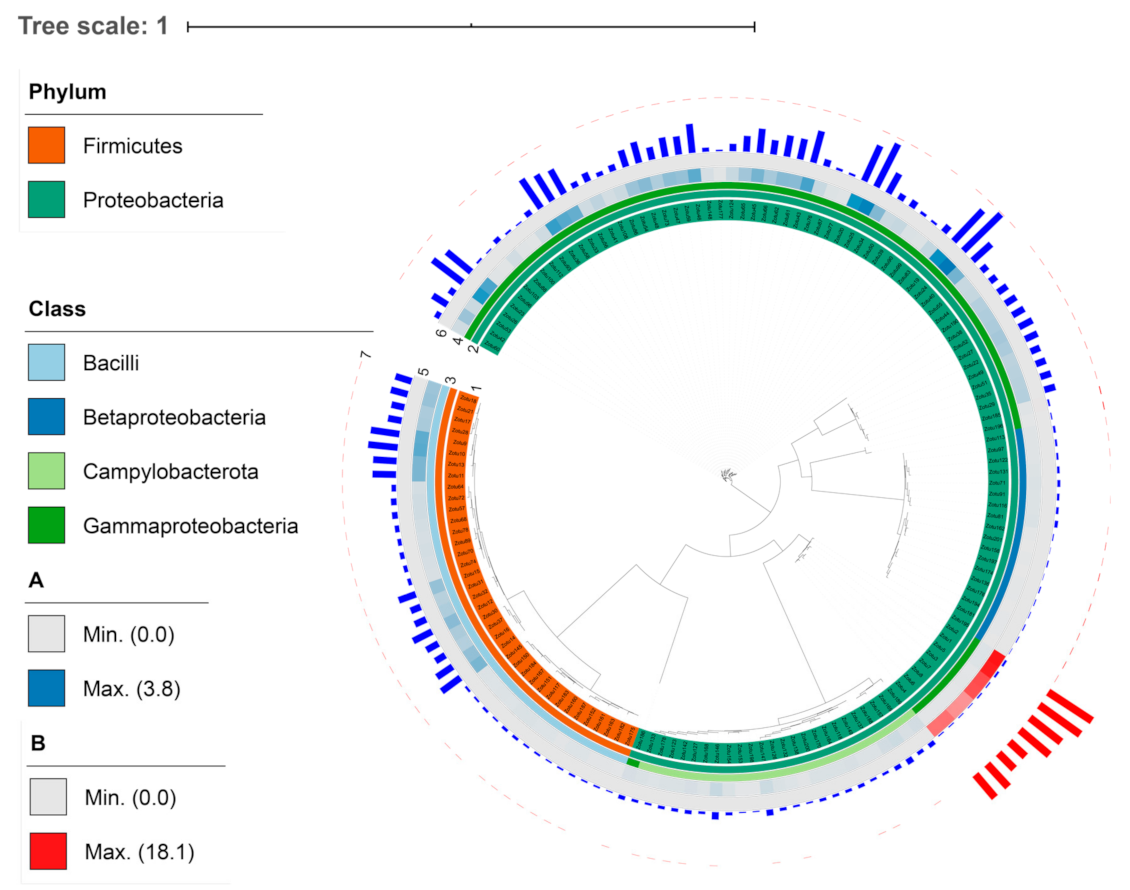
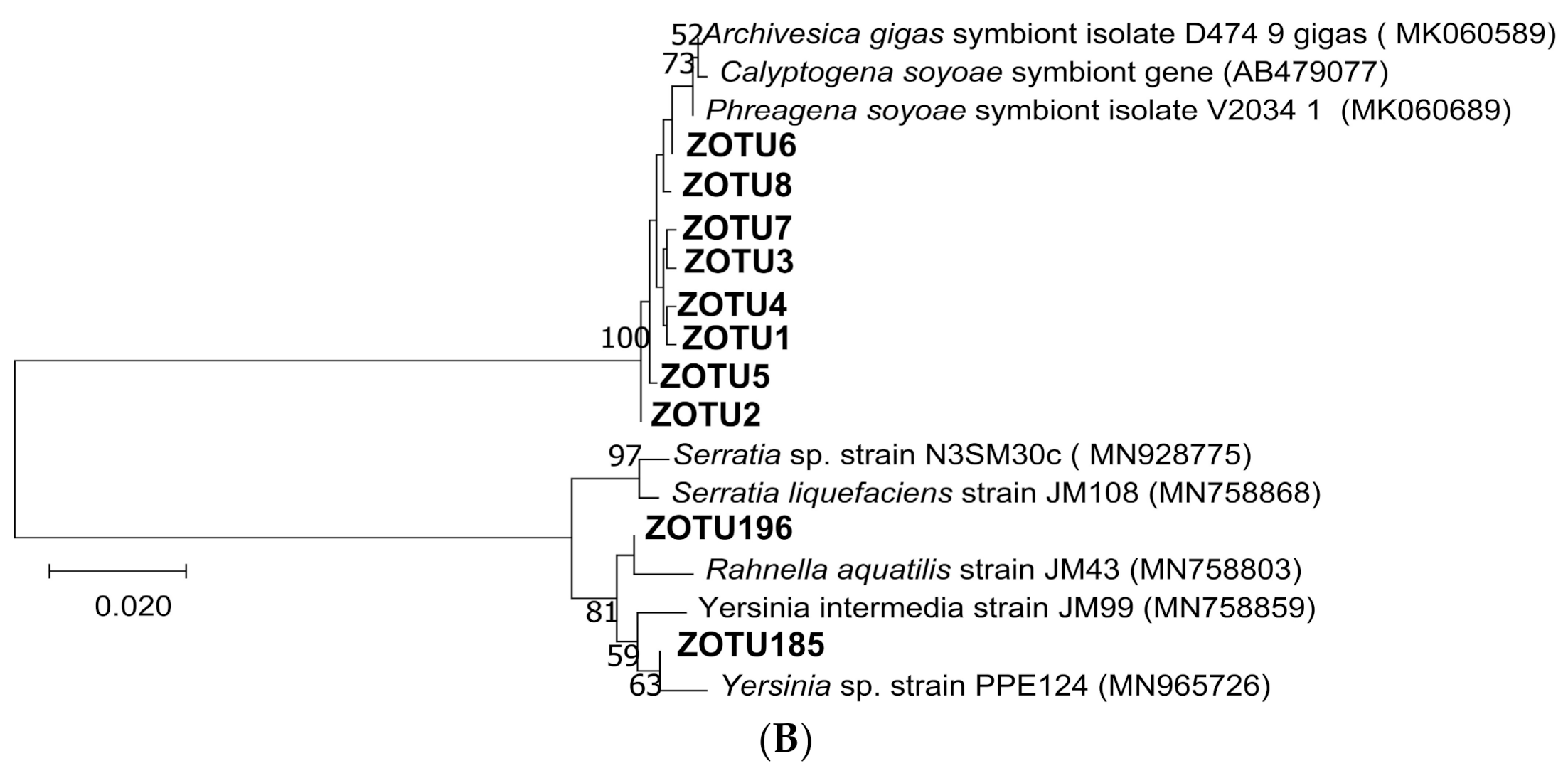
3.1. The Taxonomic and Phylogenetic Composition of the Gill-Associated Microbial Communities
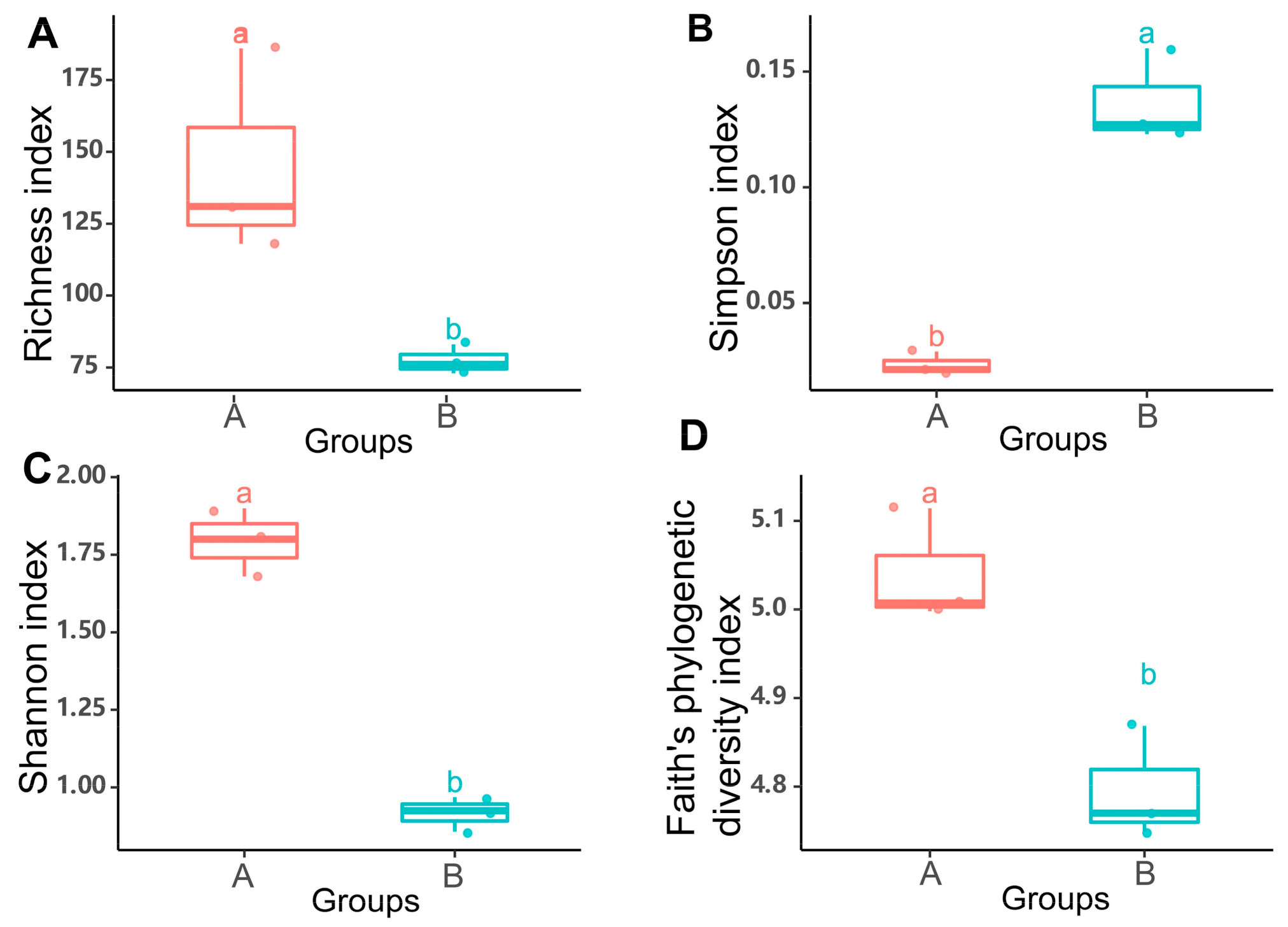
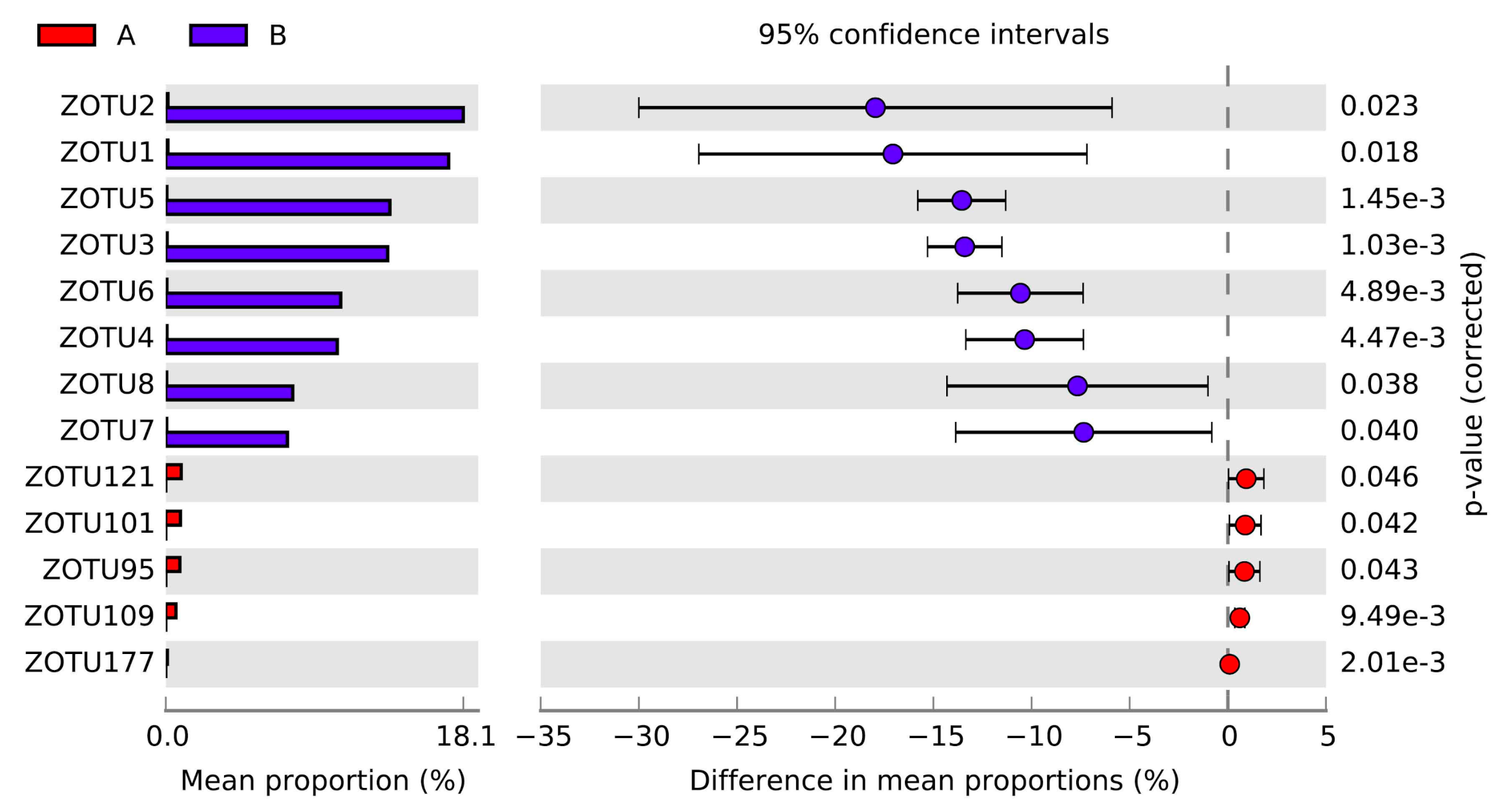
3.2. Variations of the Microbial Communities in G. haimaensis and A. marissinica
3.3. Biogeochemical Process Profiling Predicted by FAPROTAX Database
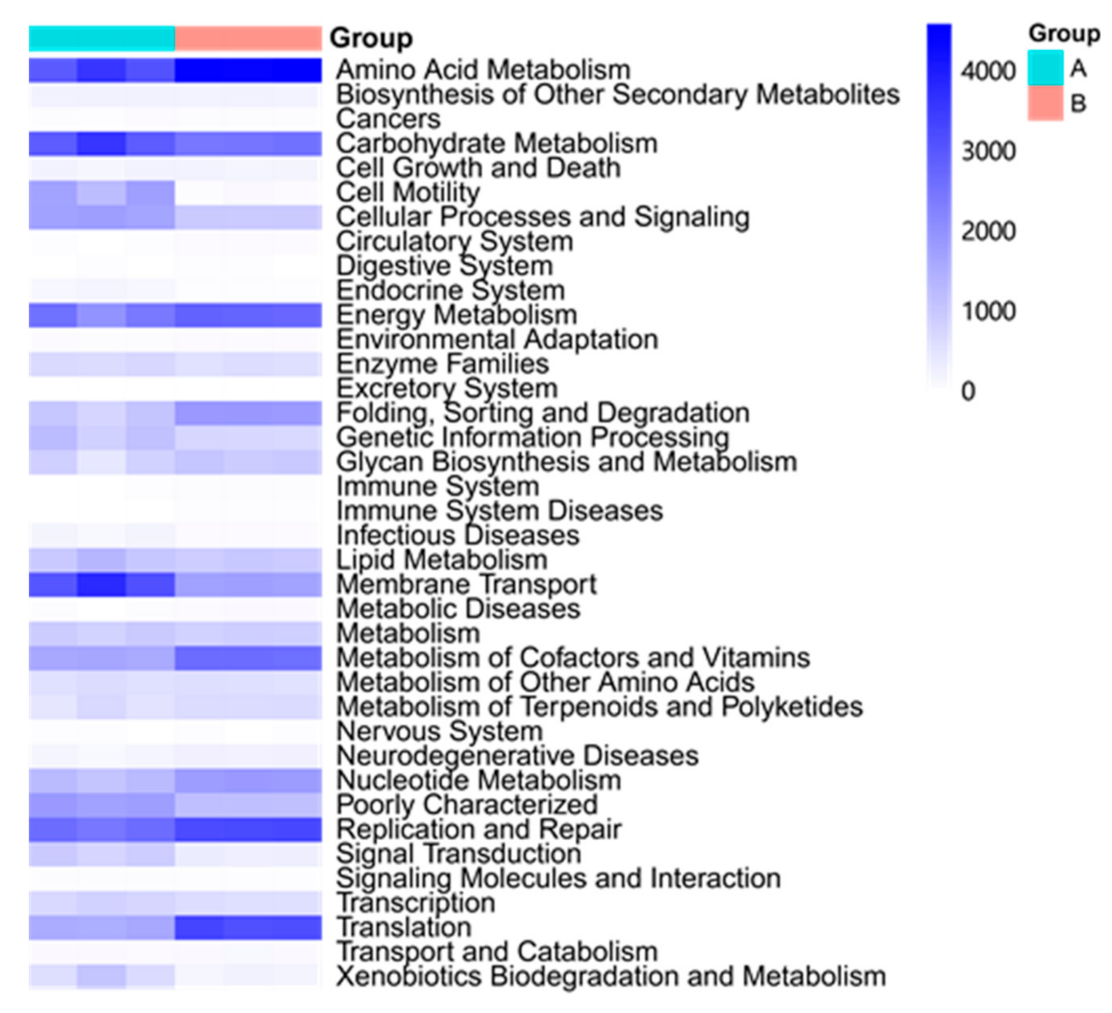
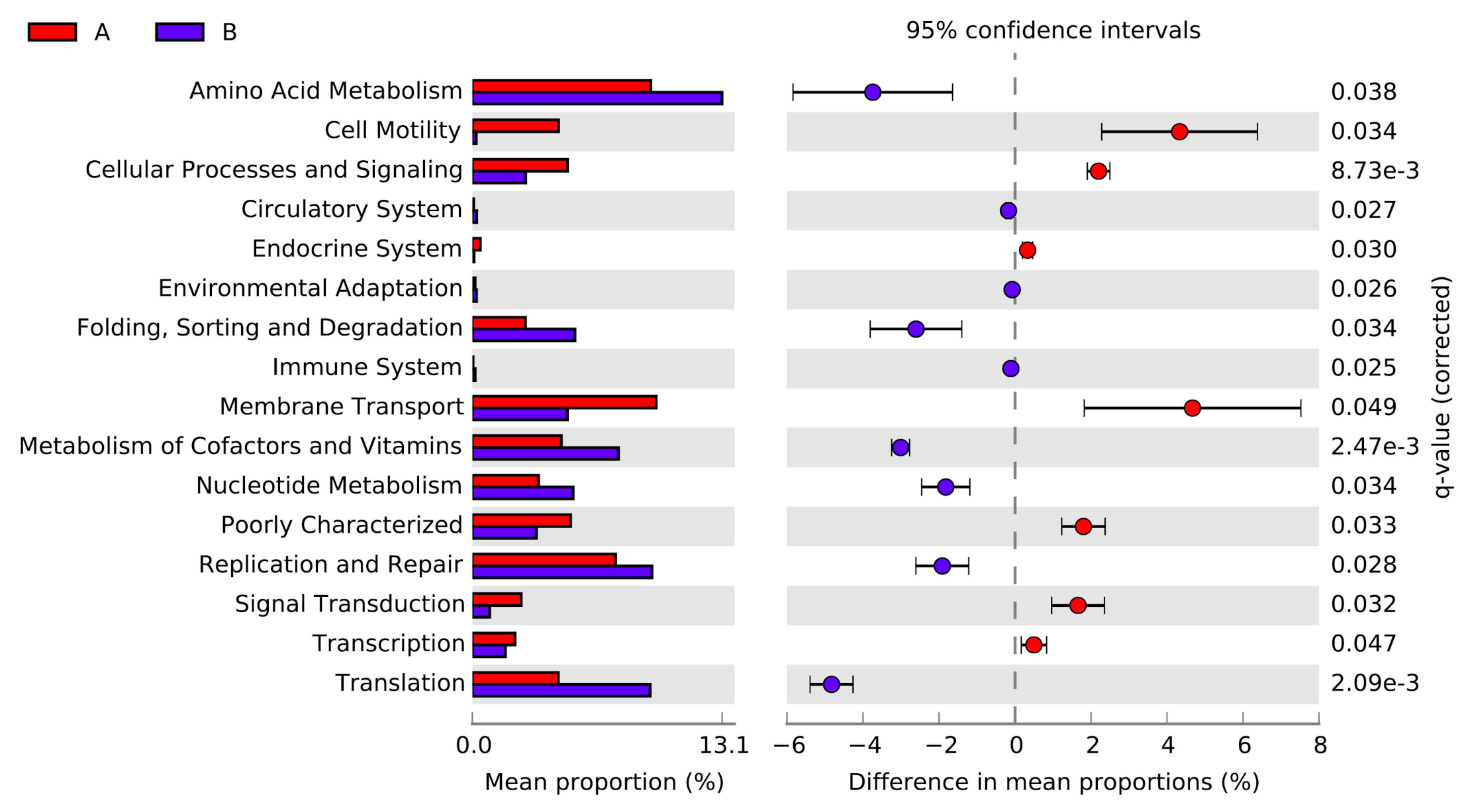
3.4. Potential Functional Profiling Analyzed by PICRUSt Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suess, E. Marine Cold Seeps: Background and Recent Advances. In Hydrocarbons, Oils and Lipids: Diversity, Origin, Chemistry and Fate; Springer: Cham, Switzerland, 2018; pp. 1–21. [Google Scholar] [CrossRef]
- Feng, D.; Qiu, J.-W.; Hu, Y.; Peckmann, J.; Guan, H.; Tong, H.; Chen, C.; Chen, J.; Gong, S.; Li, N.; et al. Cold seep systems in the South China Sea: An overview. J. Asian Earth Sci. 2018, 168, 3–16. [Google Scholar] [CrossRef]
- Chen, D.F.; Huang, Y.Y.; Yuan, X.L.; Cathles, L.M. Seep carbonates and preserved methane oxidizing archaea and sulfate reducing bacteria fossils suggest recent gas venting on the seafloor in the Northeastern South China Sea. Mar. Pet. Geol. 2005, 22, 613–621. [Google Scholar] [CrossRef]
- Guan, H.; Birgel, D.; Peckmann, J.; Liang, Q.; Feng, D.; Yang, S.; Liang, J.; Tao, J.; Wu, N.; Chen, D. Lipid biomarker patterns of authigenic carbonates reveal fluid composition and seepage intensity at Haima cold seeps, South China Sea. J. Asian Earth Sci 2018, 168, 163–172. [Google Scholar] [CrossRef]
- Boetius, A.; Ravenschlag, K.; Schubert, C.J.; Rickert, D.; Widdel, F.; Gieseke, A.; Amann, R.; Jorgensen, B.B.; Witte, U.; Pfannkuche, O. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 2000, 407, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Paull, C.K.; Heacker, B.; Commerau, R.; Freeman-Lynde, R.P.; Neumann, C.; Corso, W.P.; Golubic, S.; Hook, J.E.; Sikes, E.; Curray, J. Biological Communities at the Florida Escarpment Resemble Hydrothermal Vent Taxa. Science 1984, 226, 965–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duperron, S.; Lorion, J.; Samadi, S.; Gros, O.; Gaill, F. Symbioses between deep-sea mussels (Mytilidae: Bathymodiolinae) and chemosynthetic bacteria: Diversity, function and evolution. C. R. Biol. 2009, 332, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Xu, T.; Zhang, Y.; Mu, H.; Zhang, Y.; Lan, Y.; Fields, C.J.; Hui, J.H.L.; Zhang, W.; et al. Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes. Nat. Ecol. Evol. 2017, 1, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wang, M.; Liu, B.; Yue, X.; Li, C. Gill symbionts of the cold-seep mussel Bathymodiolus platifrons: Composition, environmental dependency and immune control. Fish. Shellfish Immunol. 2019, 86, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Fan, X.; Zhuang, G.; Liang, Q.; Wang, F. Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea. FEMS Microbiol. Ecol. 2017, 93, fix101. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Zhang, J.; Wang, M.; Cao, L.; Du, Z.; Sun, Y.; Liu, S.; Li, C.; Sun, L. High-Throughput Sequencing Reveals a Potentially Novel Sulfurovum Species Dominating the Microbial Communities of the Seawater–Sediment Interface of a Deep-Sea Cold Seep in South China Sea. Microorganisms 2020, 8, 687. [Google Scholar] [CrossRef]
- Waite, D.W.; Vanwonterghem, I.; Rinke, C.; Parks, D.H.; Zhang, Y.; Takai, K.; Sievert, S.M.; Simon, J.; Campbell, B.J.; Hanson, T.E.; et al. Addendum: Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 2018, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Duperron, S.; Bergin, C.; Zielinski, F.; Blazejak, A.; Pernthaler, A.; McKiness, Z.P.; DeChaine, E.; Cavanaugh, C.M.; Dubilier, N. A dual symbiosis shared by two mussel species, Bathymodiolus azoricus and Bathymodiolus puteoserpentis (Bivalvia: Mytilidae), from hydrothermal vents along the northern Mid-Atlantic Ridge. Environ. Microbiol. 2006, 8, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Ponnudurai, R.; Kleiner, M.; Sayavedra, L.; Petersen, J.M.; Moche, M.; Otto, A.; Becher, D.; Takeuchi, T.; Satoh, N.; Dubilier, N.; et al. Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J. 2017, 11, 463–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansorge, R.; Romano, S.; Sayavedra, L.; Kupczok, A.; Tegetmeyer, H.E.; Dubilier, N.; Petersen, J. Diversity matters: Deep-sea mussels harbor multiple symbiont strains. Nat. Microbiol. 2019, 4, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Feng, D.; Tao, J.; Qiu, J.-W. A new species of deep-sea mussel (Bivalvia: Mytilidae: Gigantidas) from the South China Sea: Morphology, phylogenetic position, and gill-associated microbes. Deep Sea Res. Part I Oceanogr. Res. Pap. 2019, 146, 79–90. [Google Scholar] [CrossRef]
- Fisher, C.R.; Brooks, J.M.; Vodenichar, J.S.; Zande, J.M.; Childress, J.J.; Burke, R.A., Jr. The Co-occurrence of Methanotrophic and Chemoautotrophic Sulfur-Oxidizing Bacterial Symbionts in a Deep-sea Mussel. Mar. Ecol. 1993, 14, 277–289. [Google Scholar] [CrossRef]
- Duperron, S.; Gros, O. Colwellia and sulfur-oxidizing bacteria: An unusual dual symbiosis in a Terua mussel (Mytilidae: Bathymodiolinae) from whale falls in the Antilles arc. Deep Sea Res. Part I Oceanogr. Res. Pap. 2016, 115, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.; Sun, J.; Zhang, W.; Xu, T.; Zhang, Y.; Chen, C.; Feng, D.; Wang, H.; Tao, J.; Qiu, J.-W.; et al. Host–Symbiont Interactions in Deep-Sea Chemosymbiotic Vesicomyid Clams: Insights From Transcriptome Sequencing. Front. Mar. Sci. 2019, 6, 680. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Okutani, T.; Liang, Q.; Qiu, J.-W. A Noteworthy New Species of the Family Vesicomyidae from the South China Sea (Bivalvia: Glossoidea). Venus 2018, 76, 29–37. [Google Scholar]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive Earth’s biogeochemical cycles. Science 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [Green Version]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Li, D.; Mi, T.; Zhao, J.; Liu, C.; Sun, C.; Zhen, Y. Characteristics of the archaeal and bacterial communities in core sediments from Southern Yap Trench via in situ sampling by the manned submersible Jiaolong. Sci. Total Environ. 2020, 703, 134884. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Fu, L.; Liu, Q.; Fu, L.; Bi, N.; Yang, Z.; Zhen, Y. Community Structure, Abundance and Potential Functions of Bacteria and Archaea in the Sansha Yongle Blue Hole, Xisha, South China Sea. Front. Microbiol. 2019, 10, 2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Xiao, X.; Zhang, Y. Microbial diversity of sediments from an inactive hydrothermal vent field, Southwest Indian Ridge. Mar. Life Sci. Technol. 2019, 2, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Feng, R.; Xu, M.; Li, J.; Huang, S.; Zhao, G.; Tu, X.; Sun, G.; Guo, J. Structure and predictive functional profiling of microbial communities in two biotrickling filters treated with continuous/discontinuous waste gases. AMB Express 2019, 9, 2. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Du, M.; Li, J.; Zhang, H.; Chen, W.; Wei, J.; Wu, Z.; Zhang, H.; Li, J.; Chen, S.; et al. Spatial distribution of seepages and associated biological communities within Haima cold seep field, South China Sea. J. Sea Res. 2020, 165, 101957. [Google Scholar] [CrossRef]
- Liang, Q.; Hu, Y.; Feng, D.; Peckmann, J.; Chen, L.; Yang, S.; Liang, J.; Tao, J.; Chen, D. Authigenic carbonates from newly discovered active cold seeps on the northwestern slope of the South China Sea: Constraints on fluid sources, formation environments, and seepage dynamics. Deep Sea Res. Part I Oceanogr. Res. Pap. 2017, 124, 31–41. [Google Scholar] [CrossRef]
- Fang, Y.; Wei, J.; Lu, H.; Liang, J.; Lu, J.a.; Fu, J.; Cao, J. Chemical and structural characteristics of gas hydrates from the Haima cold seeps in the Qiongdongnan Basin of the South China Sea. J. Asian Earth Sci. 2019, 182, 103924. [Google Scholar] [CrossRef]
- Yang, M.; Gong, L.; Sui, J.; Li, X. The complete mitochondrial genome of Calyptogena marissinica (Heterodonta: Veneroida: Vesicomyidae): Insight into the deep-sea adaptive evolution of vesicomyids. PLoS ONE 2019, 14, e0217952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.N., Jr.; Via, L.E. A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques 1993, 14, 748–750. [Google Scholar]
- Liu, Y.X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2020. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences. PeerJ 2018, 6, e4652. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package. R Package Version 2.5-2. 2018. Available online: https://CRAN.R-project.org/package=vegan (accessed on 27 October 2020).
- Zhang, J.; Liu, Y.X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- McGlynn, S.E. Energy Metabolism during Anaerobic Methane Oxidation in ANME Archaea. Microbes Environ. 2017, 32, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, K. Recent Topics on Deep-Sea Microbial Communities in Microbes and Environments. Microbes Environ. 2019, 34, 345–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.Y. Genome Diversity of Spore-Forming Firmicutes. Microbiol Spectr 2013, 1. [Google Scholar] [CrossRef] [Green Version]
- Coykendall, D.K.; Cornman, R.S.; Prouty, N.G.; Brooke, S.; Demopoulos, A.W.J.; Morrison, C.L. Molecular characterization of Bathymodiolus mussels and gill symbionts associated with chemosynthetic habitats from the U.S. Atlantic margin. PLoS ONE 2019, 14, e0211616. [Google Scholar] [CrossRef]
- Ohishi, K.; Yamamoto, M.; Tame, A.; Kusaka, C.; Nagai, Y.; Sugimura, M.; Inoue, K.; Uematsu, K.; Yoshida, T.; Ikuta, T.; et al. Long-term cultivation of the deep-sea clam Calyptogena okutanii: Changes in the abundance ofchemoautotrophic symbiont, elemental sulfur, and mucus. Biol. Bull. 2016, 230, 257–267. [Google Scholar] [CrossRef]
- Zheng, P.; Wang, M.; Li, C.; Sun, X.; Wang, X.; Sun, Y.; Sun, S. Insights into deep-sea adaptations and host-symbiont interactions: A comparative transcriptome study on Bathymodiolus mussels and their coastal relatives. Mol. Ecol. 2017, 26, 5133–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZOTU ID | The Relative Abundance | Phylum | Class | |
|---|---|---|---|---|
| G. haimaensis | A. marissinica | |||
| TOP 10 ZOTUs in G. haimaensis | ||||
| ZOTU24 | 3.83% | 0.00% | Proteobacteria | Gammaproteobacteria |
| ZOTU25 | 3.52% | 0.01% | Proteobacteria | Gammaproteobacteria |
| ZOTU19 | 3.10% | 0.02% | Proteobacteria | Gammaproteobacteria |
| ZOTU20 | 2.83% | 0.02% | Proteobacteria | Gammaproteobacteria |
| ZOTU26 | 2.74% | 0.01% | Proteobacteria | Gammaproteobacteria |
| ZOTU36 | 2.53% | 0.02% | Proteobacteria | Gammaproteobacteria |
| ZOTU43 | 2.38% | 0.02% | Proteobacteria | Gammaproteobacteria |
| ZOTU23 | 2.30% | 0.02% | Proteobacteria | Gammaproteobacteria |
| ZOTU58 | 2.22% | 0.01% | Proteobacteria | Gammaproteobacteria |
| ZOTU10 | 2.17% | 0.01% | Firmicutes | Bacilli |
| TOP 10 ZOTUs in A. marissinica | ||||
| ZOTU2 | 0.16% | 18.07% | Proteobacteria | Gammaproteobacteria |
| ZOTU1 | 0.15% | 17.38% | Proteobacteria | Gammaproteobacteria |
| ZOTU5 | 0.08% | 13.64% | Proteobacteria | Gammaproteobacteria |
| ZOTU3 | 0.10% | 13.31% | Proteobacteria | Gammaproteobacteria |
| ZOTU6 | 0.07% | 10.66% | Proteobacteria | Gammaproteobacteria |
| ZOTU4 | 0.08% | 10.39% | Proteobacteria | Gammaproteobacteria |
| ZOTU8 | 0.06% | 7.61% | Proteobacteria | Gammaproteobacteria |
| ZOTU7 | 0.05% | 7.44% | Proteobacteria | Gammaproteobacteria |
| ZOTU185 | 0.13% | 0.20% | Proteobacteria | Gammaproteobacteria |
| ZOTU196 | 0.08% | 0.16% | Proteobacteria | Gammaproteobacteria |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ling, J.; Guan, H.; Liu, L.; Tao, J.; Li, J.; Dong, J.; Zhang, S. The Diversity, Composition, and Putative Functions of Gill-Associated Bacteria of Bathymodiolin Mussel and Vesicomyid Clam from Haima Cold Seep, South China Sea. Microorganisms 2020, 8, 1699. https://doi.org/10.3390/microorganisms8111699
Ling J, Guan H, Liu L, Tao J, Li J, Dong J, Zhang S. The Diversity, Composition, and Putative Functions of Gill-Associated Bacteria of Bathymodiolin Mussel and Vesicomyid Clam from Haima Cold Seep, South China Sea. Microorganisms. 2020; 8(11):1699. https://doi.org/10.3390/microorganisms8111699
Chicago/Turabian StyleLing, Juan, Hongxiang Guan, Lihua Liu, Jun Tao, Jie Li, Junde Dong, and Si Zhang. 2020. "The Diversity, Composition, and Putative Functions of Gill-Associated Bacteria of Bathymodiolin Mussel and Vesicomyid Clam from Haima Cold Seep, South China Sea" Microorganisms 8, no. 11: 1699. https://doi.org/10.3390/microorganisms8111699