High-Throughput Sequencing Reveals a Potentially Novel Sulfurovum Species Dominating the Microbial Communities of the Seawater–Sediment Interface of a Deep-Sea Cold Seep in South China Sea

 ,
,
Abstract
:
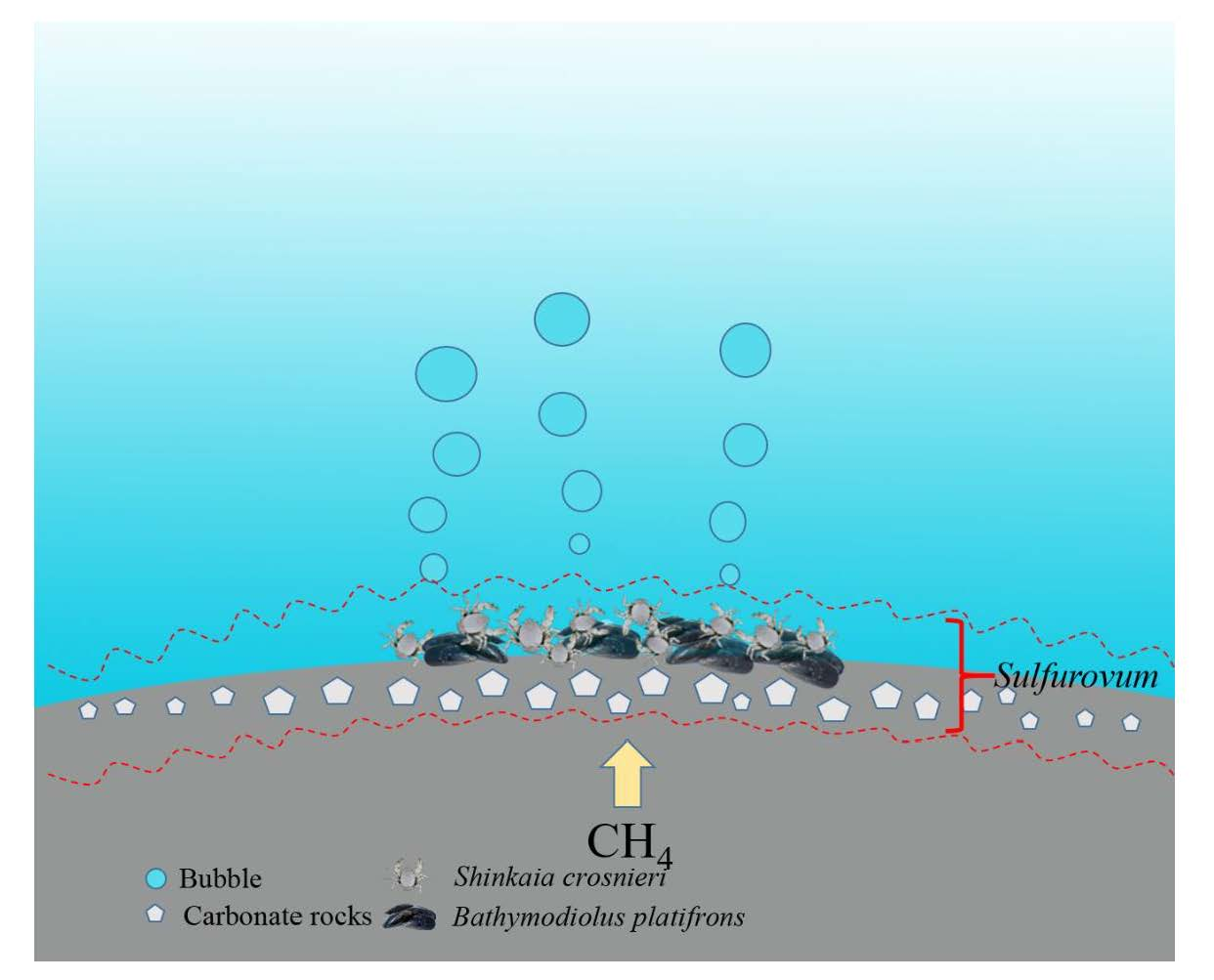{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sites and Sampling
2.2. Chemical Analysis
2.3. NGS Amplicon and Pacbio Amplicon Sequencing
2.4. Amplicon Data Analysis
2.5. Binning Based on Metagenome Data
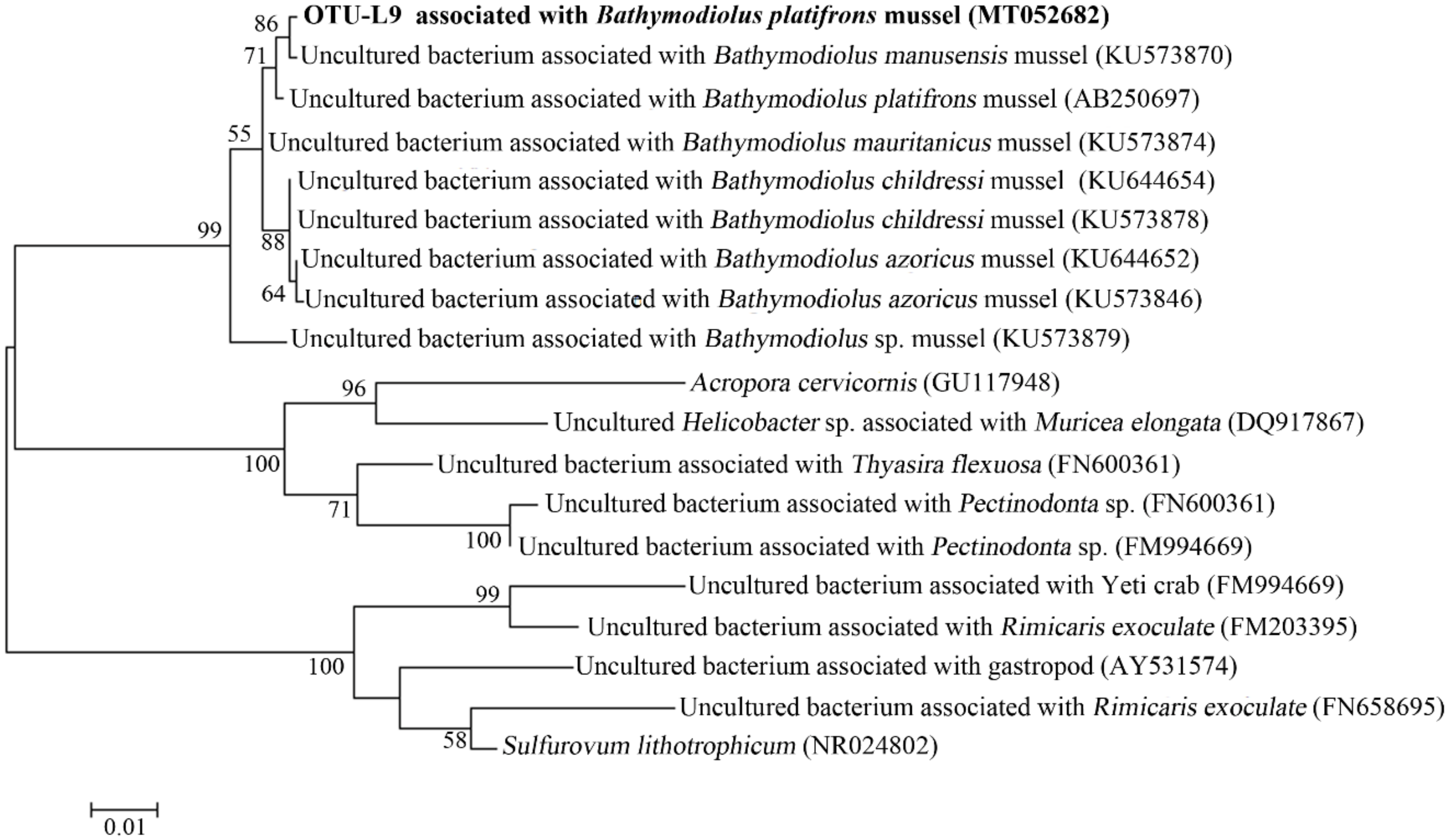
2.6. Phylogenetic Analysis
2.7. Data Availability
3. Results
3.1. Physicochemical Characteristics of Sampling Sites
3.2. Sequencing Data and Alpha Diversity
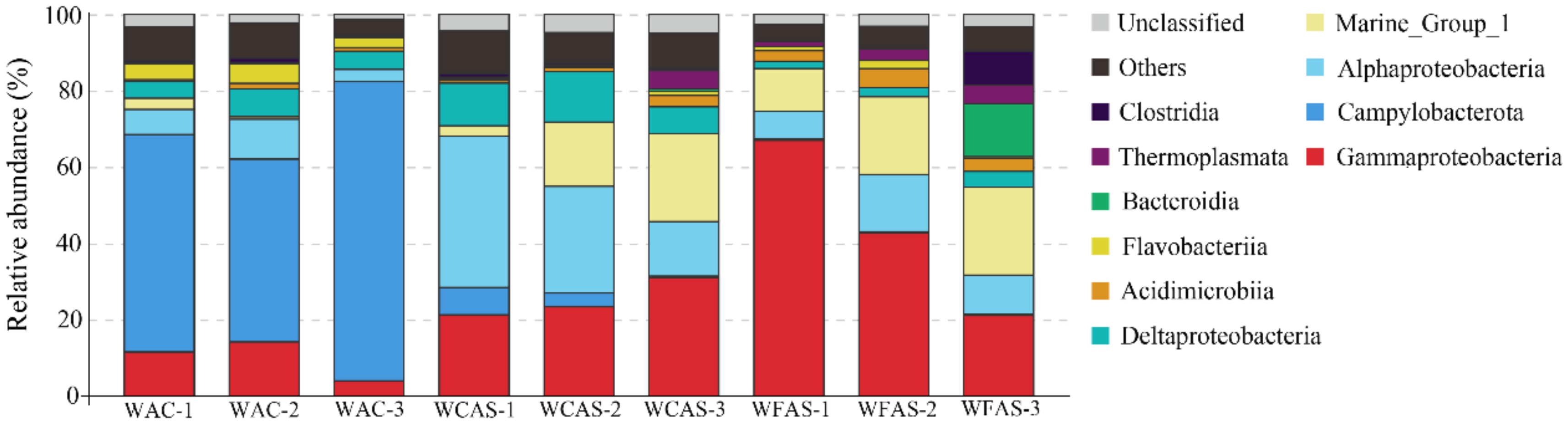
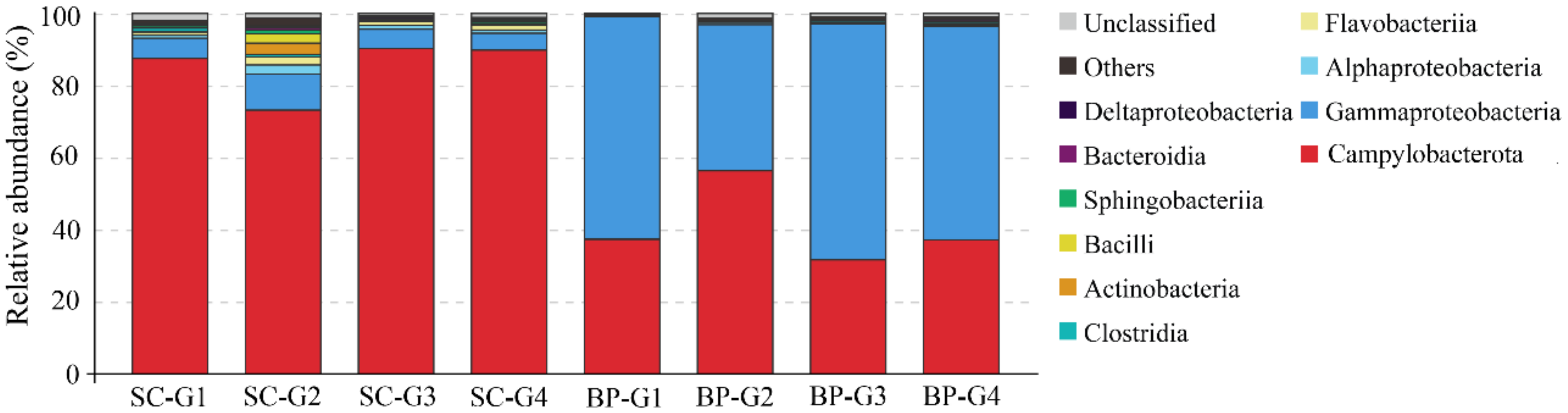
3.3. Microbial Communities in the Seawater Samples
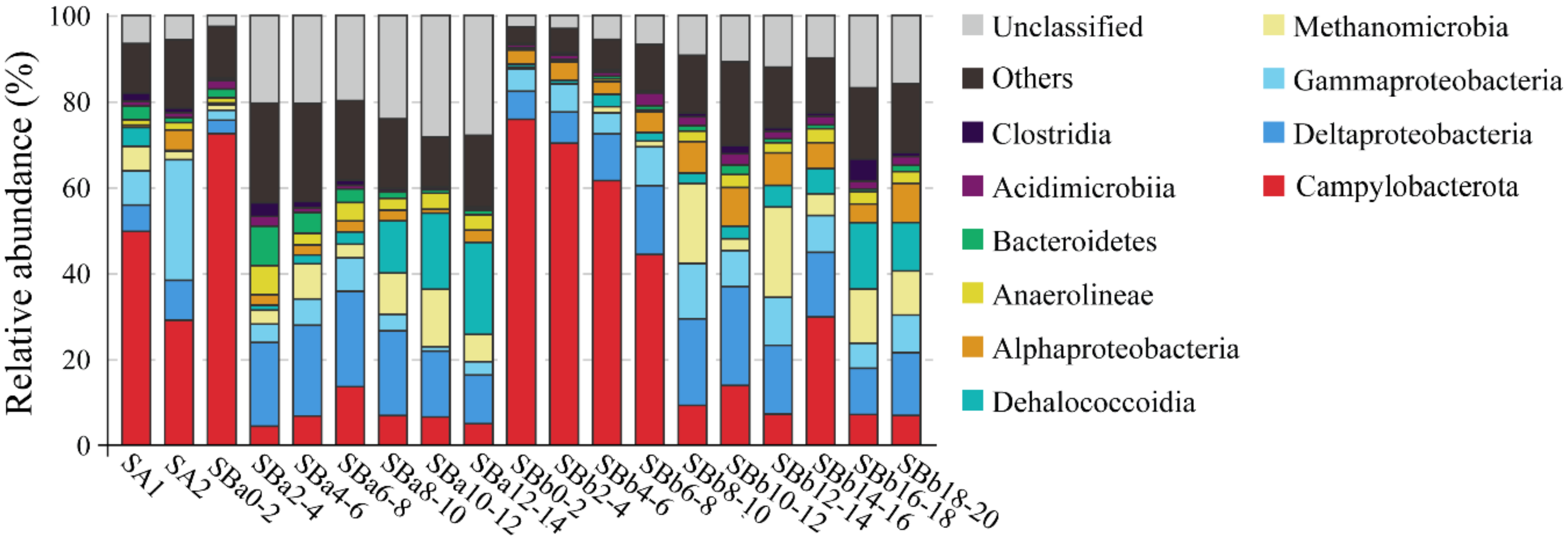
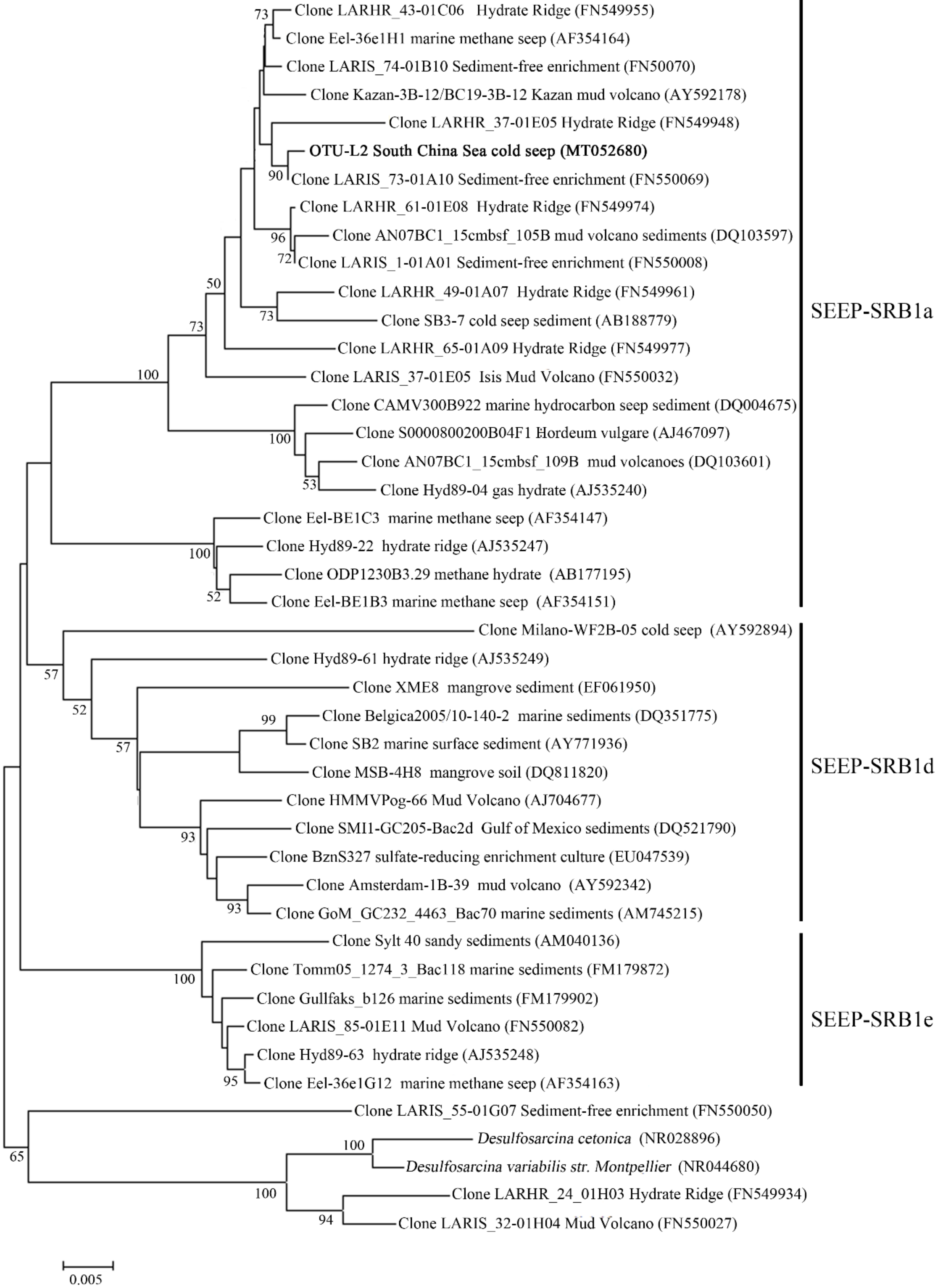
3.4. Microbial Communities in the Sediment Samples
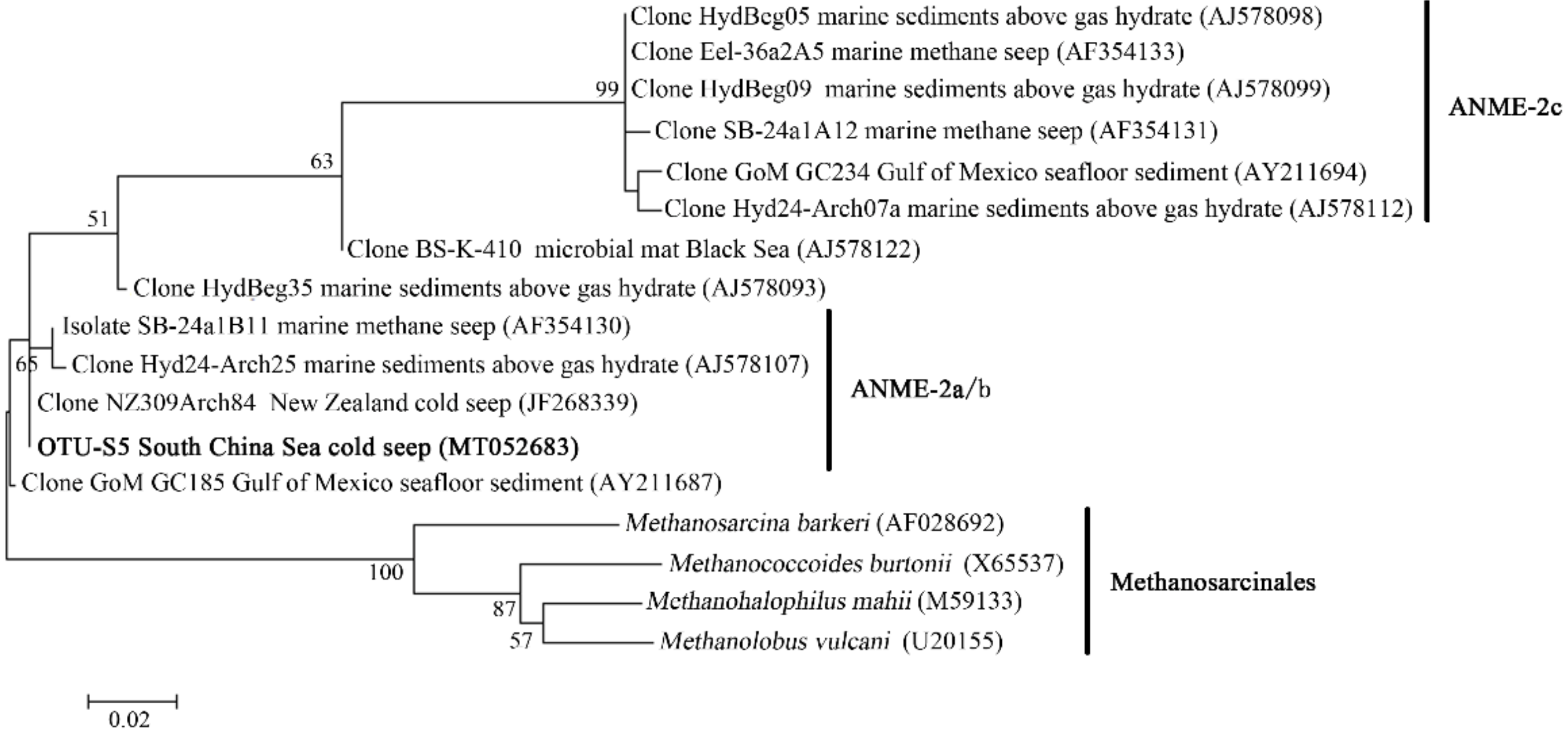
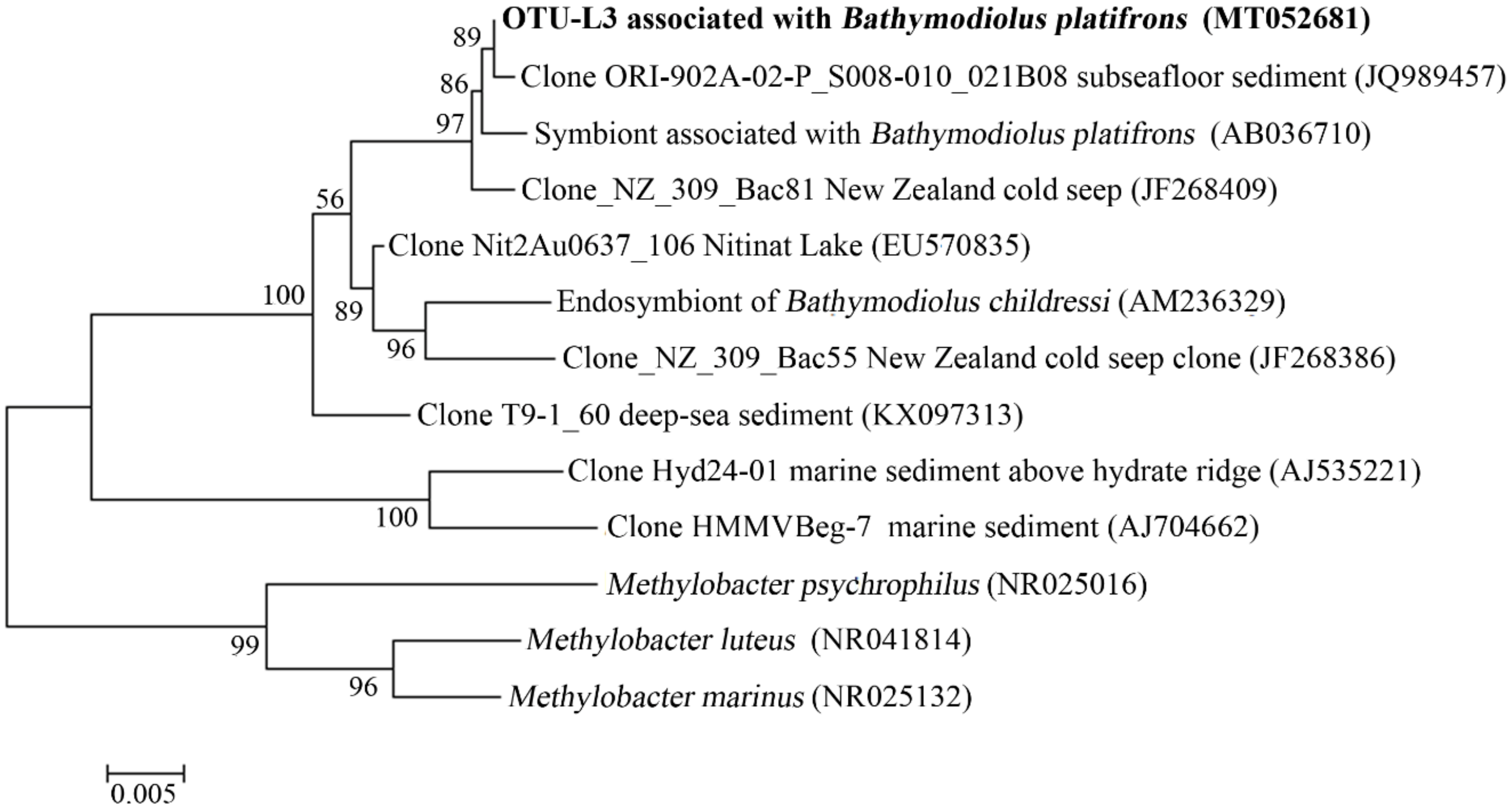
3.5. Microbial Communities of S. crosnieri and B. platifrons
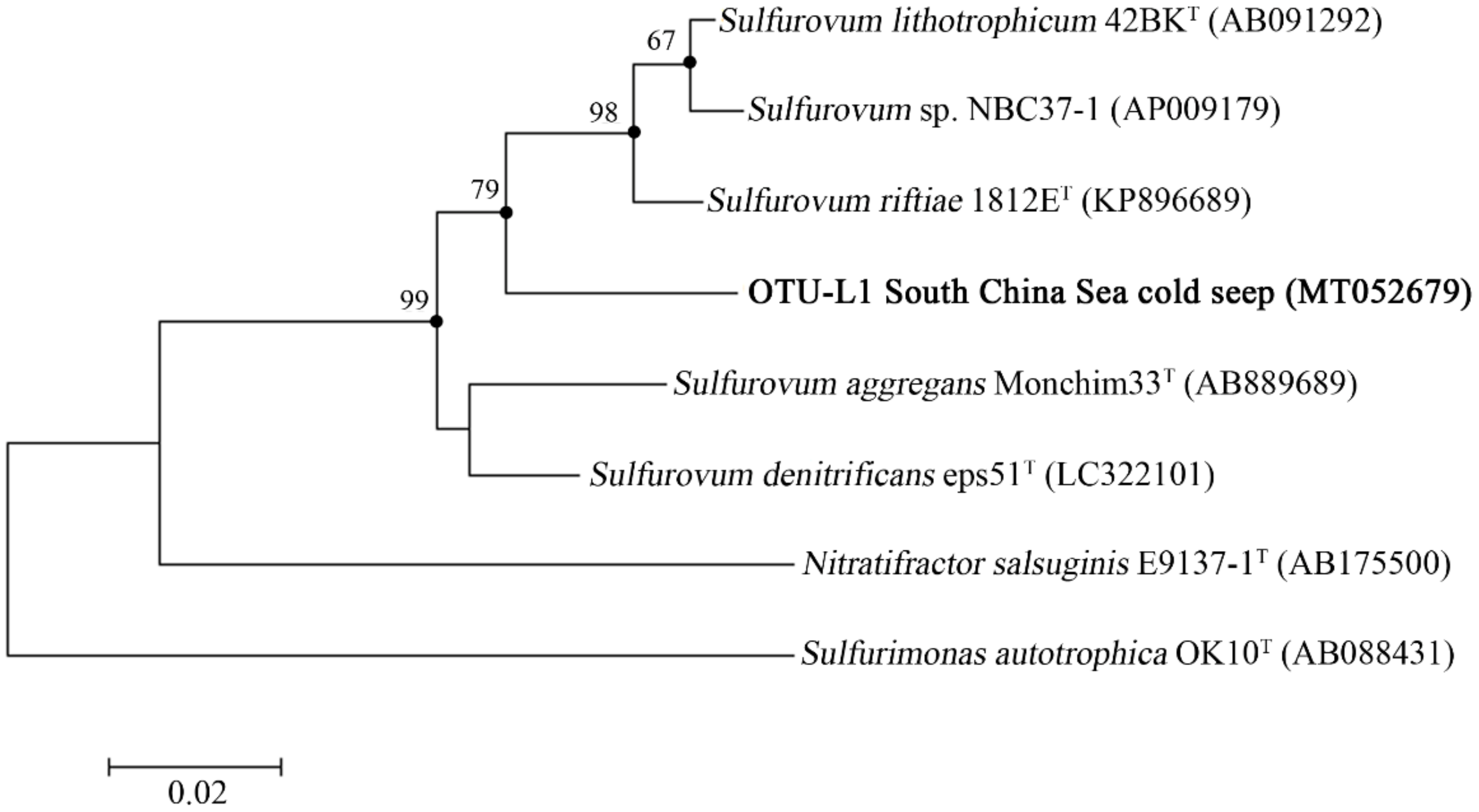
3.6. Genome Analysis of Sulfurovum sp.
4. Discussion
4.1. Microbial Communities in Cold Seep Seawater
4.2. Microbial Communities Associated with Animals
4.3. Microbial Community in Sediments
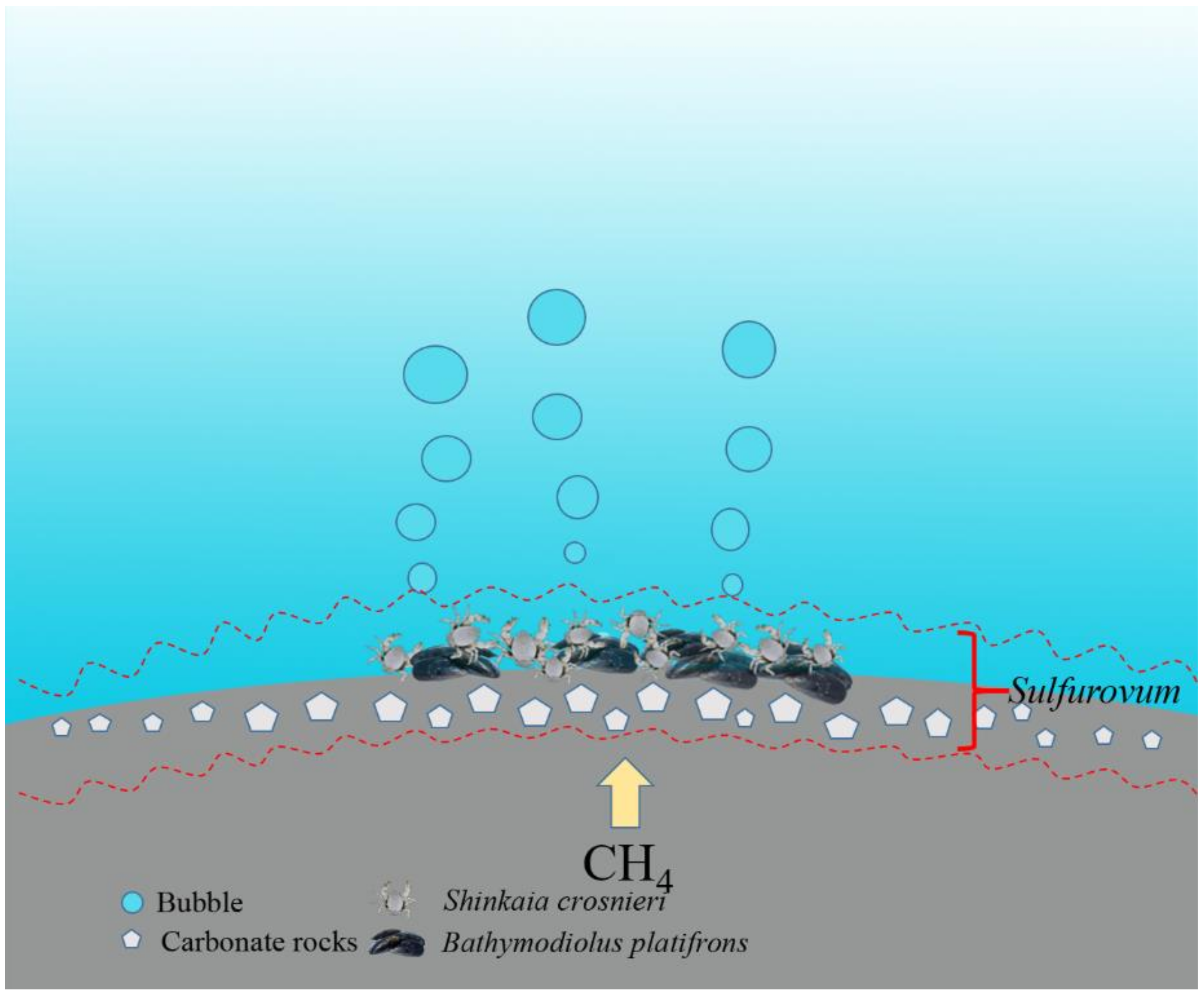
4.4. Genome Analysis of the Sulfurovum sp.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boetius, A.; Wenzhöfer, F. Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 2013, 6, 725–734. [Google Scholar] [CrossRef]
- Paull, C.K.; Hecker, B.; Commeau, R.; Freeman-Lynde, R.P.; Neumann, C.; Corso, W.P.; Golubic, S.; Hook, J.E.; Sikes, E.; Curray, J. Biological communities at the Florida Escarpment resemble hydrothermal vent taxa. Science 1984, 226, 965–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, R.K.; Scalan, R.S.; Parker, P.L.; Behrens, E.W. Seep oil and gas in gulf of Mexico slope sediment. Science 1983, 222, 619–621. [Google Scholar] [CrossRef] [PubMed]
- Suess, E. Marine cold seeps and their manifestations: Geological control, biogeochemical criteria and environmental conditions. Int. J. Earth Sci. 2014, 103, 1889–1916. [Google Scholar] [CrossRef]
- Sivan, O.; Antler, G.; Turchyn, A.V.; Marlow, J.J.; Orphan, V.J. Iron oxides stimulate sulfate-driven anaerobic methane oxidation in seeps. Proc. Natl. Acad. Sci. USA 2014, 111, E4139–E4147. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.; Fan, X.; Zhuang, G.; Liang, Q.; Wang, F. Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, A.; Cruaud, P.; Pignet, P.; Caprais, J.C.; Cambon-Bonavita, M.A.; Godfroy, A.; Toffin, L. Archaeal and anaerobic methane oxidizer communities in the Sonora Margin cold seeps, Guaymas Basin (Gulf of California). ISME J. 2013, 7, 1595. [Google Scholar] [CrossRef] [Green Version]
- Knittel, K.; Lösekann, T.; Boetius, A.; Kort, R.; Amann, R. Diversity and distribution of methanotrophic archaea at cold seeps. Appl. Environ. Microbiol. 2005, 71, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Su, X.; Chen, F.; Holland, M.; Yang, S.; Liang, J.; Su, P.; Dong, H.; Hou, W. Microbial diversity of two cold seep systems in gas hydrate-bearing sediments in the South China Sea. Mar. Environ. Res. 2019, 144, 230–239. [Google Scholar] [CrossRef]
- Gründger, F.; Carrier, V.; Svenning, M.M.; Panieri, G.; Vonnahme, T.R.; Klasek, S.; Niemann, H. Methane-fuelled biofilms predominantly composed of methanotrophic ANME-1 in Arctic gas hydrate-related sediments. Sci. Rep. 2019, 9, 9725. [Google Scholar] [CrossRef]
- Hinrichs, K.U.; Hayes, J.M.; Sylva, S.P.; Brewer, P.G.; DeLong, E.F. Methane-consuming archaebacteria in marine sediments. Nature 1999, 398, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Taubert, M.; Grob, C.; Crombie, A.; Howat, A.M.; Burns, O.J.; Weber, M.; Lott, C.; Kaster, A.K.; Vollmers, J.; Jehmlich, N.; et al. Communal metabolism by Methylococcaceae and Methylophilaceae is driving rapid aerobic methane oxidation in sediments of a shallow seep near Elba, Italy. Environ. Microbiol. 2019, 21, 3780–3795. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, M.; Liu, B.; Yue, X.; Li, C. Gill symbionts of the cold-seep mussel Bathymodiolus platifrons: Composition, environmental dependency and immune control. Fish Shellfish. Immunol. 2019, 86, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Medina-Silva, R.; Oliveira, R.R.; Trindade, F.J.; Borges, L.G.A.; Lopes Simão, T.L.; Augustin, A.H.; Valdez, F.P.; Constant, M.J.; Simundi, C.L.; Eizirik, E.; et al. Microbiota associated with tubes of Escarpia sp. from cold seeps in the southwestern Atlantic Ocean constitutes a community distinct from that of surrounding marine sediment and water. Antonie Leeuwenhoek 2018, 111, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.L.; Hatzenpichler, R.; McGlynn, S.; Chadwick, G.; Dawson, K.S.; Connon, S.A.; Orphan, V.J. Methyloprofundus sedimenti gen. nov., sp. nov., an obligate methanotroph from ocean sediment belonging to the ‘deep sea-1’ clade of marine methanotrophs. Int. J. Syst. Evol. Microbiol. 2015, 65, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Waite, D.W.; Vanwonterghem, I.; Rinke, C.; Parks, D.H.; Zhang, Y.; Takai, K.; Sievert, S.M.; Simon, J.; Campbell, B.J.; Hanson, T.E.; et al. Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. Nov.). Front. Microbiol. 2017, 8, 682. [Google Scholar] [CrossRef]
- Waite, D.W.; Vanwonterghem, I.; Rinke, C.; Parks, D.H.; Zhang, Y.; Takai, K.; Sievert, S.M.; Simon, J.; Campbell, B.J.; Hanson, T.E.; et al. Erratum: Addendum: Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. Nov.). Front. Microbiol. 2018, 9, 772. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Tian, R.M.; Zhang, W.; Cai, L.; Wong, Y.H.; Ding, W.; Qian, P.Y. Genome reduction and microbe-host interactions drive adaptation of a sulfur-oxidizing bacterium associated with a cold seep sponge. MSystems 2017, 2, e00184-16. [Google Scholar] [CrossRef] [Green Version]
- Stevens, E.W.; Bailey, J.V.; Flood, B.E.; Jones, D.S.; Gilhooly, W.P., 3rd; Joye, S.B.; Teske, A.; Mason, O.U. Barite encrustation of benthic sulfur-oxidizing bacteria at a marine cold seep. Geobiology 2015, 13, 588–603. [Google Scholar] [CrossRef] [Green Version]
- Assié, A.; Borowski, C.; van der Heijden, K.; Raggi, L.; Geier, B.; Leisch, N.; Schimak, M.P.; Dubilier, N.; Petersen, J.M. A specific and widespread association between deep-sea Bathymodiolus mussels and a novel family of Epsilonproteobacteria. Environ. Microbiol. Rep. 2016, 8, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Qiu, J.W.; Hu, Y.; Peckmanne, J.; Guan, H.; Tong, H.; Cheng, C.; Chen, J.; Gong, S.; Li, N.; et al. Cold seep systems in the South China Sea: An overview. J. Asian Earth Sci. 2018, 168, 3–16. [Google Scholar] [CrossRef]
- Cline, J.D. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 1969, 14, 454–458. [Google Scholar] [CrossRef]
- Zhang, X.; Du, Z.; Zheng, R.; Luan, Z.; Qi, F.; Cheng, K.; Wang, B.; Ye, W.; Liu, X.; Lian, C.; et al. Development of a new deep-sea hybrid Raman insertion probe and its application to the geochemistry of hydrothermal vent and cold seep fluids. Deep Sea Res. Part I 2017, 123, 1–12. [Google Scholar] [CrossRef]
- Möller, E.; Bahnweg, G.; Sandermann, H.; Geiger, H. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992, 20, 6115–6116. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, Q.L.; Zeng, Z.G.; Chen, S.; Sun, L. Microbial diversity in the deep-sea sediments of Iheya North and Iheya Ridge, Okinawa Trough. Microbiol. Res. 2015, 177, 43–52. [Google Scholar] [CrossRef]
- Lane, D.J. 16S/23S rRNA sequencing. In Nucleic Acids Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 1991; pp. 115–147. [Google Scholar]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L. Comparative metagenomics reveals insights into the deep-sea adaptation mechanism of the microorganisms in Iheya hydrothermal fields. World J. Microbiol. Biotechnol. 2017, 33, 86. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinformatics 2014, 47, 11–12. [Google Scholar] [CrossRef]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.W.; Chokhawala, H.; Schroth, G.; Luo, S.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orphan, V.J.; Hinrichs, K.U.; Ussler, W., 3rd; Paull, C.K.; Taylor, L.T.; Sylva, S.P.; Hayes, J.M.; Delong, E.F. Comparative analysis of methane-oxidizing archaea and sulfate-reducing bacteria in anoxic marine sediments. Appl. Environ. Microbiol. 2001, 67, 1922–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Mark Welch, D.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Sun, Q.L.; Zeng, Z.G.; Chen, S.; Sun, L. First comparative analysis of the community structures and carbon metabolic pathways of the bacteria associated with Alvinocaris longirostris in a hydrothermal vent of okinawa trough. PLoS ONE 2016, 11, e0154359. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Jenior, M.L.; Koumpouras, C.C.; Westcott, S.L.; Highlander, S.K. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 2016, 4, e1869. [Google Scholar] [CrossRef] [Green Version]
- Fichot, E.B.; Norman, R.S. Microbial phylogenetic profiling with the Pacific Biosciences sequencing platform. Microbiome 2013, 1, 10. [Google Scholar] [CrossRef] [Green Version]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 2013, 31, 533. [Google Scholar] [CrossRef]
- Sangwan, N.; Xia, F.; Gilbert, J.A. Recovering complete and draft population genomes from metagenome datasets. Microbiome 2016, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Yamaguchi, K.; Hanada, S. Sulfurovum denitrificans sp. nov., an obligately chemolithoautotrophic sulfur-oxidizing epsilonproteobacterium isolated from a hydrothermal field. Int. J. Syst. Evol. Microbiol. 2018, 68, 2183–2187. [Google Scholar] [CrossRef] [PubMed]
- Mino, S.; Kudo, H.; Arai, T.; Sawabe, T.; Takai, K.; Nakagawa, S. Sulfurovum aggregans sp. nov., a hydrogen-oxidizing, thiosulfate-reducing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent chimney, and an emended description of the genus Sulfurovum. Int. J. Syst. Evol. Microbiol. 2014, 64, 3195–3201. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa Trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 2004, 54, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, D.; Chung, M.; Staley, J.; Starovoytov, V.; Le Bris, N.; Vetriani, C. Sulfurovum riftiae sp. nov., a mesophilic, thiosulfate-oxidizing, nitrate-reducing chemolithoautotrophic epsilonproteobacterium isolated from the tube of the deep-sea hydrothermal vent polychaete Riftia pachyptila. Int. J. Syst. Evol. Microbiol. 2016, 66, 2697–2701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, Z.; Chen, S.; Sun, L. Bacterial communities associated with Shinkaia crosnieri from the Iheya North, Okinawa Trough: Microbial diversity and metabolic potentials. J. Mar. Syst. 2018, 180, 228–236. [Google Scholar] [CrossRef]
- Fujiyoshi, S.; Tateno, H.; Watsuji, T.; Yamaguchi, H.; Fukushima, D.; Mino, S.; Sugimura, M.; Sawabe, T.; Takai, K.; Sawayama, S.; et al. Effects of hemagglutination activity in the serum of a deep-sea vent endemic crab, Shinkaia Crosnieri, on non-symbiotic and symbiotic bacteria. Microbes Environ. 2015, 30, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Qiu, J.W.; Qian, P.Y.; Wang, Y. The vertical distribution of prokaryotes in the surface sediment of Jiaolong cold seep at the northern South China Sea. Extremophiles 2018, 22, 499–510. [Google Scholar] [CrossRef]
- Jeon, W.; Priscilla, L.; Park, G.; Lee, H.; Lee, N.; Lee, D.; Kwon, H.; Ahn, I.; Lee, C.; Lee, H.; et al. Complete genome sequence of the sulfur-oxidizing chemolithoautotrophic Sulfurovum lithotrophicum 42BKT T. Stand. Genomic Sci. 2017, 12, 54. [Google Scholar] [CrossRef] [Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Q.-L.; Zhang, J.; Wang, M.-X.; Cao, L.; Du, Z.-F.; Sun, Y.-Y.; Liu, S.-Q.; Li, C.-L.; Sun, L. High-Throughput Sequencing Reveals a Potentially Novel Sulfurovum Species Dominating the Microbial Communities of the Seawater–Sediment Interface of a Deep-Sea Cold Seep in South China Sea. Microorganisms 2020, 8, 687. https://doi.org/10.3390/microorganisms8050687
Sun Q-L, Zhang J, Wang M-X, Cao L, Du Z-F, Sun Y-Y, Liu S-Q, Li C-L, Sun L. High-Throughput Sequencing Reveals a Potentially Novel Sulfurovum Species Dominating the Microbial Communities of the Seawater–Sediment Interface of a Deep-Sea Cold Seep in South China Sea. Microorganisms. 2020; 8(5):687. https://doi.org/10.3390/microorganisms8050687
Chicago/Turabian StyleSun, Qing-Lei, Jian Zhang, Min-Xiao Wang, Lei Cao, Zeng-Feng Du, Yuan-Yuan Sun, Shi-Qi Liu, Chao-Lun Li, and Li Sun. 2020. "High-Throughput Sequencing Reveals a Potentially Novel Sulfurovum Species Dominating the Microbial Communities of the Seawater–Sediment Interface of a Deep-Sea Cold Seep in South China Sea" Microorganisms 8, no. 5: 687. https://doi.org/10.3390/microorganisms8050687