
Reducing Cardiac Injury during ST-Elevation Myocardial Infarction: A Reasoned Approach to a Multitarget Therapeutic Strategy

 , , , and
, , , and
Abstract
:1. Introduction
2. Mechanisms of Post-Infarction LVR
2.1. ncRNAs
2.2. Inflammation
2.3. ECM Homeostasis
2.4. Coronary Microvascular Dysfunction
- (1)
- Ischemia-reperfusion damages the myocardium through edema (both intracellular and interstitial), impaired vasomotility, and intravascular cell aggregates. Intracellular and interstitial edema quickly develops after coronary occlusion [91,92]. Cardiomyocytes and endothelial cells intracellular edema is the consequence of the energetic deficit and the impairment of energy-dependent ion pumps, whereas interstitial edema develops due to the increased interstitial osmolarity from the release of ions and catabolites, and the dysfunction of the endothelial barrier [93]. This latter is composed of endothelial cells, glycocalyx, and pericytes. Endothelial dysfunction contributes to edema, since adenosine (Ade) release enhances cytoskeletal derangement, followed by hyper-contracture and gap formation [94]. Another determinant of edema is represented by the degradation of the glycocalyx mediated by TNF-α promoting platelet and leukocytes adherence and aggregation [95,96,97]. During reperfusion, the rapid washout of osmotically active substances from the intravascular space increases edema formation. Notably, interstitial myocardial edema compresses capillaries and small vessels, further decreasing flow in these already dysfunctional territories.Dysfunctional vasomotility mostly depends on the impairment of endothelium-mediated vasodilatation caused by the disruption of the endothelial barrier [98]. The amount of impairment is proportional to the ischemic insult, being more pronounced in AMI than in chronic hypo-perfusion. Of note, during ischemia-reperfusion, the myocardium remains susceptible to vasoconstrictor stimuli, such as the release of α-adrenergic molecules, serotonin, and thromboxane [99]. Furthermore, the stasis and the increased expression of intercellular and vascular adhesion molecules promotes the adherence of platelet aggregates, platelet-leucocyte aggregates, and, in severe forms, of erythrocyte aggregates to the endothelium [100,101,102].
- (2)
- During AMI, atherosclerotic material with superimposed thrombotic milieu, originating from the ruptured or eroded plaque, may embolize distally to the microcirculation. Interestingly, the embolized material may aggravate reperfusion beyond the sheer mechanical obstruction mechanism [103]. In fact, the embolization of biologically active material causes patchy micro-infarcts with local inflammatory reactions, aggravating the damage caused by ischemia-reperfusion and intravascular aggregates [104].
- (3)
- In the most severe forms of AMI, the massive swelling of endothelial cells and the consequent interruption of the vascular wall leads to the leakage of circulating blood cells into the interstitial space upon reperfusion, causing IMH [105,106]. IMH represents a negative prognostic factor in patients with AMI together with the angiographic no-reflow phenomenon (NR) [107].
3. Therapeutic Strategies against Post-STEMI LVR
3.1. Conventional Pharmacological Options
3.1.1. Anti-Platelet Therapy
3.1.2. β-Adrenergic Blockers
3.1.3. RAAS Antagonists
3.1.4. Statins
3.1.5. Ezetimibe
3.2. Prospective Pharmacological Options
3.2.1. Neprilysin Inhibition
3.2.2. PCSK9 Inhibitors
3.2.3. Novel Drugs Modulating Glucose Metabolism
3.2.4. Coronary Vasodilators
3.3. Mechanical Interventions
3.3.1. Thrombus Aspiration
3.3.2. Remote Ischemic Perconditioning
3.3.3. Classic and Remote Post-Conditioning
4. Reasoned Multitarget Therapeutic Strategy against Post-STEMI LVR
4.1. “Pre-pPCI” Phase
4.2. ‘During-pPCI’ Phase
4.3. ‘Post-pPCI’ Phase
4.4. Proposal for a Randomized Clinical Trial
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Puymirat, E.; Simon, T.; Cayla, G.; Cottin, Y.; Elbaz, M.; Coste, P.; Lemesle, G.; Motreff, P.; Popovic, B.; Khalife, K.; et al. Acute myocardial infarction: Changes in patient characteristics, management, and 6-month outcomes over a period of 20 years in the FAST-MI program (French Registry of Acute ST-Elevation or Non-ST-Elevation Myocardial Infarction) 1995 to 2015. Circulation 2017, 136, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Szummer, K.; Wallentin, L.; Lindhagen, L.; Alfredsson, J.; Erlinge, D.; Held, C.; James, S.; Kellerth, T.; Lindahl, B.; Ravn-Fischer, A.; et al. Improved outcomes in patients with ST-elevation myocardial infarction during the last 20 years are related to implementation of evidence-based treatments: Experiences from the SWEDEHEART registry 1995–2014. Eur. Heart J. 2017, 38, 3056–3065. [Google Scholar] [CrossRef] [Green Version]
- Szummer, K.; Wallentin, L.; Lindhagen, L.; Alfredsson, J.; Erlinge, D.; Held, C.; James, S.; Kellerth, T.; Lindahl, B.; Ravn-Fischer, A.; et al. Relations between implementation of new treatments and improved outcomes in patients with non-ST-elevation myocardial infarction during the last 20 years: Experiences from SWEDEHEART registry 1995 to 2014. Eur. Heart J. 2018, 39, 3766–3776. [Google Scholar] [CrossRef]
- Mehta, S.R.; Yusuf, S.; Peters, R.J.; Bertrand, M.E.; Lewis, B.S.; Natarajan, M.K.; Malmberg, K.; Rupprecht, H.; Zhao, F.; Chrolavicius, S.; et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: The PCI-CURE study. Lancet 2001, 358, 527–533. [Google Scholar] [CrossRef]
- Montalescot, G.; van ‘t Hof, A.W.; Lapostolle, F.; Silvain, J.; Lassen, J.F.; Bolognese, L.; Cantor, W.J.; Cequier, A.; Chettibi, M.; Goodman, S.G.; et al. Prehospital ticagrelor in ST-segment elevation myocardial infarction. N. Engl. J. Med. 2014, 371, 1016–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalescot, G.; Wiviott, S.D.; Braunwald, E.; Murphy, S.A.; Gibson, C.M.; McCabe, C.H.; Antman, E.M. Prasugrel compared with clopidogrel in patients undergoing percutaneous coronary intervention for ST-elevation myocardial infarction (TRITON-TIMI 38): Double-blind, randomised controlled trial. Lancet 2009, 373, 723–731. [Google Scholar] [CrossRef]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Bolli, R. Myocardial ‘stunning’ in man. Circulation 1992, 86, 1671–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Frohlich, G.M.; Meier, P.; White, S.K.; Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury: Looking beyond primary PCI. Eur. Heart J. 2013, 34, 1714–1722. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Libby, P.; Gersh, B.; Yellon, D.; Böhm, M.; Lopaschuk, G.; Opie, L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 2014, 383, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Van der Bijl, P.; Abou, R.; Goedemans, L.; Gersh, B.J.; Holmes, D.R., Jr.; Ajmone Marsan, N.; Delgado, V.; Bax, J.J. Left ventricular post-infarct remodeling: Implications for systolic function improvement and outcomes in the modern era. JACC Heart Fail. 2020, 8, 131–140. [Google Scholar] [CrossRef]
- Carrabba, N.; Valenti, R.; Parodi, G.; Santoro, G.M.; Antoniucci, D. Left ventricular remodeling and heart failure in diabetic patients treated with primary angioplasty for acute myocardial infarction. Circulation 2004, 110, 1974–1979. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, G.; Montone, R.A.; Ibanez, B.; Thiele, H.; Crea, F.; Heusch, G.; Bulluck, H.; Hausenloy, D.J.; Berry, C.; Stiermaier, T.; et al. Optimized treatment of ST-elevation myocardial infarction. Circ. Res. 2019, 125, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Garcia-Dorado, D.; Botker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position paper of the european society of cardiology working group on cellular biology of the heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Botker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget strategies to reduce myocardial ischemia/reperfusion injury: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Al-Hawwas, M.; Marmagkiolis, K.; Mehta, J.L. The Impact of transcatheter aortic valve implantation and surgical aortic valve replacement on left ventricular remodeling. Am. J. Cardiol. 2017, 120, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Buono, F.; Crispo, S.; Pagano, G.; Rengo, G.; Petitto, M.; Grieco, F.; Trimarco, B.; Morisco, C. Determinants of left ventricular hypertrophy in patients with recent diagnosis of essential hypertension. J. Hypertens. 2014, 32, 166–173. [Google Scholar] [CrossRef]
- Kaesler, N.; Babler, A.; Floege, J.; Kramann, R. Cardiac remodeling in chronic kidney disease. Toxins 2020, 12, 161. [Google Scholar] [CrossRef] [Green Version]
- Riehle, C.; Abel, E.D. Insulin signaling and heart failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C.; Grassi, G. The influence of sex on left ventricular remodeling in arterial hypertension. Heart Fail. Rev. 2019, 24, 905–914. [Google Scholar] [CrossRef]
- Zannad, F.; Rossignol, P. Cardiorenal syndrome revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Bulluck, H.; Carberry, J.; Carrick, D.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Hood, S.; Watkins, S.; Lindsay, M.; Mahrous, A.; et al. Redefining adverse and reverse left ventricular remodeling by cardiovascular magnetic resonance following ST-segment-elevation myocardial infarction and their implications on long-term prognosis. Circ. Cardiovasc. Imaging 2020, 13, e009937. [Google Scholar] [CrossRef]
- Carrick, D.; Haig, C.; Rauhalammi, S.; Ahmed, N.; Mordi, I.; McEntegart, M.; Petrie, M.C.; Eteiba, H.; Lindsay, M.; Watkins, S.; et al. Pathophysiology of LV remodeling in survivors of STEMI: Inflammation, remote myocardium, and prognosis. JACC Cardiovasc. Imaging 2015, 8, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Dutka, M.; Bobiński, R.; Korbecki, J. The relevance of microRNA in post-infarction left ventricular remodelling and heart failure. Heart Fail. Rev. 2019, 24, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Stone, G.W.; Selker, H.P.; Thiele, H.; Patel, M.R.; Udelson, J.E.; Ohman, E.M.; Maehara, A.; Eitel, I.; Granger, C.B.; Jenkins, P.L.; et al. Relationship between infarct size and outcomes following primary PCI: Patient-level analysis from 10 randomized trials. J. Am. Coll. Cardiol. 2016, 67, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.P.; Grisanti, L.A. The dynamic interplay between cardiac inflammation and fibrosis. Front. Physiol. 2020, 11, 529075. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.; Ortiz, J.T.; Tejedor, P.; Lee, D.C.; Bucciarelli-Ducci, C.; Kansal, P.; Carr, J.C.; Holly, T.A.; Lloyd-Jones, D.; Klocke, F.J.; et al. Infarct size by contrast enhanced cardiac magnetic resonance is a stronger predictor of outcomes than left ventricular ejection fraction or end-systolic volume index: Prospective cohort study. Heart 2008, 94, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K. microRNAs databases: Developmental methodologies, structural and functional annotations. Interdiscip. Sci. Comput. Life Sci. 2017, 9, 357–377. [Google Scholar] [CrossRef] [PubMed]
- Rajput, C.; Tauseef, M.; Farazuddin, M.; Yazbeck, P.; Amin, M.R.; Avin Br, V.; Sharma, T.; Mehta, D. MicroRNA-150 suppression of angiopoetin-2 generation and signaling is crucial for resolving vascular injury. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Song, C.; Zhou, X.; Han, X.; Li, J.; Wang, Z.; Shang, H.; Liu, Y.; Cao, H. Mitochondria associated microRNA expression profiling of heart failure. BioMed Res. Int. 2017, 2017, 4042509. [Google Scholar] [CrossRef]
- Widlansky, M.E. miR-29 contributes to normal endothelial function and can restore it in cardiometabolic disorders. BioMed Res. Int. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Gigante, B.; Papa, L.; Bye, A.; Kunderfranco, P. MicroRNA signatures predict early major coronary events in middle-aged men and women. Cell Death Dis. 2020, 11, 74. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.L.; Guo, L.L.; Weng, Y.Q. Implication of peripheral blood miRNA-124 in predicting acute myocardial infarction. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1054–1059. [Google Scholar] [PubMed]
- Latet, S.C.; Van Herck, P.L.; Claeys, M.J.; Van Craenenbroeck, A.H.; Haine, S.E.; Vandendriessche, T.R.; Van Hoof, V.O.; Fransen, E.; De Winter, B.Y.; Van Craenenbroeck, E.M.; et al. Failed downregulation of circulating MicroRNA-155 in the early phase after ST elevation myocardial infarction is associated with adverse left ventricular remodeling. Cardiology 2017, 138, 91–96. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, Y.J.; Sara, J.D.; Liu, L.J.; Liu, L.P.; Zhao, X.; Gao, H. Circulating MicroRNA-145 is associated with acute myocardial infarction and heart failure. EMBO Mol. Med. 2017, 130, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fan, G.C. Role of microRNAs in the reperfused myocardium towards post-infarct remodelling. Cardiovasc. Res. 2012, 94, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Huang, W.; Xu, R.; Nie, Y.; Cao, X.; Meng, J.; Xu, X.; Hu, S.; Zheng, Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2150–2160. [Google Scholar] [CrossRef]
- Bayoumi, A.S.; Teoh, J.P.; Aonuma, T.; Yuan, Z.; Ruan, X.; Tang, Y.; Su, H.; Weintraub, N.L.; Kim, I.M. MicroRNA-532 protects the heart in acute myocardial infarction, and represses prss23, a positive regulator of endothelial-to-mesenchymal transition. Cardiovasc. Res. 2017, 113, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Mehta, J.L.; Li, D.; Joseph, L.; Joseph, J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ. Res. 2004, 95, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyu, K.G.; Wang, B.W.; Cheng, W.P.; Lo, H.M. MicroRNA-208a Increases myocardial endoglin expression and myocardial fibrosis in acute myocardial infarction. Can. J. Cardiol. 2015, 31, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Guo, J.; Wu, J.; Sun, Z.; Han, M.; Shan, S.W.; Deng, Z.; Yang, B.B.; Weisel, R.D.; Li, R.K. miR-17 targets tissue inhibitor of metalloproteinase 1 and 2 to modulate cardiac matrix remodeling. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4254–4265. [Google Scholar] [CrossRef] [PubMed]
- Garikipati, V.N.; Krishnamurthy, P.; Verma, S.K.; Khan, M.; Abramova, T.; Mackie, A.R.; Qin, G.; Benedict, C.; Nickoloff, E.; Johnson, J.; et al. Negative regulation of miR-375 by interleukin-10 enhances bone marrow-derived progenitor cell-mediated myocardial repair and function after myocardial infarction. Stem Cells (Dayt. Ohio) 2015, 33, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Bellera, N.; Barba, I.; Rodriguez-Sinovas, A.; Ferret, E.; Asín, M.A.; Gonzalez-Alujas, M.T.; Pérez-Rodon, J.; Esteves, M.; Fonseca, C.; Toran, N.; et al. Single intracoronary injection of encapsulated antagomir-92a promotes angiogenesis and prevents adverse infarct remodeling. J. Am. Heart Assoc. 2014, 3, e000946. [Google Scholar] [CrossRef] [Green Version]
- Arif, M.; Pandey, R.; Alam, P.; Jiang, S.; Sadayappan, S.; Paul, A.; Ahmed, R.P.H. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J. Mol. Med. (Berl. Ger.) 2017, 95, 1369–1385. [Google Scholar] [CrossRef] [Green Version]
- Sayed, D.; He, M.; Hong, C.; Gao, S.; Rane, S.; Yang, Z.; Abdellatif, M. MicroRNA-21 is a downstream effector of AKT that mediates its antiapoptotic effects via suppression of Fas ligand. J. Biol. Chem. 2010, 285, 20281–20290. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Zhu, P.; Yang, J.; Liu, X.; Dong, S.; Wang, X.; Chun, B.; Zhuang, J.; Zhang, C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc. Res. 2010, 87, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Cheng, Y.; Yang, J.; Li, J.; Liu, X.; Wang, X.; Wang, D.; Krall, T.J.; Delphin, E.S.; Zhang, C. MicroRNA expression signature and the role of microRNA-21 in the early phase of acute myocardial infarction. J. Biol. Chem. 2009, 284, 29514–29525. [Google Scholar] [CrossRef] [Green Version]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; Di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; De Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010, 31, 2765–2773. [Google Scholar] [CrossRef] [Green Version]
- Vasquez, C.; Mohandas, P.; Louie, K.L.; Benamer, N.; Bapat, A.C.; Morley, G.E. Enhanced fibroblast-myocyte interactions in response to cardiac injury. Circ. Res. 2010, 107, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, B. Long noncoding RNAs in pathological cardiac remodeling: A review of the update literature. BioMed Res. Int. 2019, 2019, 7159592. [Google Scholar] [CrossRef]
- Salgado-Somoza, A.; Zhang, L.; Vausort, M.; Devaux, Y. The circular RNA MICRA for risk stratification after myocardial infarction. BioMed Res. Int. 2017, 17, 33–36. [Google Scholar] [CrossRef]
- Vausort, M.; Salgado-Somoza, A.; Zhang, L.; Leszek, P.; Scholz, M.; Teren, A.; Burkhardt, R.; Thiery, J.; Wagner, D.R.; Devaux, Y. Myocardial infarction-associated circular RNA predicting left ventricular dysfunction. J. Am. Coll. Cardiol. 2016, 68, 1247–1248. [Google Scholar] [CrossRef] [PubMed]
- Montone, R.A.; Vetrugno, V.; Camilli, M.; Russo, M.; Fracassi, F.; Khan, S.Q.; Doshi, S.N.; Townend, J.N.; Ludman, P.F.; Trani, C.; et al. Macrophage infiltrates in coronary plaque erosion and cardiovascular outcome in patients with acute coronary syndrome. Atherosclerosis 2020, 311, 158–166. [Google Scholar] [CrossRef]
- Ruggio, A.; Pedicino, D.; Flego, D.; Vergallo, R.; Severino, A.; Lucci, C.; Niccoli, G.; Trani, C.; Burzotta, F.; Aurigemma, C.; et al. Correlation between CD4(+)CD28(null) T lymphocytes, regulatory T cells and plaque rupture: An optical coherence tomography study in acute coronary syndromes. Int. J. Cardiol. 2019, 276, 289–292. [Google Scholar] [CrossRef]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Westman, P.C.; Lipinski, M.J.; Luger, D.; Waksman, R.; Bonow, R.O.; Wu, E.; Epstein, S.E. Inflammation as a driver of adverse left ventricular remodeling after acute myocardial infarction. J. Am. Coll. Cardiol. 2016, 67, 2050–2060. [Google Scholar] [CrossRef]
- Cuijpers, I.; Simmonds, S.J.; van Bilsen, M.; Czarnowska, E.; González Miqueo, A.; Heymans, S.; Kuhn, A.R.; Mulder, P.; Ratajska, A.; Jones, E.A.V.; et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. High Blood Press. Cardiovasc. Prev. Off. J. Ital. Soc. Hypertens. 2020, 115, 39. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Vujacic-Mirski, K. Regulation of vascular function and inflammation via cross talk of reactive oxygen and nitrogen species from mitochondria or NADPH oxidase-implications for diabetes progression. Int. J. Mol. Sci. 2020, 21, 3405. [Google Scholar] [CrossRef] [PubMed]
- Fiordelisi, A.; Iaccarino, G. NFkappaB is a key player in the crosstalk between inflammation and cardiovascular diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Mancusi, C.; Izzo, R.; di Gioia, G.; Losi, M.A.; Barbato, E.; Morisco, C. Insulin Resistance the hinge between hypertension and type 2 diabetes. High Blood Press. Cardiovasc. Prev. 2020, 27, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Chaikijurajai, T.; Tang, W.H.W. Reappraisal of inflammatory biomarkers in heart failure. Curr. Heart Fail. Rep. 2020, 17, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, C.; Gan, F.; Wang, Y.; Ding, L.; Hua, W. Plasma NT pro-BNP, hs-CRP and big-ET levels at admission as prognostic markers of survival in hospitalized patients with dilated cardiomyopathy: A single-center cohort study. BMC Cardiovasc. Disord. 2014, 14, 67. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Kontos, M.C.; Grizzard, J.D.; Biondi-Zoccai, G.G.; Van Tassell, B.W.; Robati, R.; Roach, L.M.; Arena, R.A.; Roberts, C.S.; Varma, A.; et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). Am. J. Cardiol. 2010, 105, 1371–1377. [Google Scholar] [CrossRef] [Green Version]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: Results of the anti-TNF therapy against congestive heart failure (ATTACH) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Torre-Amione, G.; Anker, S.D.; Bourge, R.C.; Colucci, W.S.; Greenberg, B.H.; Hildebrandt, P.; Keren, A.; Motro, M.; Moyé, L.A.; Otterstad, J.E.; et al. Results of a non-specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): A placebo-controlled randomised trial. Lancet 2008, 371, 228–236. [Google Scholar] [CrossRef]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Abbate, A.; Salloum, F.N.; Vecile, E.; Das, A.; Hoke, N.N.; Straino, S.; Biondi-Zoccai, G.G.; Houser, J.E.; Qureshi, I.Z.; Ownby, E.D.; et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 2008, 117, 2670–2683. [Google Scholar] [CrossRef] [Green Version]
- Ørn, S.; Ueland, T.; Manhenke, C.; Sandanger, Ø.; Godang, K.; Yndestad, A.; Mollnes, T.E.; Dickstein, K.; Aukrust, P. Increased interleukin-1β levels are associated with left ventricular hypertrophy and remodelling following acute ST segment elevation myocardial infarction treated by primary percutaneous coronary intervention. J. Intern. Med. 2012, 272, 267–276. [Google Scholar] [CrossRef]
- Gajarsa, J.J.; Kloner, R.A. Left ventricular remodeling in the post-infarction heart: A review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail. Rev. 2011, 16, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H. Activation mechanisms of matrix metalloproteinases. Biol. Chem. 1997, 378, 151–160. [Google Scholar]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulos, D.P.; Moyssakis, I.; Makris, T.K.; Poulakou, M.; Stavroulakis, G.; Perrea, D.; Votteas, V.E. Clinical significance of matrix metalloproteinases activity in acute myocardial infarction. Eur. Cytokine Netw. 2005, 16, 152–160. [Google Scholar]
- Webb, C.S.; Bonnema, D.D.; Ahmed, S.H.; Leonardi, A.H.; McClure, C.D.; Clark, L.L.; Stroud, R.E.; Corn, W.C.; Finklea, L.; Zile, M.R.; et al. Specific temporal profile of matrix metalloproteinase release occurs in patients after myocardial infarction: Relation to left ventricular remodeling. Circulation 2006, 114, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Hua, Q. Correlations between serum inflammation factors and left ventricular remodeling in acute ST segment elevation myocardial infarction. Yonsei Med. J. 2012, 53, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef]
- Hayashidani, S.; Tsutsui, H.; Ikeuchi, M.; Shiomi, T.; Matsusaka, H.; Kubota, T.; Imanaka-Yoshida, K.; Itoh, T.; Takeshita, A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1229–H1235. [Google Scholar] [CrossRef] [Green Version]
- Claassens, D.M.F.; Vos, G.J.A.; Bergmeijer, T.O.; Hermanides, R.S.; van ‘t Hof, A.W.J.; van der Harst, P.; Barbato, E.; Morisco, C.; Tjon Joe Gin, R.M.; Asselbergs, F.W.; et al. A genotype-guided strategy for oral P2Y(12) inhibitors in primary PCI. N. Engl. J. Med. 2019, 381, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, F.; Gargiulo, G.; Paolillo, R.; Ferrone, M.; Cimino, S.; Giugliano, G.; Schiattarella, G.G.; Verde, N.; Stabile, E.; Perrino, C.; et al. Impact of chronic kidney disease on platelet aggregation in patients with acute coronary syndrome. J. Cardiovasc. Med. (Hagerstown Md.) 2020, 21, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Strisciuglio, T.; Franco, D.; Di Gioia, G.; De Biase, C.; Morisco, C.; Trimarco, B.; Barbato, E. Impact of genetic polymorphisms on platelet function and response to anti platelet drugs. Cardiovasc. Diagn. Ther. 2018, 8, 610–620. [Google Scholar] [CrossRef]
- Chen, Y.; Dong, S.; He, M.; Qi, T.; Zhu, W. Angiotensin-converting enzyme insertion/deletion polymorphism and risk of myocardial infarction in an updated meta-analysis based on 34993 participants. Gene 2013, 522, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Dharma, S.; Sari, N.Y.; Parlautan, A.; Sukmawan, R.; Wijaya, S.; Ekawati, E.; Santoso, A. The 3q25 rs2305619 polymorphism is associated with coronary microvascular obstruction following primary angioplasty for acute ST-segment-elevation myocardial infarction. Circ. Cardiovasc. Interv. 2019, 12, e008228. [Google Scholar] [CrossRef]
- Gigante, B.; Bellis, A.; Visconti, R.; Marino, M.; Morisco, C.; Trimarco, V.; Galasso, G.; Piscione, F.; De Luca, N.; Prince, J.A.; et al. Retrospective analysis of coagulation factor II receptor (F2R) sequence variation and coronary heart disease in hypertensive patients. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1213–1219. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, P.; Izzo, R.; Mancusi, C.; Buono, F.; Ziviello, F.; Spinelli, L.; Esposito, G.; Di Gioia, G.; Barbato, E.; Strisciuglio, T.; et al. Impact of drug-eluting stents on left ventricular wall motion after successful reperfusion of first anterior ST elevation myocardial infarction. Minerva Cardiol. Angiol. 2021, 69, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Britten, M.B.; Zeiher, A.M.; Schächinger, V. Microvascular dysfunction in angiographically normal or mildly diseased coronary arteries predicts adverse cardiovascular long-term outcome. Coron. Artery Dis. 2004, 15, 259–264. [Google Scholar] [CrossRef]
- Niccoli, G.; Scalone, G.; Lerman, A.; Crea, F. Coronary microvascular obstruction in acute myocardial infarction. Eur. Heart J. 2016, 37, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Aty, H.; Cocker, M.; Meek, C.; Tyberg, J.V.; Friedrich, M.G. Edema as a very early marker for acute myocardial ischemia: A cardiovascular magnetic resonance study. J. Am. Coll. Cardiol. 2009, 53, 1194–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Dorado, D.; Oliveras, J.; Gili, J.; Sanz, E.; Pérez-Villa, F.; Barrabés, J.; Carreras, M.J.; Solares, J.; Soler-Soler, J. Analysis of myocardial oedema by magnetic resonance imaging early after coronary artery occlusion with or without reperfusion. Cardiovasc. Res. 1993, 27, 1462–1469. [Google Scholar] [CrossRef]
- Noll, T.; Muhs, A.; Besselmann, M.; Watanabe, H.; Piper, H.M. Initiation of hyperpermeability in energy-depleted coronary endothelial monolayers. Am. J. Physiol. 1995, 268, H1462–H1470. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.F.; Chappell, D.; Jacob, M. Endothelial glycocalyx and coronary vascular permeability: The fringe benefit. Basic Res. Cardiol. 2010, 105, 687–701. [Google Scholar] [CrossRef]
- Chappell, D.; Brettner, F.; Doerfler, N.; Jacob, M.; Rehm, M.; Bruegger, D.; Conzen, P.; Jacob, B.; Becker, B.F. Protection of glycocalyx decreases platelet adhesion after ischaemia/reperfusion: An animal study. Eur. J. Anaesthesiol. 2014, 31, 474–481. [Google Scholar] [CrossRef]
- Chappell, D.; Dörfler, N.; Jacob, M.; Rehm, M.; Welsch, U.; Conzen, P.; Becker, B.F. Glycocalyx protection reduces leukocyte adhesion after ischemia/reperfusion. Shock 2010, 34, 133–139. [Google Scholar] [CrossRef]
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B.F. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res. Cardiol. 2009, 104, 78–89. [Google Scholar] [CrossRef]
- Ehring, T.; Krajcar, M.; Baumgart, D.; Kompa, S.; Hümmelgen, M.; Heusch, G. Cholinergic and alpha-adrenergic coronary vasomotion [corrected] with increasing ischemia-reperfusion injury. Am. J. Physiol. 1995, 268, H886–H894. [Google Scholar] [CrossRef]
- Baumgart, D.; Haude, M.; Görge, G.; Liu, F.; Ge, J.; Grosse-Eggebrecht, C.; Erbel, R.; Heusch, G. Augmented alpha-adrenergic constriction of atherosclerotic human coronary arteries. Circulation 1999, 99, 2090–2097. [Google Scholar] [CrossRef] [Green Version]
- Driesen, R.B.; Zalewski, J.; Vanden Driessche, N.; Vermeulen, K.; Bogaert, J.; Sipido, K.R.; Van de Werf, F.; Claus, P. Histological correlate of a cardiac magnetic resonance imaged microvascular obstruction in a porcine model of ischemia-reperfusion. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2012, 21, 129–131. [Google Scholar] [CrossRef]
- Sheridan, F.M.; Dauber, I.M.; McMurtry, I.F.; Lesnefsky, E.J.; Horwitz, L.D. Role of leukocytes in coronary vascular endothelial injury due to ischemia and reperfusion. Circ. Res. 1991, 69, 1566–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogaki, S.; Sawa, Y.; Sano, T.; Matsushita, T.; Ohata, T.; Kurotobi, S.; Tojo, S.J.; Matsuda, H.; Okada, S. Selectin on activated platelets enhances neutrophil endothelial adherence in myocardial reperfusion injury. Cardiovasc. Res. 1999, 43, 968–973. [Google Scholar] [CrossRef] [Green Version]
- Porto, I.; Biasucci, L.M.; De Maria, G.L.; Leone, A.M.; Niccoli, G.; Burzotta, F.; Trani, C.; Tritarelli, A.; Vergallo, R.; Liuzzo, G.; et al. Intracoronary microparticles and microvascular obstruction in patients with ST elevation myocardial infarction undergoing primary percutaneous intervention. Eur. Heart J. 2012, 33, 2928–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörge, H.; Neumann, T.; Behrends, M.; Skyschally, A.; Schulz, R.; Kasper, C.; Erbel, R.; Heusch, G. Perfusion-contraction mismatch with coronary microvascular obstruction: Role of inflammation. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2587–H2592. [Google Scholar] [CrossRef]
- Beek, A.M.; Nijveldt, R.; van Rossum, A.C. Intramyocardial hemorrhage and microvascular obstruction after primary percutaneous coronary intervention. Int. J. Cardiovasc. Imaging 2010, 26, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Higginson, L.A.; White, F.; Heggtveit, H.A.; Sanders, T.M.; Bloor, C.M.; Covell, J.W. Determinants of myocardial hemorrhage after coronary reperfusion in the anesthetized dog. Circulation 1982, 65, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Betgem, R.P.; de Waard, G.A.; Nijveldt, R.; Beek, A.M.; Escaned, J.; van Royen, N. Intramyocardial haemorrhage after acute myocardial infarction. Nat. Rev. Cardiol. 2015, 12, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Daubert, M.A.; White, J.A.; Al-Khalidi, H.R.; Velazquez, E.J.; Rao, S.V.; Crowley, A.L.; Zeymer, U.; Kasprzak, J.D.; Guetta, V.; Krucoff, M.W.; et al. Cardiac remodeling after large ST-elevation myocardial infarction in the current therapeutic era. Am. Heart J. 2020, 223, 87–97. [Google Scholar] [CrossRef]
- Funaro, S.; La Torre, G.; Madonna, M.; Galiuto, L.; Scarà, A.; Labbadia, A.; Canali, E.; Mattatelli, A.; Fedele, F.; Alessandrini, F.; et al. Incidence, determinants, and prognostic value of reverse left ventricular remodelling after primary percutaneous coronary intervention: Results of the acute myocardial infarction contrast imaging (AMICI) multicenter study. Eur. Heart J. 2009, 30, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, L.; Morisco, C.; Assante di Panzillo, E.; Izzo, R.; Trimarco, B. Reverse left ventricular remodeling after acute myocardial infarction: The prognostic impact of left ventricular global torsion. Int. J. Cardiovasc. Imaging 2013, 29, 787–795. [Google Scholar] [CrossRef]
- Jolly, S.S.; Cairns, J.A.; Yusuf, S.; Rokoss, M.J.; Gao, P.; Meeks, B.; Kedev, S.; Stankovic, G.; Moreno, R.; Gershlick, A.; et al. Outcomes after thrombus aspiration for ST elevation myocardial infarction: 1-year follow-up of the prospective randomised TOTAL trial. Lancet 2016, 387, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Jolly, S.S.; James, S.; Džavík, V.; Cairns, J.A.; Mahmoud, K.D.; Zijlstra, F.; Yusuf, S.; Olivecrona, G.K.; Renlund, H.; Gao, P.; et al. Thrombus aspiration in ST-segment-elevation myocardial infarction: An individual patient meta-analysis: Thrombectomy trialists collaboration. Circulation 2017, 135, 143–152. [Google Scholar] [CrossRef]
- Lagerqvist, B.; Fröbert, O.; Olivecrona, G.K.; Gudnason, T.; Maeng, M.; Alström, P.; Andersson, J.; Calais, F.; Carlsson, J.; Collste, O.; et al. Outcomes 1 year after thrombus aspiration for myocardial infarction. N. Engl. J. Med. 2014, 371, 1111–1120. [Google Scholar] [CrossRef] [Green Version]
- Sardu, C.; Barbieri, M.; Balestrieri, M.L.; Siniscalchi, M.; Paolisso, P.; Calabrò, P.; Minicucci, F.; Signoriello, G.; Portoghese, M.; Mone, P.; et al. Thrombus aspiration in hyperglycemic ST-elevation myocardial infarction (STEMI) patients: Clinical outcomes at 1-year follow-up. Cardiovasc. Diabetol. 2018, 17, 152. [Google Scholar] [CrossRef] [Green Version]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The task force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39, 119–177. [Google Scholar] [CrossRef] [Green Version]
- Turer, A.T.; Hill, J.A. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am. J. Cardiol. 2010, 106, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Steg, P.G.; James, S.; Harrington, R.A.; Ardissino, D.; Becker, R.C.; Cannon, C.P.; Emanuelsson, H.; Finkelstein, A.; Husted, S.; Katus, H.; et al. Ticagrelor versus clopidogrel in patients with ST-elevation acute coronary syndromes intended for reperfusion with primary percutaneous coronary intervention: A platelet inhibition and patient outcomes (PLATO) trial subgroup analysis. Circulation 2010, 122, 2131–2141. [Google Scholar] [CrossRef] [Green Version]
- Valgimigli, M.; Bueno, H.; Byrne, R.A.; Collet, J.P.; Costa, F.; Jeppsson, A.; Juni, P.; Kastrati, A.; Kolh, P.; Mauri, L.; et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: The task force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2018, 39, 213–260. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef] [Green Version]
- Motovska, Z.; Hlinomaz, O.; Kala, P.; Hromadka, M.; Knot, J.; Varvarovsky, I.; Dusek, J.; Jarkovsky, J.; Miklik, R.; Rokyta, R.; et al. 1-Year outcomes of patients undergoing primary angioplasty for myocardial infarction treated with prasugrel versus ticagrelor. J. Am. Coll. Cardiol. 2018, 71, 371–381. [Google Scholar] [CrossRef]
- Motovska, Z.; Hlinomaz, O.; Miklik, R.; Hromadka, M.; Varvarovsky, I.; Dusek, J.; Knot, J.; Jarkovsky, J.; Kala, P.; Rokyta, R.; et al. Prasugrel Versus ticagrelor in patients with acute myocardial infarction treated with primary percutaneous coronary intervention: Multicenter randomized PRAGUE-18 study. Circulation 2016, 134, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Bonello, L.; Laine, M.; Kipson, N.; Mancini, J.; Helal, O.; Fromonot, J.; Gariboldi, V.; Condo, J.; Thuny, F.; Frere, C.; et al. Ticagrelor increases adenosine plasma concentration in patients with an acute coronary syndrome. J. Am. Coll. Cardiol. 2014, 63, 872–877. [Google Scholar] [CrossRef]
- Vilahur, G.; Gutiérrez, M.; Casani, L.; Varela, L.; Capdevila, A.; Pons-Lladó, G.; Carreras, F.; Carlsson, L.; Hidalgo, A.; Badimon, L. Protective effects of ticagrelor on myocardial injury after infarction. Circulation 2016, 134, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoeven, N.W.; Janssens, G.N.; Everaars, H.; Nap, A.; Lemkes, J.S.; de Waard, G.A.; van de Ven, P.M.; van Rossum, A.C.; Escaned, J.; Mejia-Renteria, H.; et al. Platelet inhibition, endothelial function, and clinical outcome in patients presenting with ST-segment-elevation myocardial infarction randomized to ticagrelor versus prasugrel maintenance therapy: Long-term follow-up of the REDUCE-MVI trial. J. Am. Heart Assoc. 2020, 9, e014411. [Google Scholar] [CrossRef] [PubMed]
- Zeymer, U.; Hohlfeld, T.; Vom Dahl, J.; Erbel, R.; Munzel, T.; Zahn, R.; Roitenberg, A.; Breitenstein, S.; Pap, A.F.; Trenk, D. Prospective, randomised trial of the time dependent antiplatelet effects of 500 mg and 250 mg acetylsalicylic acid i. v. and 300 mg p. o. in ACS (ACUTE). Thromb. Haemost. 2017, 117, 625–635. [Google Scholar] [CrossRef]
- Heestermans, A.A.; van Werkum, J.W.; Taubert, D.; Seesing, T.H.; von Beckerath, N.; Hackeng, C.M.; Schomig, E.; Verheugt, F.W.; ten Berg, J.M. Impaired bioavailability of clopidogrel in patients with a ST-segment elevation myocardial infarction. Thromb. Res. 2008, 122, 776–781. [Google Scholar] [CrossRef]
- Parodi, G.; Valenti, R.; Bellandi, B.; Migliorini, A.; Marcucci, R.; Comito, V.; Carrabba, N.; Santini, A.; Gensini, G.F.; Abbate, R.; et al. Comparison of prasugrel and ticagrelor loading doses in ST-segment elevation myocardial infarction patients: RAPID (Rapid Activity of Platelet Inhibitor Drugs) primary PCI study. J. Am. Coll. Cardiol. 2013, 61, 1601–1606. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, D.L.; Stone, G.W.; Mahaffey, K.W.; Gibson, C.M.; Steg, P.G.; Hamm, C.W.; Price, M.J.; Leonardi, S.; Gallup, D.; Bramucci, E.; et al. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N. Engl. J. Med. 2013, 368, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- Franchi, F.; Rollini, F.; Rivas, A.; Wali, M.; Briceno, M.; Agarwal, M.; Shaikh, Z.; Nawaz, A.; Silva, G.; Been, L.; et al. Platelet inhibition with cangrelor and crushed ticagrelor in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention. Circulation 2019, 139, 1661–1670. [Google Scholar] [CrossRef]
- Abo-Aly, M.; George, B.; Shokri, E.; Chelvarajan, L.; El-Helw, M.; Smyth, S.S.; Abdel-Latif, A. Cangrelor in addition to standard therapy reduces cardiac damage and inflammatory markers in patients with ST-segment elevation myocardial infarction. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef]
- Mehilli, J.; Kastrati, A.; Schulz, S.; Frungel, S.; Nekolla, S.G.; Moshage, W.; Dotzer, F.; Huber, K.; Pache, J.; Dirschinger, J.; et al. Abciximab in patients with acute ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention after clopidogrel loading: A randomized double-blind trial. Circulation 2009, 119, 1933–1940. [Google Scholar] [CrossRef]
- Gargiulo, G.; Esposito, G.; Avvedimento, M.; Nagler, M.; Minuz, P.; Campo, G.; Gragnano, F.; Manavifar, N.; Piccolo, R.; Tebaldi, M.; et al. Cangrelor, tirofiban, and chewed or standard prasugrel regimens in patients with ST-segment-elevation myocardial infarction: Primary results of the FABOLUS-FASTER trial. Circulation 2020, 142, 441–454. [Google Scholar] [CrossRef]
- Gargiulo, G.; Esposito, G.; Cirillo, P.; Nagler, M.; Minuz, P.; Campo, G.; Gragnano, F.; Manavifar, N.; Piccolo, R.; Avvedimento, M.; et al. Facilitation through aggrastat or cangrelor bolus and infusion over prasugrel: A multicenter randomized open-label trial in patientS with ST-elevation myocardial infarction referred for primary percutaneous intervention (FABOLUS FASTER) trial: Design and rationale: The FABOLUS FASTER trial. J. Cardiovasc. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The beta(2)-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through G(i)-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.Z.; Zheng, M.; Koch, W.J.; Lefkowitz, R.J.; Kobilka, B.K.; Xiao, R.P. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 1607–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Woo, A.Y.; Yang, D.; Cheng, H.; Crow, M.T.; Xiao, R.P. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 10833–10839. [Google Scholar] [CrossRef]
- Zhu, W.Z.; Wang, S.Q.; Chakir, K.; Yang, D.; Zhang, T.; Brown, J.H.; Devic, E.; Kobilka, B.K.; Cheng, H.; Xiao, R.P. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J. Clin. Investig. 2003, 111, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Li, Y.; Jiang, S.; Xie, Y.P.; Guo, A.; Kutschke, W.; Zimmerman, K.; Weiss, R.M.; Miller, F.J.; Anderson, M.E.; et al. β-Adrenergic receptor antagonists ameliorate myocyte T-tubule remodeling following myocardial infarction. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2531–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo-Gonzalez, M.; Galán-Arriola, C.; Rossello, X.; González-Del-Hoyo, M.; Vilchez, J.P.; Higuero-Verdejo, M.I.; García-Ruiz, J.M.; López-Martín, G.J.; Sánchez-González, J.; Oliver, E.; et al. Metoprolol blunts the time-dependent progression of infarct size. Basic Res. Cardiol. 2020, 115, 55. [Google Scholar] [CrossRef]
- Qin, W.; Zhang, L.; Li, Z.; Xiao, D.; Zhang, Y.; Yang, H.; Zhang, H.; Xu, C.; Zhang, Y. Metoprolol protects against myocardial infarction by inhibiting miR-1 expression in rats. J. Pharm. Pharmacol. 2020, 72, 76–83. [Google Scholar] [CrossRef]
- Ibanez, B.; Macaya, C.; Sánchez-Brunete, V.; Pizarro, G.; Fernández-Friera, L.; Mateos, A.; Fernández-Ortiz, A.; García-Ruiz, J.M.; García-Álvarez, A.; Iñiguez, A.; et al. Effect of early metoprolol on infarct size in ST-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: The effect of metoprolol in cardioprotection during an acute myocardial infarction (METOCARD-CNIC) trial. Circulation 2013, 128, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- García-Ruiz, J.M.; Fernández-Jiménez, R.; García-Alvarez, A.; Pizarro, G.; Galán-Arriola, C.; Fernández-Friera, L.; Mateos, A.; Nuno-Ayala, M.; Aguero, J.; Sánchez-González, J.; et al. Impact of the timing of metoprolol administration during STEMI on infarct size and ventricular function. J. Am. Coll. Cardiol. 2016, 67, 2093–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roolvink, V.; Ibáñez, B.; Ottervanger, J.P.; Pizarro, G.; van Royen, N.; Mateos, A.; Dambrink, J.E.; Escalera, N.; Lipsic, E.; Albarran, A.; et al. Early intravenous beta-blockers in patients with ST-segment elevation myocardial infarction before primary percutaneous coronary intervention. J. Am. Coll. Cardiol. 2016, 67, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Maczewski, M.; Borys, M.; Kacprzak, P.; Gdowski, T.; Kowalewski, M.; Wojciechowski, D. Late ventricular remodeling in non-reperfused acute myocardial infarction in humans is predicted by angiotensin II type 1 receptor density on blood platelets. Int. J. Cardiol. 2008, 127, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Maczewski, M.; Borys, M.; Kacprzak, P.; Gdowski, T.; Wojciechowski, D. Angiotensin II AT1 receptor density on blood platelets predicts early left ventricular remodelling in non-reperfused acute myocardial infarction in humans. Eur. J. Heart Fail. 2006, 8, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Bellis, A.; Sorriento, D.; Fiordelisi, A.; Izzo, R.; Sadoshima, J.; Mauro, C.; Cerasuolo, F.; Mancusi, C.; Barbato, E.; Pilato, E.; et al. Autocrine bradykinin release promotes ischemic preconditioning-induced cytoprotection in bovine aortic endothelial cells. Int. J. Mol. Sci. 2020, 21, 2965. [Google Scholar] [CrossRef]
- Sheng, Z.; Yao, Y.; Li, Y.; Yan, F.; Huang, J.; Ma, G. Bradykinin preconditioning improves therapeutic potential of human endothelial progenitor cells in infarcted myocardium. PLoS ONE 2013, 8, e81505. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Hamamoto, H.; Honda, Y.; Hirose, M.; Furukawa, S.; Kimura, E. Changes in components of kinin system and hemodynamics in acute myocardial infarction. Am. Heart J. 1978, 95, 619–626. [Google Scholar] [CrossRef]
- Schaefer, U.; Kurz, T.; Bonnemeier, H.; Dendorfer, A.; Hartmann, F.; Schunkert, H.; Richardt, G. Intracoronary enalaprilat during angioplasty for acute myocardial infarction: Alleviation of postischaemic neurohumoral and inflammatory stress? J. Intern. Med. 2007, 261, 188–200. [Google Scholar] [CrossRef]
- Prasad, A.; Husain, S.; Quyyumi, A.A. Abnormal flow-mediated epicardial vasomotion in human coronary arteries is improved by angiotensin-converting enzyme inhibition: A potential role of bradykinin. J. Am. Coll. Cardiol. 1999, 33, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.; Peto, R.; Flather, M.; Parish, S.; Sleight, P.; Conway, M.; Perez, J.E.; Pipilis, A.; Baigent, C.; Barnett, D. ISIS-4: A randomised factorial trial assessing early oral captopril, oral mononitrate, and intravenous magnesium sulphate in 58,050 patients with suspected acute myocardial infarction. ISIS-4 (Fourth International Study of Infarct Survival) Collaborative Group. Lancet 1995, 345, 669–685. [Google Scholar]
- Franzosi, M.G. Indications for ACE inhibitors in the early treatment of acute myocardial infarction: Systematic overview of individual data from 100,000 patients in randomized trials. ACE Inhibitor Myocardial Infarction Collaborative Group. Circulation 1998, 97, 2202–2212. [Google Scholar] [CrossRef] [Green Version]
- Ball, S.G.; Hall, A.S.; Murray, G.D. ACE inhibition, atherosclerosis and myocardial infarction--the AIRE Study in practice. Acute infarction ramipril efficacy study. Eur. Heart J. 1994, 15 (Suppl. SB), 20–25. [Google Scholar] [CrossRef]
- Kober, L.; Torp-Pedersen, C.; Carlsen, J.E.; Bagger, H.; Eliasen, P.; Lyngborg, K.; Videbaek, J.; Cole, D.S.; Auclert, L.; Pauly, N.C. A clinical trial of the angiotensin-converting-enzyme inhibitor trandolapril in patients with left ventricular dysfunction after myocardial infarction. Trandolapril Cardiac Evaluation (TRACE) Study Group. N. Engl. J. Med. 1995, 333, 1670–1676. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Braunwald, E.; Moye, L.A.; Basta, L.; Brown, E.J., Jr.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C.; et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, M.A.; Greaves, S.C.; Arnold, J.M.; Glynn, R.J.; LaMotte, F.S.; Lee, R.T.; Menapace, F.J., Jr.; Rapaport, E.; Ridker, P.M.; Rouleau, J.L.; et al. Early versus delayed angiotensin-converting enzyme inhibition therapy in acute myocardial infarction. The healing and early afterload reducing therapy trial. Circulation 1997, 95, 2643–2651. [Google Scholar] [CrossRef]
- Oishi, Y.; Ozono, R.; Yoshizumi, M.; Akishita, M.; Horiuchi, M.; Oshima, T. AT2 receptor mediates the cardioprotective effects of AT1 receptor antagonist in post-myocardial infarction remodeling. Life Sci. 2006, 80, 82–88. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; McMurray, J.J.; Velazquez, E.J.; Rouleau, J.L.; Kober, L.; Maggioni, A.P.; Solomon, S.D.; Swedberg, K.; Van de Werf, F.; White, H.; et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N. Engl. J. Med. 2003, 349, 1893–1906. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, E.; Kataoka, K.; Dong, Y.F.; Nakamura, T.; Fukuda, M.; Tokutomi, Y.; Matsuba, S.; Nako, H.; Nakagata, N.; Kaneko, T.; et al. Aliskiren enhances the protective effects of valsartan against cardiovascular and renal injury in endothelial nitric oxide synthase-deficient mice. Hypertension 2009, 54, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Shin, S.H.; Shah, A.; Skali, H.; Desai, A.; Kober, L.; Maggioni, A.P.; Rouleau, J.L.; Kelly, R.Y.; Hester, A.; et al. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur. Heart J. 2011, 32, 1227–1234. [Google Scholar] [CrossRef]
- Iraqi, W.; Rossignol, P.; Angioi, M.; Fay, R.; Nuée, J.; Ketelslegers, J.M.; Vincent, J.; Pitt, B.; Zannad, F. Extracellular cardiac matrix biomarkers in patients with acute myocardial infarction complicated by left ventricular dysfunction and heart failure: Insights from the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS) study. Circulation 2009, 119, 2471–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Montalescot, G.; Pitt, B.; Lopez de Sa, E.; Hamm, C.W.; Flather, M.; Verheugt, F.; Shi, H.; Turgonyi, E.; Orri, M.; Vincent, J.; et al. Early eplerenone treatment in patients with acute ST-elevation myocardial infarction without heart failure: The randomized double-blind reminder study. Eur. Heart J. 2014, 35, 2295–2302. [Google Scholar] [CrossRef] [Green Version]
- Beygui, F.; Cayla, G.; Roule, V.; Roubille, F.; Delarche, N.; Silvain, J.; Van Belle, E.; Belle, L.; Galinier, M.; Motreff, P.; et al. Early aldosterone blockade in acute myocardial infarction: The ALBATROSS randomized clinical trial. J. Am. Coll. Cardiol. 2016, 67, 1917–1927. [Google Scholar] [CrossRef]
- Girerd, N.; Collier, T.; Pocock, S.; Krum, H.; McMurray, J.J.; Swedberg, K.; Van Veldhuisen, D.J.; Vincent, J.; Pitt, B.; Zannad, F. Clinical benefits of eplerenone in patients with systolic heart failure and mild symptoms when initiated shortly after hospital discharge: Analysis from the EMPHASIS-HF trial. Eur. Heart J. 2015, 36, 2310–2317. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; Group, E.-H.S. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Stienen, S.; Ferreira, J.P.; Bär, C.; Thum, T.; Barros, A.; Pitt, B.; Girerd, N.; Rossignol, P.; Zannad, F. Serum microRNAs and antifibrotic response to eplerenone in acute myocardial infarction complicated by systolic dysfunction. Int. J. Cardiol. 2021. [Google Scholar] [CrossRef]
- Aung, N.; Sanghvi, M.M.; Piechnik, S.K.; Neubauer, S.; Munroe, P.B.; Petersen, S.E. The effect of blood lipids on the left ventricle: A mendelian randomization study. J. Am. Coll. Cardiol. 2020, 76, 2477–2488. [Google Scholar] [CrossRef] [PubMed]
- Buono, F.; Spinelli, L.; Giallauria, F.; Assante di Panzillo, E.; Di Marino, S.; Ferrara, F.; Vigorito, C.; Trimarco, B.; Morisco, C. Usefulness of satisfactory control of low-density lipoprotein cholesterol to predict left ventricular remodeling after a first ST-elevation myocardial infarction successfully reperfused. Am. J. Cardiol. 2011, 107, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M., Jr.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N. Engl. J. Med. 2008, 359, 2195–2207. [Google Scholar] [CrossRef] [Green Version]
- Navarese, E.P.; Kowalewski, M.; Andreotti, F.; van Wely, M.; Camaro, C.; Kolodziejczak, M.; Gorny, B.; Wirianta, J.; Kubica, J.; Kelm, M.; et al. Meta-analysis of time-related benefits of statin therapy in patients with acute coronary syndrome undergoing percutaneous coronary intervention. Am. J. Cardiol. 2014, 113, 1753–1764. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Fayyad, R.; Szarek, M.; DeMicco, D.; Olsson, A.G. Early, intensive statin treatment reduces ‘hard’ cardiovascular outcomes after acute coronary syndrome. Eur. J. Prev. Cardiol. 2017, 24, 1294–1296. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, J.; Choi, D.; Lee, C.J.; Lee, S.H.; Ko, Y.G.; Hong, M.K.; Kim, B.K.; Oh, S.J.; Jeon, D.W.; et al. Efficacy of high-dose atorvastatin loading before primary percutaneous coronary intervention in ST-segment elevation myocardial infarction: The STATIN STEMI trial. JACC Cardiovasc. Interv. 2010, 3, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Iwakura, K.; Ito, H.; Kawano, S.; Okamura, A.; Kurotobi, T.; Date, M.; Inoue, K.; Fujii, K. Chronic pre-treatment of statins is associated with the reduction of the no-reflow phenomenon in the patients with reperfused acute myocardial infarction. Eur. Heart J. 2006, 27, 534–539. [Google Scholar] [CrossRef] [Green Version]
- Marenzi, G.; Cosentino, N.; Cortinovis, S.; Milazzo, V.; Rubino, M.; Cabiati, A.; De Metrio, M.; Moltrasio, M.; Lauri, G.; Campodonico, J.; et al. Myocardial infarct size in patients on long-term statin therapy undergoing primary percutaneous coronary intervention for ST-elevation myocardial infarction. Am. J. Cardiol. 2015, 116, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Berwanger, O.; Santucci, E.V.; de Barros, E.S.P.G.M.; Jesuíno, I.A.; Damiani, L.P.; Barbosa, L.M.; Santos, R.H.N.; Laranjeira, L.N.; Egydio, F.M.; Borges de Oliveira, J.A.; et al. Effect of loading dose of atorvastatin prior to planned percutaneous coronary intervention on major adverse cardiovascular events in acute coronary syndrome: The SECURE-PCI randomized clinical trial. JAMA 2018, 319, 1331–1340. [Google Scholar] [CrossRef]
- Xie, W.; Li, P.; Wang, Z.; Chen, J.; Lin, Z.; Liang, X.; Mo, Y. Rosuvastatin may reduce the incidence of cardiovascular events in patients with acute coronary syndromes receiving percutaneous coronary intervention by suppressing miR-155/SHIP-1 signaling pathway. Cardiovasc. Ther. 2014, 32, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Bianconi, V.; Sahebkar, A.; Atkin, S.L.; Pirro, M. The regulation and importance of monocyte chemoattractant protein-1. Curr. Opin. Hematol. 2018, 25, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Clinical implications for statin pleiotropy. Curr. Opin. Lipidol. 2005, 16, 624–629. [Google Scholar] [CrossRef]
- Verdoia, M.; Pergolini, P.; Rolla, R.; Nardin, M.; Schaffer, A.; Barbieri, L.; Daffara, V.; Marino, P.; Bellomo, G.; Suryapranata, H.; et al. Impact of high-dose statins on vitamin D levels and platelet function in patients with coronary artery disease. Thromb. Res. 2017, 150, 90–95. [Google Scholar] [CrossRef]
- Rumley, A.; Lowe, G.D.; Sweetnam, P.M.; Yarnell, J.W.; Ford, R.P. Factor VIII, von Willebrand factor and the risk of major ischaemic heart disease in the Caerphilly Heart Study. Br. J. Haematol. 1999, 105, 110–116. [Google Scholar] [CrossRef]
- Biedermann, J.S.; Kruip, M.; van der Meer, F.J.; Rosendaal, F.R.; Leebeek, F.W.G.; Cannegieter, S.C.; Lijfering, W.M. Rosuvastatin use improves measures of coagulation in patients with venous thrombosis. Eur. Heart J. 2018, 39, 1740–1747. [Google Scholar] [CrossRef]
- Satoh, M.; Tabuchi, T.; Minami, Y.; Takahashi, Y.; Itoh, T.; Nakamura, M. Expression of let-7i is associated with Toll-like receptor 4 signal in coronary artery disease: Effect of statins on let-7i and Toll-like receptor 4 signal. Immunobiology 2012, 217, 533–539. [Google Scholar] [CrossRef]
- Frostegård, J.; Zhang, Y.; Sun, J.; Yan, K.; Liu, A. Oxidized low-density lipoprotein (OxLDL)-treated dendritic cells promote activation of t cells in human atherosclerotic plaque and blood, which is repressed by statins: microRNA let-7c is integral to the effect. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Ming, J.Y.; Fiskesund, R.; Ninio, E.; Karabina, S.A.; Bergmark, C.; Frostegård, A.G.; Frostegård, J. Induction of dendritic cell-mediated T-cell activation by modified but not native low-density lipoprotein in humans and inhibition by annexin a5: Involvement of heat shock proteins. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Hou, R.; Ma, A.; Wang, T.; Wu, M.; Zhu, X.; Yang, S.; Xiao, X. Atorvastatin upregulates the expression of miR-126 in apolipoprotein e-knockout mice with carotid atherosclerotic plaque. Cell. Mol. Neurobiol. 2017, 37, 29–36. [Google Scholar] [CrossRef]
- Minami, Y.; Satoh, M.; Maesawa, C.; Takahashi, Y.; Tabuchi, T.; Itoh, T.; Nakamura, M. Effect of atorvastatin on microRNA 221/222 expression in endothelial progenitor cells obtained from patients with coronary artery disease. Eur. J. Clin. Investig. 2009, 39, 359–367. [Google Scholar] [CrossRef]
- Mohajeri, M.; Banach, M.; Atkin, S.L.; Butler, A.E.; Ruscica, M.; Watts, G.F.; Sahebkar, A. MicroRNAs: Novel molecular targets and response modulators of statin therapy. Trends Pharmacol. Sci. 2018, 39, 967–981. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Theroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe added to statin therapy after acute coronary syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giugliano, R.P.; Cannon, C.P.; Blazing, M.A.; Nicolau, J.C.; Corbalan, R.; Spinar, J.; Park, J.G.; White, J.A.; Bohula, E.A.; Braunwald, E.; et al. Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and safety in patients with versus without diabetes mellitus: Results from IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation 2018, 137, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.A.; Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; White, J.A.; Lokhnygina, Y.; Reist, C.; Im, K.; Bohula, E.A.; Isaza, D.; et al. Reduction in total cardiovascular events with ezetimibe/simvastatin post-acute coronary syndrome: The IMPROVE-IT trial. J. Am. Coll. Cardiol. 2016, 67, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Bohula, E.A.; Wiviott, S.D.; Giugliano, R.P.; Blazing, M.A.; Park, J.G.; Murphy, S.A.; White, J.A.; Mach, F.; Van de Werf, F.; Dalby, A.J.; et al. Prevention of stroke with the addition of ezetimibe to statin therapy in patients with acute coronary syndrome in IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation 2017, 136, 2440–2450. [Google Scholar] [CrossRef]
- Bohula, E.A.; Morrow, D.A.; Giugliano, R.P.; Blazing, M.A.; He, P.; Park, J.G.; Murphy, S.A.; White, J.A.; Kesaniemi, Y.A.; Pedersen, T.R.; et al. Atherothrombotic risk stratification and ezetimibe for secondary prevention. J. Am. Coll. Cardiol. 2017, 69, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Yasue, H.; Yoshimura, M.; Ogawa, H.; Jougasaki, M.; Matsumura, T.; Mukoyama, M.; Nakao, K. Increased plasma levels of brain natriuretic peptide in patients with acute myocardial infarction. Circulation 1993, 88, 82–91. [Google Scholar] [CrossRef] [Green Version]
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, F.L.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: Mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J. Neprilysin inhibitors and bradykinin. Front. Med. 2018, 5, 257. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.W.; Claggett, B.L.; O’Meara, E.; Prescott, M.F.; Pfeffer, M.A.; Shah, S.J.; Redfield, M.M.; Zannad, F.; Chiang, L.M.; Rizkala, A.R.; et al. Effect of sacubitril/valsartan on biomarkers of extracellular matrix regulation in patients with HFpEF. J. Am. Coll. Cardiol. 2020, 76, 503–514. [Google Scholar] [CrossRef]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef] [Green Version]
- Vaskova, E.; Ikeda, G.; Tada, Y.; Wahlquist, C.; Mercola, M.; Yang, P.C. Sacubitril/valsartan improves cardiac function and decreases myocardial fibrosis via downregulation of exosomal miR-181a in a rodent chronic myocardial infarction model. J. Am. Heart Assoc. 2020, 9, e015640. [Google Scholar] [CrossRef]
- DeVore, A.D.; Braunwald, E.; Morrow, D.A.; Duffy, C.I.; Ambrosy, A.P.; Chakraborty, H.; McCague, K.; Rocha, R.; Velazquez, E.J. Initiation of angiotensin-neprilysin inhibition after acute decompensated heart failure: Secondary analysis of the open-label extension of the PIONEER-HF trial. JAMA Cardiol. 2020, 5, 202–207. [Google Scholar] [CrossRef]
- Guo, Y.; Yan, B.; Gui, Y.; Tang, Z.; Tai, S.; Zhou, S. Physiology and role of PCSK9 in vascular disease: Potential impact of localized PCSK9 in vascular wall. J. Cell. Physiol. 2021, 236, 2333–2351. [Google Scholar] [CrossRef]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Ngo Sock, E.T.; Ong, H.; Mayer, G. PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. J. Cell. Physiol. 2015, 35, 2517–2525. [Google Scholar] [CrossRef]
- Gencer, B.; Mach, F. Lipid management in ACS: Should we go lower faster? Atherosclerosis 2018, 275, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Navarese, E.P.; Kolodziejczak, M.; Winter, M.P.; Alimohammadi, A.; Lang, I.M.; Buffon, A.; Lip, G.Y.; Siller-Matula, J.M. Association of PCSK9 with platelet reactivity in patients with acute coronary syndrome treated with prasugrel or ticagrelor: The PCSK9-REACT study. Int. J. Cardiol. 2017, 227, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, X.; Liu, S.; Shahanawaz, J.; Theus, S.; Fan, Y.; Deng, X.; Zhou, S.; Mehta, J.L. PCSK9 expression in the ischaemic heart and its relationship to infarct size, cardiac function, and development of autophagy. Cardiovasc. Res. 2018, 114, 1738–1751. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; De Ferrari, G.M.; Giugliano, R.P.; Huber, K.; Lewis, B.S.; Ferreira, J.; Kuder, J.F.; Murphy, S.A.; Wiviott, S.D.; Kurtz, C.E.; et al. Clinical benefit of evolocumab by severity and extent of coronary artery disease. Circulation 2018, 138, 756–766. [Google Scholar] [CrossRef]
- Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; Jukema, J.W.; Lecorps, G.; Mahaffey, K.W.; Moryusef, A.; et al. Intensity of statin treatment after acute coronary syndrome, residual risk, and its modification by alirocumab: Insights from the ODYSSEY OUTCOMES trial. N. Engl. J. Med. 2021, 28, 33–43. [Google Scholar] [CrossRef]
- Koskinas, K.C.; Windecker, S.; Buhayer, A.; Gencer, B.; Pedrazzini, G.; Mueller, C.; Cook, S.; Muller, O.; Matter, C.M.; Räber, L.; et al. Design of the randomized, placebo-controlled evolocumab for early reduction of LDL-cholesterol levels in patients with acute coronary syndromes (EVOPACS) trial. Clin. Cardiol. 2018, 41, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Koskinas, K.C.; Windecker, S.; Pedrazzini, G.; Mueller, C.; Cook, S.; Matter, C.M.; Muller, O.; Häner, J.; Gencer, B.; Crljenica, C.; et al. Evolocumab for early reduction of LDL cholesterol levels in patients with acute coronary syndromes (EVOPACS). J. Am. Coll. Cardiol. 2019, 74, 2452–2462. [Google Scholar] [CrossRef]
- Leucker, T.M.; Blaha, M.J.; Jones, S.R.; Vavuranakis, M.A.; Williams, M.S.; Lai, H.; Schindler, T.H.; Latina, J.; Schulman, S.P.; Gerstenblith, G. Effect of evolocumab on atherogenic lipoproteins during the peri- and early postinfarction period: A placebo-controlled, randomized trial. Circulation 2020, 142, 419–421. [Google Scholar] [CrossRef]
- Trankle, C.R.; Wohlford, G.; Buckley, L.F.; Kadariya, D.; Ravindra, K.; Markley, R.; Park, T.S.; Potere, N.; Van Tassell, B.W.; Abbate, A. Alirocumab in acute myocardial infarction: Results from the virginia commonwealth university alirocumab response trial (VCU-AlirocRT). J. Cardiovasc. Pharmacol. 2019, 74, 266–269. [Google Scholar] [CrossRef]
- Ridker, P.M. Mortality differences associated with treatment responses in CANTOS and FOURIER: Insights and implications. Circulation 2018, 137, 1763–1766. [Google Scholar] [CrossRef]
- Van Craeyveld, E.; Jacobs, F.; Gordts, S.C.; De Geest, B. Low-density lipoprotein receptor gene transfer in hypercholesterolemic mice improves cardiac function after myocardial infarction. Gene Ther. 2012, 19, 860–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: A systematic review and meta-analysis. JAMA 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, B.J.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.K.; Boekholdt, S.M.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Navarese, E.P.; Kolodziejczak, M.; Kereiakes, D.J.; Tantry, U.S.; O’Connor, C.; Gurbel, P.A. Proprotein convertase subtilisin/kexin type 9 monoclonal antibodies for acute coronary syndrome: A narrative review. Ann. Intern. Med. 2016, 164, 600–607. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Shmookler Reis, R.J.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Banach, M. Lipoprotein(a) and inhibitors of proprotein convertase subtilisin/kexin type 9. J. Thorac. Dis. 2017, 9, E78–E82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) enhances platelet activation, thrombosis, and myocardial infarct expansion by binding to platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Minana, G.; Nunez, J.; Bayes-Genis, A.; Revuelta-Lopez, E.; Rios-Navarro, C.; Nunez, E.; Chorro, F.J.; Lopez-Lereu, M.P.; Monmeneu, J.V.; Lupon, J.; et al. Role of PCSK9 in the course of ejection fraction change after ST-segment elevation myocardial infarction: A pilot study. ESC Heart Fail. 2020. [Google Scholar] [CrossRef]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Gerstein, H.C. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: A systematic overview. Lancet 2000, 355, 773–778. [Google Scholar] [CrossRef]
- Kosiborod, M.; Rathore, S.S.; Inzucchi, S.E.; Masoudi, F.A.; Wang, Y.; Havranek, E.P.; Krumholz, H.M. Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction: Implications for patients with and without recognized diabetes. Circulation 2005, 111, 3078–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norhammar, A.M.; Ryden, L.; Malmberg, K. Admission plasma glucose. Independent risk factor for long-term prognosis after myocardial infarction even in nondiabetic patients. Diabetes Care 1999, 22, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Wahab, N.N.; Cowden, E.A.; Pearce, N.J.; Gardner, M.J.; Merry, H.; Cox, J.L. Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era? J. Am. Coll. Cardiol. 2002, 40, 1748–1754. [Google Scholar] [CrossRef] [Green Version]
- Bellis, A.; Mauro, C.; Barbato, E.; Ceriello, A.; Cittadini, A.; Morisco, C. Stress-induced hyperglycaemia in non-diabetic patients with acute coronary syndrome: From molecular mechanisms to new therapeutic perspectives. Int. J. Mol. Sci. 2021, 22, 775. [Google Scholar] [CrossRef]
- Eitel, I.; Hintze, S.; de Waha, S.; Fuernau, G.; Lurz, P.; Desch, S.; Schuler, G.; Thiele, H. Prognostic impact of hyperglycemia in nondiabetic and diabetic patients with ST-elevation myocardial infarction: Insights from contrast-enhanced magnetic resonance imaging. Int. J. Mol. Sci. 2012, 5, 708–718. [Google Scholar] [CrossRef] [Green Version]
- Lønborg, J.; Vejlstrup, N.; Kelbæk, H.; Nepper-Christensen, L.; Jørgensen, E.; Helqvist, S.; Holmvang, L.; Saunamäki, K.; Bøtker, H.E.; Kim, W.Y.; et al. Impact of acute hyperglycemia on myocardial infarct size, area at risk, and salvage in patients with STEMI and the association with exenatide treatment: Results from a randomized study. Diabetes 2014, 63, 2474–2485. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.W.; Wong, V.W.; McLean, M. The Hyperglycemia: Intensive insulin infusion in infarction (HI-5) study: A randomized controlled trial of insulin infusion therapy for myocardial infarction. Diabetes Care 2006, 29, 765–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmberg, K.; Ryden, L.; Wedel, H.; Birkeland, K.; Bootsma, A.; Dickstein, K.; Efendic, S.; Fisher, M.; Hamsten, A.; Herlitz, J.; et al. Intense metabolic control by means of insulin in patients with diabetes mellitus and acute myocardial infarction (DIGAMI 2): Effects on mortality and morbidity. Eur. Heart J. 2005, 26, 650–661. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Zhou, N.; Nan, Y.; Zhang, L.; Li, Y.; Hao, X.; Xiong, L.; Lau, W.B.; Ma, X.L.; Wang, H.; et al. Effective glycaemic control critically determines insulin cardioprotection against ischaemia/reperfusion injury in anaesthetized dogs. Cardiovasc. Res. 2014, 103, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Malmberg, K. Prospective randomised study of intensive insulin treatment on long term survival after acute myocardial infarction in patients with diabetes mellitus. DIGAMI (Diabetes Mellitus, Insulin Glucose Infusion in Acute Myocardial Infarction) Study Group. BMJ 1997, 314, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, K.; Norhammar, A.; Wedel, H.; Ryden, L. Glycometabolic state at admission: Important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: Long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation 1999, 99, 2626–2632. [Google Scholar]
- Ritsinger, V.; Malmberg, K.; Martensson, A.; Ryden, L.; Wedel, H.; Norhammar, A. Intensified insulin-based glycaemic control after myocardial infarction: Mortality during 20 year follow-up of the randomised Diabetes Mellitus Insulin Glucose Infusion in Acute Myocardial Infarction (DIGAMI 1) trial. Lancet Diabetes Endocrinol. 2014, 2, 627–633. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef] [Green Version]
- Lonborg, J.; Vejlstrup, N.; Kelbaek, H.; Botker, H.E.; Kim, W.Y.; Mathiasen, A.B.; Jorgensen, E.; Helqvist, S.; Saunamaki, K.; Clemmensen, P.; et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur. Heart J. 2012, 33, 1491–1499. [Google Scholar] [CrossRef]
- Woo, J.S.; Kim, W.; Ha, S.J.; Kim, J.B.; Kim, S.J.; Kim, W.S.; Seon, H.J.; Kim, K.S. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: Results of exenatide myocardial protection in revascularization study. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2252–2260. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.R.; Chen, Y.D.; Tian, F.; Yang, N.; Cheng, L.Q.; Hu, S.Y.; Wang, J.; Yang, J.J.; Wang, S.F.; Gu, X.F. Effects of liraglutide on reperfusion injury in patients with ST-segment-elevation myocardial infarction. Circ. Cardiovasc. Imaging 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.R.; Hu, S.Y.; Chen, Y.D.; Zhang, Y.; Qian, G.; Wang, J.; Yang, J.J.; Wang, Z.F.; Tian, F.; Ning, Q.X. Effects of liraglutide on left ventricular function in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am. Heart J. 2015, 170, 845–854. [Google Scholar] [CrossRef]
- Chen, W.R.; Shen, X.Q.; Zhang, Y.; Chen, Y.D.; Hu, S.Y.; Qian, G.; Wang, J.; Yang, J.J.; Wang, Z.F.; Tian, F. Effects of liraglutide on left ventricular function in patients with non-ST-segment elevation myocardial infarction. Endocrine 2016, 52, 516–526. [Google Scholar] [CrossRef]
- Chang, G.; Zhang, D.; Yu, H.; Zhang, P.; Wang, Y.; Zheng, A.; Qin, S. Cardioprotective effects of exenatide against oxidative stress-induced injury. Int. J. Mol. Med. 2013, 32, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, J.; Hu, L.; Liang, M.; Wang, X.; Feng, S.; Shen, J.; Luan, X. Liraglutide regulates the viability of pancreatic α-cells and pancreatic β-cells through cAMP-PKA signal pathway. Life Sci. 2018, 195, 87–94. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J.; Cavender, M.A.; Abd El Aziz, M.; Drucker, D.J. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation 2017, 136, 849–870. [Google Scholar] [CrossRef]
- Verbrugge, F.H. Role of SGLT2 inhibitors in patients with diabetes mellitus and heart failure. Curr. Heart Fail. Rep. 2017, 14, 275–283. [Google Scholar] [CrossRef]
- Verma, S.; Mazer, C.D.; Yan, A.T.; Mason, T.; Garg, V.; Teoh, H.; Zuo, F.; Quan, A.; Farkouh, M.E.; Fitchett, D.H.; et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes mellitus and coronary artery disease: The EMPA-HEART cardiolink-6 randomized clinical trial. Cardiovasc. Diabetol. 2019, 140, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Furtado, R.H.M.; Bonaca, M.P.; Raz, I.; Zelniker, T.A.; Mosenzon, O.; Cahn, A.; Kuder, J.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; et al. Dapagliflozin and cardiovascular outcomes in patients with type 2 diabetes mellitus and previous myocardial infarction. Circulation 2019, 139, 2516–2527. [Google Scholar] [CrossRef]
- Fudim, M.; White, J.; Pagidipati, N.J.; Lokhnygina, Y.; Wainstein, J.; Murin, J.; Iqbal, N.; Öhman, P.; Lopes, R.D.; Reicher, B.; et al. Effect of once-weekly exenatide in patients with type 2 diabetes mellitus with and without heart failure and heart failure-related outcomes: Insights from the EXSCEL trial. Circulation 2019, 140, 1613–1622. [Google Scholar] [CrossRef]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B., Sr.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Husain, M.; Bain, S.C. Semaglutide (SUSTAIN and PIONEER) reduces cardiovascular events in type 2 diabetes across varying cardiovascular risk. Diabetes Obes. Metab. 2020, 22, 442–451. [Google Scholar] [CrossRef]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Marso, S.P.; Baeres, F.M.M.; Bain, S.C.; Goldman, B.; Husain, M.; Nauck, M.A.; Poulter, N.R.; Pratley, R.E.; Thomsen, A.B.; Buse, J.B. Effects of liraglutide on cardiovascular outcomes in patients with diabetes with or without heart failure. J. Am. Coll. Cardiol. 2020, 75, 1128–1141. [Google Scholar] [CrossRef]
- Scirica, B.M.; Braunwald, E.; Raz, I.; Cavender, M.A.; Morrow, D.A.; Jarolim, P.; Udell, J.A.; Mosenzon, O.; Im, K.; Umez-Eronini, A.A.; et al. Heart failure, saxagliptin, and diabetes mellitus: Observations from the SAVOR-TIMI 53 randomized trial. Circulation 2014, 130, 1579–1588. [Google Scholar] [CrossRef] [Green Version]
- Tripolt, N.J.; Kolesnik, E.; Pferschy, P.N.; Verheyen, N.; Ablasser, K.; Sailer, S.; Alber, H.; Berger, R.; Kaulfersch, C.; Leitner, K.; et al. Impact of EMpagliflozin on cardiac function and biomarkers of heart failure in patients with acute MYocardial infarction-The EMMY trial. Am. Heart J. 2020, 221, 39–47. [Google Scholar] [CrossRef]
- Cohen, M.V.; Downey, J.M. Adenosine: Trigger and mediator of cardioprotection. Basic Res. Cardiol. 2008, 103, 203–215. [Google Scholar] [CrossRef]
- Johnston-Cox, H.A.; Yang, D.; Ravid, K. Physiological implications of adenosine receptor-mediated platelet aggregation. J. Cell. Physiol. 2011, 226, 46–51. [Google Scholar] [CrossRef]
- Heusch, G. Adenosine and maximum coronary vasodilation in humans: Myth and misconceptions in the assessment of coronary reserve. Basic Res. Cardiol. 2010, 105, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.Q.; Sato, H.; Williams, M.W.; Fernandez, A.Z.; Vinten-Johansen, J. Adenosine A2-receptor activation inhibits neutrophil-mediated injury to coronary endothelium. Am. J. Physiol. 1996, 271, H1456–H1464. [Google Scholar] [CrossRef] [PubMed]
- Mahaffey, K.W.; Puma, J.A.; Barbagelata, N.A.; DiCarli, M.F.; Leesar, M.A.; Browne, K.F.; Eisenberg, P.R.; Bolli, R.; Casas, A.C.; Molina-Viamonte, V.; et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: Results of a multicenter, randomized, placebo-controlled trial: The acute myocardial infarction study of adenosine (AMISTAD) trial. J. Am. Coll. Cardiol. 1999, 34, 1711–1720. [Google Scholar] [CrossRef]
- Ross, A.M.; Gibbons, R.J.; Stone, G.W.; Kloner, R.A.; Alexander, R.W.; Investigators, A.-I. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (AMISTAD-II). J. Am. Coll. Cardiol. 2005, 45, 1775–1780. [Google Scholar] [CrossRef] [Green Version]
- Kloner, R.A.; Forman, M.B.; Gibbons, R.J.; Ross, A.M.; Alexander, R.W.; Stone, G.W. Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: The AMISTAD-2 trial. Eur. Heart J. 2006, 27, 2400–2405. [Google Scholar] [CrossRef] [Green Version]
- Niccoli, G.; Rigattieri, S.; De Vita, M.R.; Valgimigli, M.; Corvo, P.; Fabbiocchi, F.; Romagnoli, E.; De Caterina, A.R.; La Torre, G.; Lo Schiavo, P.; et al. Open-label, randomized, placebo-controlled evaluation of intracoronary adenosine or nitroprusside after thrombus aspiration during primary percutaneous coronary intervention for the prevention of microvascular obstruction in acute myocardial infarction: The REOPEN-AMI study (Intracoronary Nitroprusside Versus Adenosine in Acute Myocardial Infarction). JACC Cardiovasc. Interv. 2013, 6, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Nazir, S.A.; McCann, G.P.; Greenwood, J.P.; Kunadian, V.; Khan, J.N.; Mahmoud, I.Z.; Blackman, D.J.; Been, M.; Abrams, K.R.; Shipley, L.; et al. Strategies to attenuate micro-vascular obstruction during P-PCI: The randomized reperfusion facilitated by local adjunctive therapy in ST-elevation myocardial infarction trial. Eur. Heart J. 2016, 37, 1910–1919. [Google Scholar] [CrossRef] [Green Version]
- Kondo, H.; Suzuki, T.; Fukutomi, T.; Suzuki, S.; Hayase, M.; Ito, S.; Ojio, S.; Ehara, M.; Takeda, Y.; Itoh, M. Effects of percutaneous coronary arterial thrombectomy during acute myocardial infarction on left ventricular remodeling. Am. J. Cardiol. 2004, 93, 527–531. [Google Scholar] [CrossRef]
- De Luca, L.; Sardella, G.; Davidson, C.J.; De Persio, G.; Beraldi, M.; Tommasone, T.; Mancone, M.; Nguyen, B.L.; Agati, L.; Gheorghiade, M.; et al. Impact of intracoronary aspiration thrombectomy during primary angioplasty on left ventricular remodelling in patients with anterior ST elevation myocardial infarction. Heart 2006, 92, 951–957. [Google Scholar] [CrossRef]
- Sardella, G.; Mancone, M.; Bucciarelli-Ducci, C.; Agati, L.; Scardala, R.; Carbone, I.; Francone, M.; Di Roma, A.; Benedetti, G.; Conti, G.; et al. Thrombus aspiration during primary percutaneous coronary intervention improves myocardial reperfusion and reduces infarct size: The EXPIRA (thrombectomy with export catheter in infarct-related artery during primary percutaneous coronary intervention) prospective, randomized trial. J. Am. Coll. Cardiol. 2009, 53, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Carlo, M.; Aquaro, G.D.; Palmieri, C.; Guerra, E.; Misuraca, L.; Giannini, C.; Lombardi, M.; Berti, S.; Petronio, A.S. A prospective randomized trial of thrombectomy versus no thrombectomy in patients with ST-segment elevation myocardial infarction and thrombus-rich lesions: MUSTELA (MUltidevice Thrombectomy in Acute ST-Segment ELevation Acute Myocardial Infarction) trial. JACC Cardiovasc. Interv. 2012, 5, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Liistro, F.; Grotti, S.; Angioli, P.; Falsini, G.; Ducci, K.; Baldassarre, S.; Sabini, A.; Brandini, R.; Capati, E.; Bolognese, L. Impact of thrombus aspiration on myocardial tissue reperfusion and left ventricular functional recovery and remodeling after primary angioplasty. Circ. Cardiovasc. Interv. 2009, 2, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Tokuno, S.; Hinokiyama, K.; Tokuno, K.; Löwbeer, C.; Hansson, L.O.; Valen, G. Spontaneous ischemic events in the brain and heart adapt the hearts of severely atherosclerotic mice to ischemia. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randhawa, P.K.; Jaggi, A.S. Opioids in remote ischemic preconditioning-induced cardioprotection. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 112–121. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Jaggi, A.S. Unraveling the role of adenosine in remote ischemic preconditioning-induced cardioprotection. Life Sci. 2016, 155, 140–146. [Google Scholar] [CrossRef]
- Krieg, T.; Qin, Q.; Philipp, S.; Alexeyev, M.F.; Cohen, M.V.; Downey, J.M. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2606–H2611. [Google Scholar] [CrossRef] [Green Version]
- Gedik, N.; Kottenberg, E.; Thielmann, M.; Frey, U.H.; Jakob, H.; Peters, J.; Heusch, G.; Kleinbongard, P. Potential humoral mediators of remote ischemic preconditioning in patients undergoing surgical coronary revascularization. Sci. Rep. 2017, 7, 12660. [Google Scholar] [CrossRef] [Green Version]
- Giricz, Z.; Varga, Z.V.; Baranyai, T.; Sipos, P.; Pálóczi, K.; Kittel, Á.; Buzás, E.I.; Ferdinandy, P. Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J. Mol. Cell. Cardiol. 2014, 68, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minghua, W.; Zhijian, G.; Chahua, H.; Qiang, L.; Minxuan, X.; Luqiao, W.; Weifang, Z.; Peng, L.; Biming, Z.; Lingling, Y.; et al. Plasma exosomes induced by remote ischaemic preconditioning attenuate myocardial ischaemia/reperfusion injury by transferring miR-24. Cell Death Dis. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Riquelme, J.A.; Zheng, Y.; Vicencio, J.M.; Lavandero, S. Endothelial cells release cardioprotective exosomes that may contribute to ischaemic preconditioning. Sci. Rep. 2018, 8, 15885. [Google Scholar] [CrossRef]
- Ma, F.; Liu, H.; Shen, Y.; Zhang, Y.; Pan, S. Platelet-derived microvesicles are involved in cardio-protective effects of remote preconditioning. Int. J. Clin. Exp. Pathol. 2015, 8, 10832–10839. [Google Scholar]
- Yellon, D.M.; Liu, M.; Wang, Y.; Zhu, Q.; Zhao, J.; Wang, Y.; Shang, M.; Liu, M.; Wu, Y.; Song, J.; et al. Protective effects of circulating microvesicles derived from ischemic preconditioning on myocardial ischemia/reperfusion injury in rats by inhibiting endoplasmic reticulum stress. Sci. Rep. 2018, 23, 436–448. [Google Scholar] [CrossRef]
- Hu, Q.; Luo, W.; Huang, L.; Huang, R.; Chen, R. Apoptosis-related microRNA changes in the right atrium induced by remote ischemic perconditioning during valve replacement surgery. Sci. Rep. 2016, 6, 18959. [Google Scholar] [CrossRef]
- Bartekova, M.; Jelemensky, M.; Dhalla, N.S. Emerging role of non-coding RNAs and extracellular vesicles in cardioprotection by remote ischemic conditioning of the heart. Rev. Cardiovasc. Med. 2019, 20, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Botker, H.E.; Kharbanda, R.; Schmidt, M.R.; Bottcher, M.; Kaltoft, A.K.; Terkelsen, C.J.; Munk, K.; Andersen, N.H.; Hansen, T.M.; Trautner, S.; et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: A randomised trial. Lancet 2010, 375, 727–734. [Google Scholar] [CrossRef]
- Gaspar, A.; Lourenço, A.P. Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Cardiovasc. Res. 2018, 113, 14. [Google Scholar] [CrossRef]
- White, S.K.; Frohlich, G.M.; Sado, D.M.; Maestrini, V.; Fontana, M.; Treibel, T.A.; Tehrani, S.; Flett, A.S.; Meier, P.; Ariti, C.; et al. Remote ischemic conditioning reduces myocardial infarct size and edema in patients with ST-segment elevation myocardial infarction. JACC Cardiovasc. Interv. 2015, 8, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, R.K.; Mortensen, U.M.; White, P.A.; Kristiansen, S.B.; Schmidt, M.R.; Hoschtitzky, J.A.; Vogel, M.; Sorensen, K.; Redington, A.N.; MacAllister, R. Transient limb ischemia induces remote ischemic preconditioning in vivo. Basic Res. Cardiol. 2002, 106, 2881–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitel, I.; Stiermaier, T.; Rommel, K.P.; Fuernau, G.; Sandri, M.; Mangner, N.; Linke, A.; Erbs, S.; Lurz, P.; Boudriot, E.; et al. Cardioprotection by combined intrahospital remote ischaemic perconditioning and postconditioning in ST-elevation myocardial infarction: The randomized LIPSIA CONDITIONING trial. Eur. Heart J. 2015, 36, 3049–3057. [Google Scholar] [CrossRef]
- Rentoukas, I.; Giannopoulos, G.; Kaoukis, A.; Kossyvakis, C.; Raisakis, K.; Driva, M.; Panagopoulou, V.; Tsarouchas, K.; Vavetsi, S.; Pyrgakis, V.; et al. Cardioprotective role of remote ischemic periconditioning in primary percutaneous coronary intervention: Enhancement by opioid action. JACC Cardiovasc. Interv. 2010, 3, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verouhis, D.; Sörensson, P.; Gourine, A.; Henareh, L.; Persson, J.; Saleh, N.; Settergren, M.; Sundqvist, M.; Tornvall, P.; Witt, N.; et al. Effect of remote ischemic conditioning on infarct size in patients with anterior ST-elevation myocardial infarction. Am. Heart J. 2016, 181, 66–73. [Google Scholar] [CrossRef]
- Yellon, D.M.; Ackbarkhan, A.K.; Balgobin, V.; Bulluck, H.; Deelchand, A.; Dhuny, M.R.; Domah, N.; Gaoneadry, D.; Jagessur, R.K.; Joonas, N.; et al. Remote ischemic conditioning reduces myocardial infarct size in STEMI patients treated by thrombolysis. J. Am. Coll. Cardiol. 2015, 65, 2764–2765. [Google Scholar] [CrossRef] [Green Version]
- Prunier, F.; Angoulvant, D.; Saint Etienne, C.; Vermes, E.; Gilard, M.; Piot, C.; Roubille, F.; Elbaz, M.; Ovize, M.; Bière, L.; et al. The RIPOST-MI study, assessing remote ischemic perconditioning alone or in combination with local ischemic postconditioning in ST-segment elevation myocardial infarction. Basic Res. Cardiol. 2014, 109, 400. [Google Scholar] [CrossRef]
- Sloth, A.D.; Schmidt, M.R.; Munk, K.; Kharbanda, R.K.; Redington, A.N.; Schmidt, M.; Pedersen, L.; Sørensen, H.T.; Bøtker, H.E. Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur. Heart J. 2014, 35, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Stiermaier, T.; Jensen, J.O.; Rommel, K.P.; de Waha-Thiele, S.; Fuernau, G.; Desch, S.; Thiele, H.; Eitel, I. Combined intrahospital remote ischemic perconditioning and postconditioning improves clinical outcome in ST-elevation myocardial infarction. Circ. Res. 2019, 124, 1482–1491. [Google Scholar] [CrossRef]
- Gorog, D.A.; Farag, M.; Spinthakis, N.; Yellon, D.M.; Bøtker, H.E.; Kharbanda, R.K.; Hausenloy, D.J. Effect of remote ischaemic conditioning on platelet reactivity and endogenous fibrinolysis in ST-elevation myocardial infarction: A substudy of the CONDI-2/ERIC-PPCI randomized controlled trial. Cardiovasc. Res. 2021, 117, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- He, Z.; Davidson, S.M. The importance of clinically relevant background therapy in cardioprotective studies. Basic Res. Cardiol. 2020, 115, 69. [Google Scholar] [CrossRef]
- Engstrøm, T.; Kelbæk, H.; Helqvist, S.; Høfsten, D.E.; Kløvgaard, L.; Clemmensen, P.; Holmvang, L.; Jørgensen, E.; Pedersen, F.; Saunamaki, K.; et al. Effect of ischemic postconditioning during primary percutaneous coronary intervention for patients with ST-segment elevation myocardial infarction: A randomized clinical trial. JAMA Cardiol. 2017, 2, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Traverse, J.H.; Swingen, C.M.; Henry, T.D.; Fox, J.; Wang, Y.L.; Chavez, I.J.; Lips, D.L.; Lesser, J.R.; Pedersen, W.R.; Burke, N.M.; et al. NHLBI-sponsored randomized trial of postconditioning during primary percutaneous coronary intervention for ST-elevation myocardial infarction. Circ. Res. 2019, 124, 769–778. [Google Scholar] [CrossRef]
- Crimi, G.; Pica, S.; Raineri, C.; Bramucci, E.; De Ferrari, G.M.; Klersy, C.; Ferlini, M.; Marinoni, B.; Repetto, A.; Romeo, M.; et al. Remote ischemic post-conditioning of the lower limb during primary percutaneous coronary intervention safely reduces enzymatic infarct size in anterior myocardial infarction: A randomized controlled trial. JACC Cardiovasc. Interv. 2013, 6, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Gersh, B.J.; Stone, G.W.; White, H.D.; Holmes, D.R., Jr. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: Is the slope of the curve the shape of the future? JAMA 2005, 293, 979–986. [Google Scholar] [CrossRef]
- Rezkalla, S.H.; Kloner, R.A. Ischemic preconditioning and preinfarction angina in the clinical arena. Nat. Clin. Pract. Cardiovasc. Med. 2004, 1, 96–102. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote ischemic preconditioning and outcomes of cardiac surgery. N. Engl. J. Med. 2015, 373, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Böning, A.; Niemann, B.; et al. A multicenter trial of remote ischemic preconditioning for heart surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef]
- Musiolik, J.; van Caster, P.; Skyschally, A.; Boengler, K.; Gres, P.; Schulz, R.; Heusch, G. Reduction of infarct size by gentle reperfusion without activation of reperfusion injury salvage kinases in pigs. Cardiovasc. Res. 2010, 85, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Loubeyre, C.; Morice, M.C.; Lefèvre, T.; Piéchaud, J.F.; Louvard, Y.; Dumas, P. A randomized comparison of direct stenting with conventional stent implantation in selected patients with acute myocardial infarction. J. Am. Coll. Cardiol. 2002, 39, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Kleinbongard, P.; Böse, D.; Levkau, B.; Haude, M.; Schulz, R.; Erbel, R. Coronary microembolization: From bedside to bench and back to bedside. Circulation 2009, 120, 1822–1836. [Google Scholar] [CrossRef]
- Skyschally, A.; Walter, B.; Heusch, G. Coronary microembolization during early reperfusion: Infarct extension, but protection by ischaemic postconditioning. Eur. Heart J. 2013, 34, 3314–3321. [Google Scholar] [CrossRef]
- Nijveldt, R.; Beek, A.M.; Hirsch, A.; Stoel, M.G.; Hofman, M.B.; Umans, V.A.; Algra, P.R.; Twisk, J.W.; van Rossum, A.C. Functional recovery after acute myocardial infarction: Comparison between angiography, electrocardiography, and cardiovascular magnetic resonance measures of microvascular injury. J. Am. Coll. Cardiol. 2008, 52, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Botker, H.E.; Engstrom, T.; Erlinge, D.; Heusch, G.; Ibanez, B.; Kloner, R.A.; Ovize, M.; Yellon, D.M.; Garcia-Dorado, D. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: Trials and tribulations. Eur. Heart J. 2017, 38, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Nazir, S.A.; Khan, J.N.; Mahmoud, I.Z.; Greenwood, J.P.; Blackman, D.J.; Kunadian, V.; Been, M.; Abrams, K.R.; Wilcox, R.; Adgey, A.A.J.; et al. The REFLO-STEMI (REperfusion Facilitated by LOcal Adjunctive Therapy in ST-Elevation Myocardial Infarction) Trial: A Randomised Controlled Trial Comparing Intracoronary Administration of Adenosine or Sodium Nitroprusside with Control for Attenuation of Microvascular Obstruction During Primary Percutaneous Coronary Intervention; NIHR Journals Library: Southampton, UK, 2016. [Google Scholar] [CrossRef]
- Peart, J.N.; Gross, E.R.; Gross, G.J. Opioid-induced preconditioning: Recent advances and future perspectives. Vasc. Pharmacol. 2005, 42, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, S.; Yamada, T.; Watanabe, T.; Morita, T.; Furukawa, Y.; Kawasaki, M.; Kikuchi, A.; Kawai, T.; Seo, M.; Abe, M.; et al. Effect of empagliflozin as an add-on therapy on decongestion and renal function in patients with diabetes hospitalized for acute decompensated heart failure: A prospective randomized controlled study. Circ. Heart Fail. 2021, 14, e007048. [Google Scholar] [CrossRef] [PubMed]





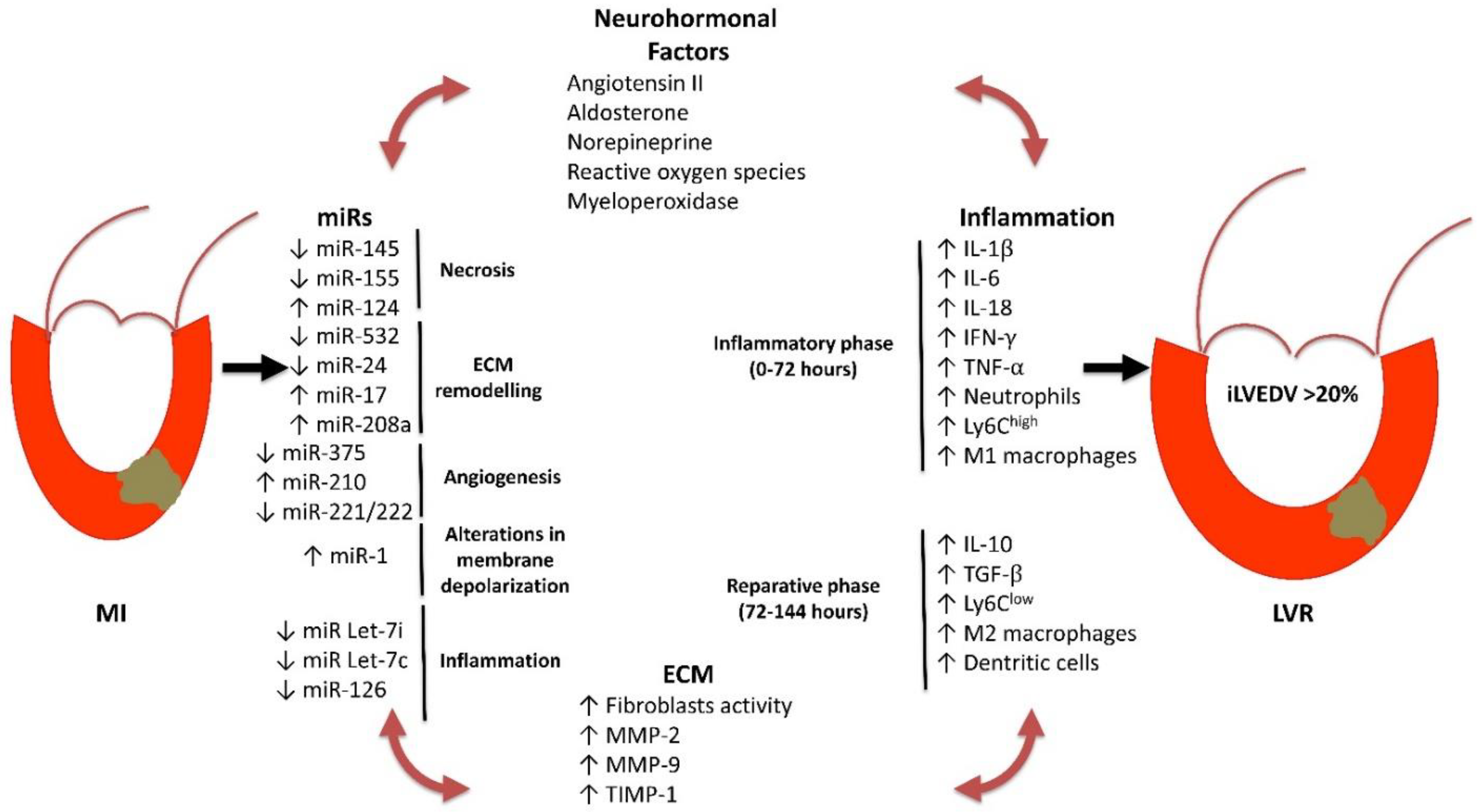{kind=link}
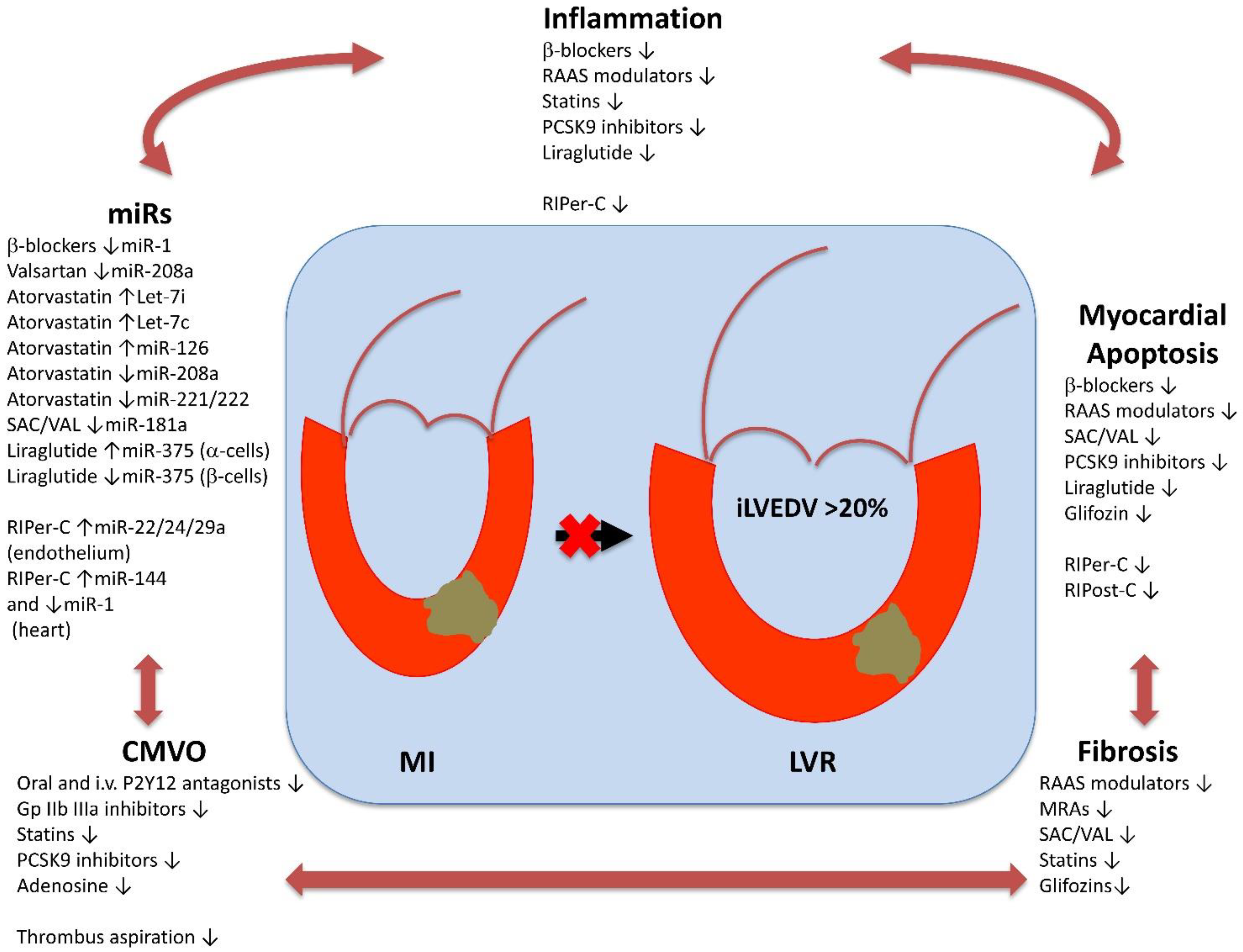{kind=link}
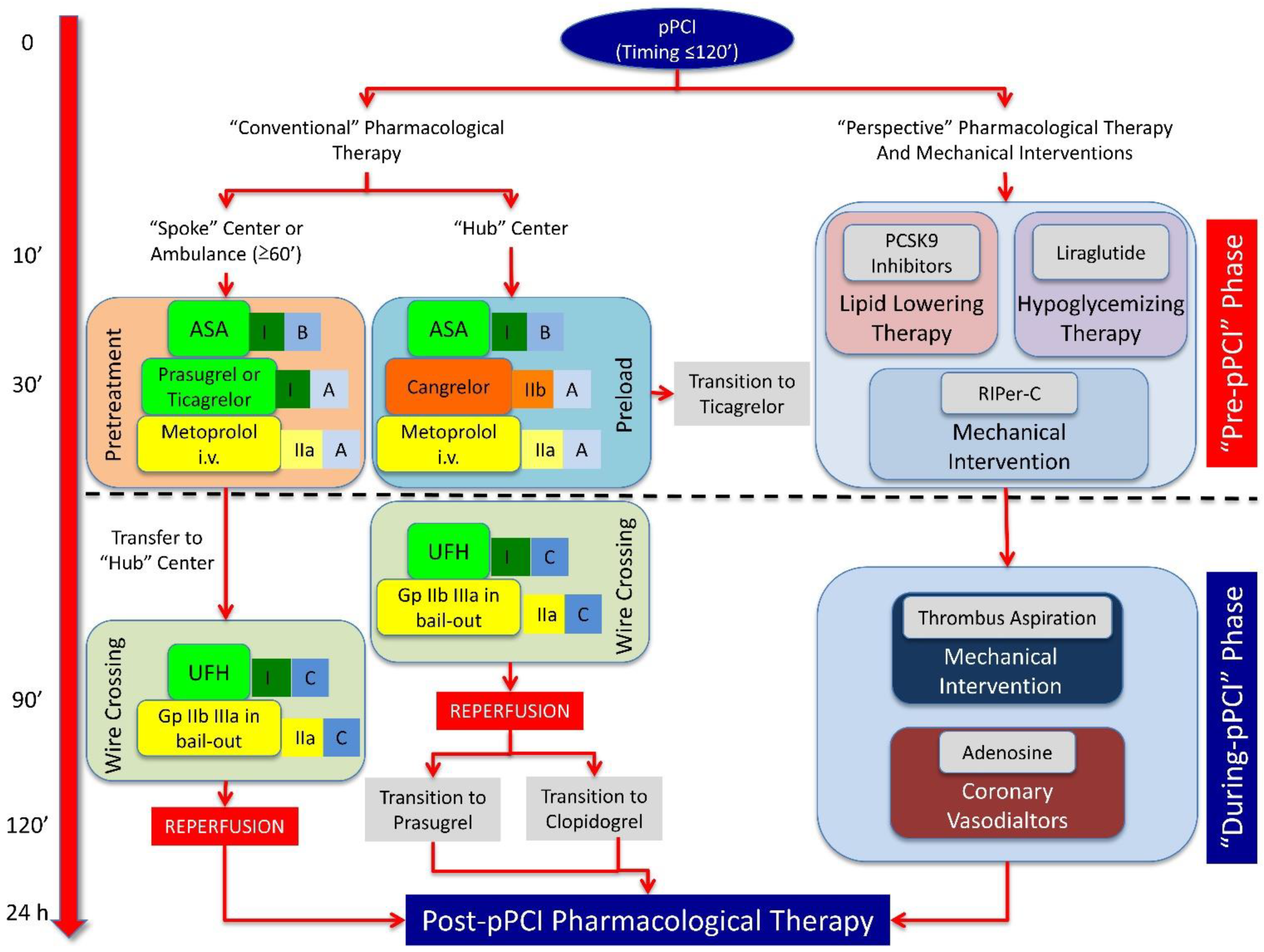{kind=link}
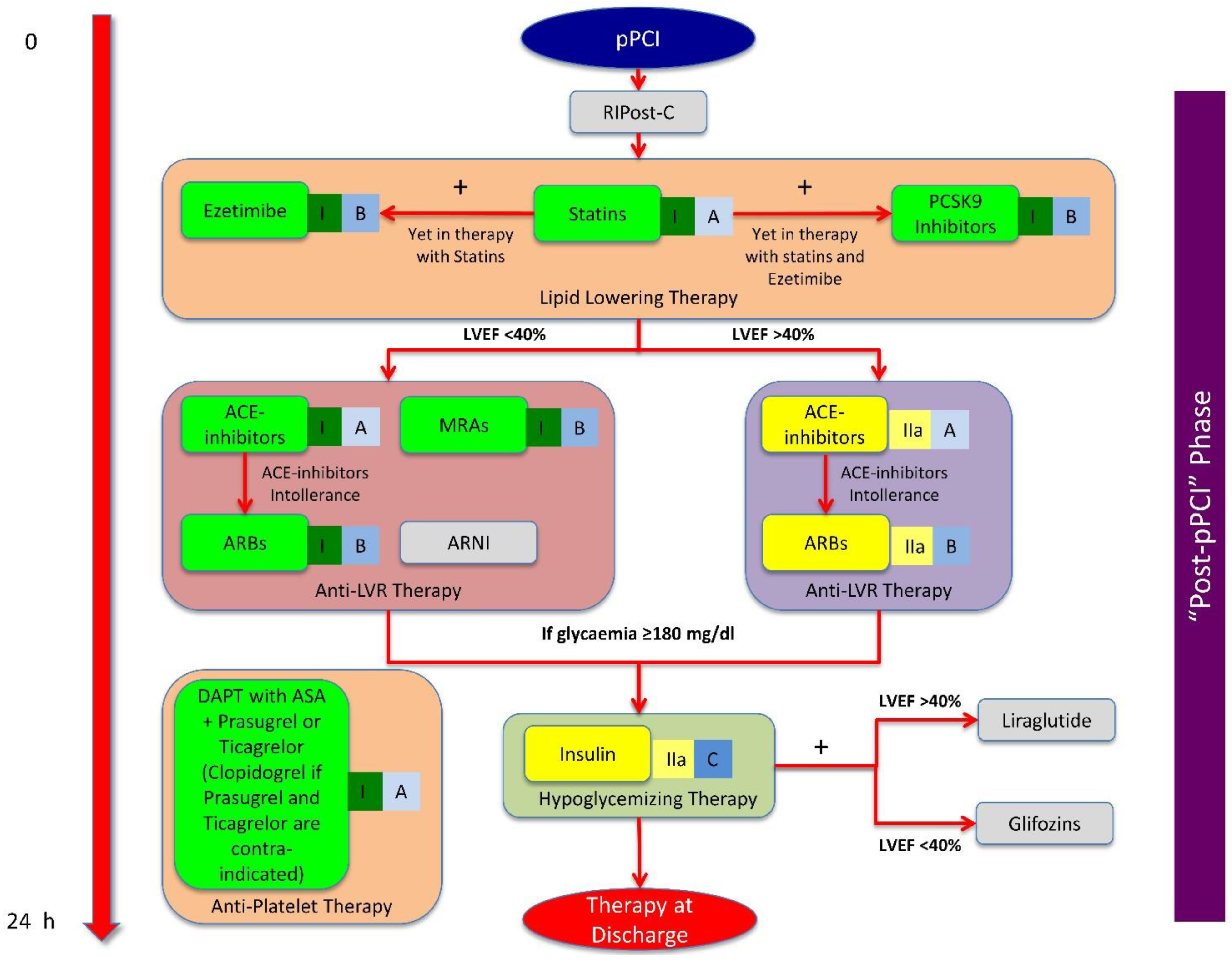{kind=link}
| miRs | Target | Physiological Effects | Model (Organ/Cell Line) | Drug and Mechanical Intervention | Drug Effects on miR Expression |
|---|---|---|---|---|---|
| miR-1 | ↓ Cx43 expression | ↑ Arrhythmias | Rat (heart/cardiomyocytes, fibroblasts) | Metoprolol, RIPer-C | ↓ |
| miR-17 | ↓ TIMPs | ↑ MMPs activity | Mouse (heart) | Not known | Not known |
| miR-21 | ↓ Apoptosis | ↓ IS extension | Rat (heart/cardiomyocytes) | Not known | Not known |
| miR-24 | ↓ TGF-β, fibronectin, collagen type 1 | ↓ Fibrosis in area at risk | Mouse (heart/cardiomyocytes) | RIPer-C | ↑ |
| miR-126 | ↓ VCAM-1 | ↓ Plaque formation | Mouse (carotid) | Atorvastatin | ↑ |
| miR-181a | ↓ Expression of TGF-β receptor III | ↑ Fibrosis, hypertrophy | Rodent (Pluripotent stem cells, cardiomyocytes) | SAC/VAL | ↓ |
| miR-208 | ↑ Endoglin | ↑ Cardiac fibrosis | Rat (heart) | Valsartan, Atorvastatin | ↓ |
| miR-210 | ↑ Micro-vessel density | ↑ LV contractility | Rat (heart/cardiomyocytes) | Not known | Not known |
| miR-221/222 | ↓ eNOS activity | ↓ Angiogenesis | Human (EPCs) | Atorvastatin | ↓ |
| miR-375 | ↓ PDK1, PI3K and Akt activity | ↓ Cell survival | Mouse (heart/pancreatic β-cells) | Liraglutide | ↓ |
| miR-532 | ↓ prss activity | ↓ Synthesis of ECM proteins | Mouse (heart) | Not known | Not known |
| Let-7c | ↓ T cell proliferation | ↓ Plaque rupture | Human (dendrocytes) | Atorvastatin | ↑ |
| Let-7i | ↓ Toll-like receptor | ↓ Activation of atherosclerotic plaque | Human (monocyte) | Atorvastatin | ↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellis, A.; Di Gioia, G.; Mauro, C.; Mancusi, C.; Barbato, E.; Izzo, R.; Trimarco, B.; Morisco, C. Reducing Cardiac Injury during ST-Elevation Myocardial Infarction: A Reasoned Approach to a Multitarget Therapeutic Strategy. J. Clin. Med. 2021, 10, 2968. https://doi.org/10.3390/jcm10132968
Bellis A, Di Gioia G, Mauro C, Mancusi C, Barbato E, Izzo R, Trimarco B, Morisco C. Reducing Cardiac Injury during ST-Elevation Myocardial Infarction: A Reasoned Approach to a Multitarget Therapeutic Strategy. Journal of Clinical Medicine. 2021; 10(13):2968. https://doi.org/10.3390/jcm10132968
Chicago/Turabian StyleBellis, Alessandro, Giuseppe Di Gioia, Ciro Mauro, Costantino Mancusi, Emanuele Barbato, Raffaele Izzo, Bruno Trimarco, and Carmine Morisco. 2021. "Reducing Cardiac Injury during ST-Elevation Myocardial Infarction: A Reasoned Approach to a Multitarget Therapeutic Strategy" Journal of Clinical Medicine 10, no. 13: 2968. https://doi.org/10.3390/jcm10132968