
Complement Blockade Is a Promising Therapeutic Approach in a Subset of Critically Ill Adult Patients with Complement-Mediated Hemolytic Uremic Syndromes

 , ,
, , {kind=link}
{kind=link}
Abstract
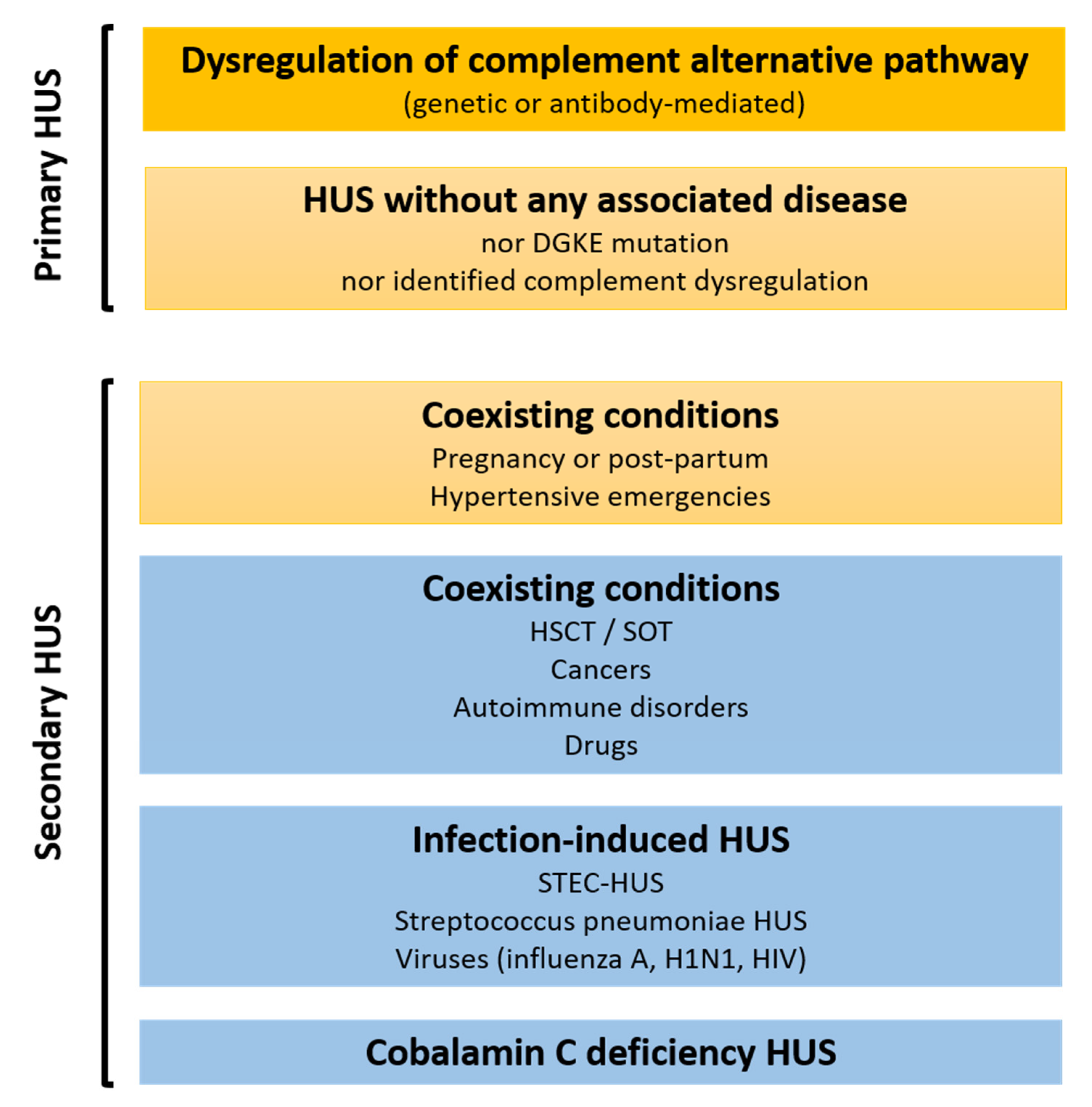
:1. Introduction
2. A Role for Complement in Secondary HUS Pathophysiology?
3. A Role for Complement in Primary HUS Pathophysiology
4. When to Consider Exploration of Complement Regulation in Critically Ill Patients with TMA Features?
5. What Is the Impact of Complement Dysregulation in HUS Patients Care?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic Uraemic Syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef]
- George, J.N.; Nester, C.M. Syndromes of Thrombotic Micro-angiopathy. N. Engl. J. Med. 2014, 371, 1847–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhouri, F.; Hourmant, M.; Campistol, J.M.; Cataland, S.R.; Espinosa, M.; Gaber, A.O.; Menne, J.; Minetti, E.E.; Provôt, F.; Rondeau, E.; et al. Terminal Complement Inhibitor Eculizumab in Adult Patients with Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial. Am. J. Kidney Dis. 2016, 68, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic-Uremic Syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, S.A.M.E.G.; van Paassen, P. The Syndromes of Thrombotic Microangiopathy: A Critical Appraisal on Complement Dysregulation. J. Clin. Med. 2021, 10, 3034. [Google Scholar] [CrossRef]
- Cavero, T.; Rabasco, C.; López, A.; Román, E.; Ávila, A.; Sevillano, Á.; Huerta, A.; Rojas-Rivera, J.; Fuentes, C.; Blasco, M.; et al. Eculizumab in Secondary Atypical Haemolytic Uraemic Syndrome. Nephrol. Dial. Transplant. 2017, 32, 466–474. [Google Scholar] [CrossRef]
- Le Clech, A.; Simon-Tillaux, N.; Provôt, F.; Delmas, Y.; Vieira-Martins, P.; Limou, S.; Halimi, J.-M.; Le Quintrec, M.; Lebourg, L.; Grangé, S.; et al. Atypical and Secondary Hemolytic Uremic Syndromes Have a Distinct Presentation and No Common Genetic Risk Factors. Kidney Int. 2019, 95, 1443–1452. [Google Scholar] [CrossRef]
- Langer, R.M.; Van Buren, C.T.; Katz, S.M.; Kahan, B.D. De Novo Hemolytic Uremic Syndrome after Kidney Transplantation in Patients Treated with Cyclosporine-Sirolimus Combination. Transplantation 2002, 73, 756–760. [Google Scholar] [CrossRef]
- Satoskar, A.A.; Pelletier, R.; Adams, P.; Nadasdy, G.M.; Brodsky, S.; Pesavento, T.; Henry, M.; Nadasdy, T. De Novo Thrombotic Microangiopathy in Renal Allograft Biopsies-Role of Antibody-Mediated Rejection. Am. J. Transplant. 2010, 10, 1804–1811. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Agodoa, L.Y.; Yuan, C.M.; Abbott, K.C. Thrombotic Microangiopathy after Renal Transplantation in the United States. Am. J. Kidney Dis. 2003, 42, 1058–1068. [Google Scholar] [CrossRef]
- Le Quintrec, M.; Zuber, J.; Moulin, B.; Kamar, N.; Jablonski, M.; Lionet, A.; Chatelet, V.; Mousson, C.; Mourad, G.; Bridoux, F.; et al. Complement Genes Strongly Predict Recurrence and Graft Outcome in Adult Renal Transplant Recipients with Atypical Hemolytic and Uremic Syndrome. Am. J. Transplant. 2013, 13, 663–675. [Google Scholar] [CrossRef]
- Le Quintrec, M.; Lionet, A.; Kamar, N.; Karras, A.; Barbier, S.; Buchler, M.; Fakhouri, F.; Provost, F.; Fridman, W.H.; Thervet, E.; et al. Complement Mutation-Associated de Novo Thrombotic Microangiopathy Following Kidney Transplantation. Am. J. Transplant. 2008, 8, 1694–1701. [Google Scholar] [CrossRef]
- Zuber, J.; Le Quintrec, M.; Krid, S.; Bertoye, C.; Gueutin, V.; Lahoche, A.; Heyne, N.; Ardissino, G.; Chatelet, V.; Noël, L.-H.; et al. Eculizumab for Atypical Hemolytic Uremic Syndrome Recurrence in Renal Transplantation. Am. J. Transplant. 2012, 12, 3337–3354. [Google Scholar] [CrossRef]
- Zuber, J.; Frimat, M.; Caillard, S.; Kamar, N.; Gatault, P.; Petitprez, F.; Couzi, L.; Jourde-Chiche, N.; Chatelet, V.; Gaisne, R.; et al. Use of Highly Individualized Complement Blockade Has Revolutionized Clinical Outcomes after Kidney Transplantation and Renal Epidemiology of Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2019, 30, 2449–2463. [Google Scholar] [CrossRef]
- Marks, W.H.; Mamode, N.; Montgomery, R.A.; Stegall, M.D.; Ratner, L.E.; Cornell, L.D.; Rowshani, A.T.; Colvin, R.B.; Dain, B.; Boice, J.A.; et al. Safety and Efficacy of Eculizumab in the Prevention of Antibody-Mediated Rejection in Living-Donor Kidney Transplant Recipients Requiring Desensitization Therapy: A Randomized Trial. Am. J. Transplant. 2019, 19, 2876–2888. [Google Scholar] [CrossRef] [Green Version]
- Fakhouri, F.; Roumenina, L.; Provot, F.; Sallée, M.; Caillard, S.; Couzi, L.; Essig, M.; Ribes, D.; Dragon-Durey, M.-A.; Bridoux, F.; et al. Pregnancy-Associated Hemolytic Uremic Syndrome Revisited in the Era of Complement Gene Mutations. J. Am. Soc. Nephrol. 2010, 21, 859–867. [Google Scholar] [CrossRef] [Green Version]
- Bruel, A.; Kavanagh, D.; Noris, M.; Delmas, Y.; Wong, E.K.S.; Bresin, E.; Provôt, F.; Brocklebank, V.; Mele, C.; Remuzzi, G.; et al. Hemolytic Uremic Syndrome in Pregnancy and Postpartum. Clin. J. Am. Soc. Nephrol. 2017, 12, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Huerta, A.; Arjona, E.; Portoles, J.; Lopez-Sanchez, P.; Rabasco, C.; Espinosa, M.; Cavero, T.; Blasco, M.; Cao, M.; Manrique, J.; et al. A Retrospective Study of Pregnancy-Associated Atypical Hemolytic Uremic Syndrome. Kidney Int. 2018, 93, 450–459. [Google Scholar] [CrossRef]
- Fakhouri, F.; Scully, M.; Provôt, F.; Blasco, M.; Coppo, P.; Noris, M.; Paizis, K.; Kavanagh, D.; Pène, F.; Quezada, S.; et al. Management of Thrombotic Microangiopathy in Pregnancy and Postpartum: Report from an International Working Group. Blood 2020, 136, 2103–2117. [Google Scholar] [CrossRef]
- Timmermans, S.A.M.E.G.; Werion, A.; Spaanderman, M.E.A.; Reutelingsperger, C.P.; Damoiseaux, J.G.M.C.; Morelle, J.; van Paassen, P. The Natural Course of Pregnancies in Women with Primary Atypical Haemolytic Uraemic Syndrome and Asymptomatic Relatives. Br. J. Haematol. 2020, 190, 442–449. [Google Scholar] [CrossRef]
- Gaggl, M.; Aigner, C.; Csuka, D.; Szilágyi, Á.; Prohászka, Z.; Kain, R.; Haninger, N.; Knechtelsdorfer, M.; Sunder-Plassmann, R.; Sunder-Plassmann, G.; et al. Maternal and Fetal Outcomes of Pregnancies in Women with Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2018, 29, 1020–1029. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Palomo, M.; Blasco, M.; Molina, P.; Lozano, M.; Praga, M.; Torramade-Moix, S.; Martinez-Sanchez, J.; Cid, J.; Escolar, G.; Carreras, E.; et al. Complement Activation and Thrombotic Microangiopathies. Clin. J. Am. Soc. Nephrol. 2019, 14, 1719–1732. [Google Scholar] [CrossRef]
- Kelly, R.J.; Höchsmann, B.; Szer, J.; Kulasekararaj, A.; de Guibert, S.; Röth, A.; Weitz, I.C.; Armstrong, E.; Risitano, A.M.; Patriquin, C.J.; et al. Eculizumab in Pregnant Patients with Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2015, 373, 1032–1039. [Google Scholar] [CrossRef] [Green Version]
- Servais, A.; Devillard, N.; Frémeaux-Bacchi, V.; Hummel, A.; Salomon, L.; Contin-Bordes, C.; Gomer, H.; Legendre, C.; Delmas, Y. Atypical Haemolytic Uraemic Syndrome and Pregnancy: Outcome with Ongoing Eculizumab. Nephrol. Dial. Transplant. 2016, 31, 2122–2130. [Google Scholar] [CrossRef]
- Cañigral, C.; Moscardó, F.; Castro, C.; Pajares, A.; Lancharro, A.; Solves, P.; de la Rubia, J.; Carpio, N.; Sanz, M.A. Eculizumab for the Treatment of Pregnancy-Related Atypical Hemolytic Uremic Syndrome. Ann. Hematol. 2014, 93, 1421–1422. [Google Scholar] [CrossRef]
- De Sousa Amorim, E.; Blasco, M.; Quintana, L.; Sole, M.; de Cordoba, S.R.; Campistol, J.M. Eculizumab in Pregnancy-Associated Atypical Hemolytic Uremic Syndrome: Insights for Optimizing Management. J. Nephrol. 2015, 28, 641–645. [Google Scholar] [CrossRef]
- Delmas, Y.; Bordes, C.; Loirat, C.; Frémeaux-Bacchi, V.; Combe, C. Post-Partum Atypical Haemolytic-Uraemic Syndrome Treated with Eculizumab: Terminal Complement Activity Assessment in Clinical Practice. Clin. Kidney J. 2013, 6, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Zschiedrich, S.; Prager, E.P.; Kuehn, E.W. Successful Treatment of the Postpartum Atypical Hemolytic Uremic Syndrome with Eculizumab. Ann. Intern. Med. 2013, 159, 76. [Google Scholar] [CrossRef] [PubMed]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the SFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Baert, J.; McCarey, C.; Berkane, N.; Martinez de Tejada, B.; Vial, Y.; Rieder, W. The Role of SFlt1/PlGF Ratio in the Assessment of Preeclampsia and Pregnancy-Related Hypertensive Disorders. Swiss Med. Wkly. 2021, 151, w20533. [Google Scholar] [CrossRef]
- El Karoui, K.; Boudhabhay, I.; Petitprez, F.; Vieira-Martins, P.; Fakhouri, F.; Zuber, J.; Aulagnon, F.; Matignon, M.; Rondeau, E.; Mesnard, L.; et al. Impact of Hypertensive Emergency and Rare Complement Variants on the Presentation and Outcome of Atypical Hemolytic Uremic Syndrome. Haematologica 2019, 104, 2501–2511. [Google Scholar] [CrossRef]
- Cavero, T.; Arjona, E.; Soto, K.; Caravaca-Fontán, F.; Rabasco, C.; Bravo, L.; de la Cerda, F.; Martín, N.; Blasco, M.; Ávila, A.; et al. Severe and Malignant Hypertension Are Common in Primary Atypical Hemolytic Uremic Syndrome. Kidney Int. 2019, 96, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Rubin, S.; Cremer, A.; Boulestreau, R.; Rigothier, C.; Kuntz, S.; Gosse, P. Malignant Hypertension: Diagnosis, Treatment and Prognosis with Experience from the Bordeaux Cohort. J. Hypertens. 2019, 37, 316–324. [Google Scholar] [CrossRef]
- van den Born, B.-J.H.; Koopmans, R.P.; Groeneveld, J.O.; van Montfrans, G.A. Ethnic Disparities in the Incidence, Presentation and Complications of Malignant Hypertension. J. Hypertens. 2006, 24, 2299–2304. [Google Scholar] [CrossRef]
- Shantsila, A.; Shantsila, E.; Beevers, D.G.; Lip, G.Y.H. Predictors of 5-Year Outcomes in Malignant Phase Hypertension: The West Birmingham Malignant Hypertension Registry. J. Hypertens. 2017, 35, 2310–2314. [Google Scholar] [CrossRef]
- Larsen, C.P.; Wilson, J.D.; Best-Rocha, A.; Beggs, M.L.; Hennigar, R.A. Genetic Testing of Complement and Coagulation Pathways in Patients with Severe Hypertension and Renal Microangiopathy. Mod. Pathol. 2018, 31, 488–494. [Google Scholar] [CrossRef] [Green Version]
- Polgreen, L.A.; Suneja, M.; Tang, F.; Carter, B.L.; Polgreen, P.M. Increasing Trend in Admissions for Malignant Hypertension and Hypertensive Encephalopathy in the United States. Hypertension 2015, 65, 1002–1007. [Google Scholar] [CrossRef] [Green Version]
- van den Born, B.-J.H.; Lip, G.Y.H.; Brguljan-Hitij, J.; Cremer, A.; Segura, J.; Morales, E.; Mahfoud, F.; Amraoui, F.; Persu, A.; Kahan, T.; et al. ESC Council on Hypertension Position Document on the Management of Hypertensive Emergencies. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.A.; Lip, G.Y.H.; Beevers, D.G. Improving Survival of Malignant Hypertension Patients over 40 Years. Am. J. Hypertens. 2009, 22, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Lip, G.Y.; Beevers, M.; Beevers, D.G. Complications and Survival of 315 Patients with Malignant-Phase Hypertension. J. Hypertens. 1995, 13, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.A.M.E.G.; Wérion, A.; Damoiseaux, J.G.M.C.; Morelle, J.; Reutelingsperger, C.P.; van Paassen, P. Diagnostic and Risk Factors for Complement Defects in Hypertensive Emergency and Thrombotic Microangiopathy. Hypertension 2020, 75, 422–430. [Google Scholar] [CrossRef] [PubMed]
- van den Born, B.J.H.; Honnebier, U.P.F.; Koopmans, R.P.; van Montfrans, G.A. Microangiopathic Hemolysis and Renal Failure in Malignant Hypertension. Hypertension 2005, 45, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, S. Acute Severe Hypertension. N. Engl. J. Med. 2020, 382, e11. [Google Scholar]
- González, R.; Morales, E.; Segura, J.; Ruilope, L.M.; Praga, M. Long-Term Renal Survival in Malignant Hypertension. Nephrol. Dial. Transplant. 2010, 25, 3266–3272. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, S.A.M.E.G.; Abdul-Hamid, M.A.; Potjewijd, J.; Theunissen, R.O.M.F.I.H.; Damoiseaux, J.G.M.C.; Reutelingsperger, C.P.; van Paassen, P.; Limburg Renal Registry. C5b9 Formation on Endothelial Cells Reflects Complement Defects among Patients with Renal Thrombotic Microangiopathy and Severe Hypertension. J. Am. Soc. Nephrol. 2018, 29, 2234–2243. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, S.A.M.E.G.; Abdul-Hamid, M.A.; Vanderlocht, J.; Damoiseaux, J.G.M.C.; Reutelingsperger, C.P.; van Paassen, P.; Limburg Renal Registry. Patients with Hypertension-Associated Thrombotic Microangiopathy May Present with Complement Abnormalities. Kidney Int. 2017, 91, 1420–1425. [Google Scholar] [CrossRef]
- Sullivan, M.; Erlic, Z.; Hoffmann, M.M.; Arbeiter, K.; Patzer, L.; Budde, K.; Hoppe, B.; Zeier, M.; Lhotta, K.; Rybicki, L.A.; et al. Epidemiological Approach to Identifying Genetic Predispositions for Atypical Hemolytic Uremic Syndrome. Ann. Hum. Genet. 2010, 74, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Bresin, E.; Rurali, E.; Caprioli, J.; Sanchez-Corral, P.; Fremeaux-Bacchi, V.; Rodriguez de Cordoba, S.; Pinto, S.; Goodship, T.H.J.; Alberti, M.; Ribes, D.; et al. Combined Complement Gene Mutations in Atypical Hemolytic Uremic Syndrome Influence Clinical Phenotype. J. Am. Soc. Nephrol. 2013, 24, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Fremeaux-Bacchi, V.; Fakhouri, F.; Garnier, A.; Bienaimé, F.; Dragon-Durey, M.-A.; Ngo, S.; Moulin, B.; Servais, A.; Provot, F.; Rostaing, L.; et al. Genetics and Outcome of Atypical Hemolytic Uremic Syndrome: A Nationwide French Series Comparing Children and Adults. Clin. J. Am. Soc. Nephrol. 2013, 8, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative Role of Genetic Complement Abnormalities in Sporadic and Familial AHUS and Their Impact on Clinical Phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Bu, F.; Borsa, N.G.; Jones, M.B.; Takanami, E.; Nishimura, C.; Hauer, J.J.; Azaiez, H.; Black-Ziegelbein, E.A.; Meyer, N.C.; Kolbe, D.L.; et al. High-Throughput Genetic Testing for Thrombotic Microangiopathies and C3 Glomerulopathies. J. Am. Soc. Nephrol. 2016, 27, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Osborne, A.J.; Breno, M.; Borsa, N.G.; Bu, F.; Frémeaux-Bacchi, V.; Gale, D.P.; van den Heuvel, L.P.; Kavanagh, D.; Noris, M.; Pinto, S.; et al. Statistical Validation of Rare Complement Variants Provides Insights into the Molecular Basis of Atypical Hemolytic Uremic Syndrome and C3 Glomerulopathy. J. Immunol. 2018, 200, 2464–2478. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, F.; Ardissino, G.; Ariceta, G.; Fakhouri, F.; Scully, M.; Isbel, N.; Lommelé, Å.; Kupelian, V.; Gasteyger, C.; Greenbaum, L.A.; et al. Clinical and Genetic Predictors of Atypical Hemolytic Uremic Syndrome Phenotype and Outcome. Kidney Int. 2018, 94, 408–418. [Google Scholar] [CrossRef]
- Fakhouri, F.; Frémeaux-Bacchi, V. Thrombotic Microangiopathy in AHUS and beyond: Clinical Clues from Complement Genetics. Nat. Rev. Nephrol. 2021, 17, 543–553. [Google Scholar] [CrossRef]
- Warwicker, P.; Donne, R.L.; Goodship, J.A.; Goodship, T.H.; Howie, A.J.; Kumararatne, D.S.; Thompson, R.A.; Taylor, C.M. Familial Relapsing Haemolytic Uraemic Syndrome and Complement Factor H Deficiency. Nephrol. Dial. Transplant. 1999, 14, 1229–1233. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.; Kemp, E.J.; Liszewski, M.K.; Goodship, J.A.; Lampe, A.K.; Decorte, R.; Müslümanoğlu, M.H.; Kavukcu, S.; Filler, G.; Pirson, Y.; et al. Mutations in Human Complement Regulator, Membrane Cofactor Protein (CD46), Predispose to Development of Familial Hemolytic Uremic Syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 12966–12971. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Brioschi, S.; Caprioli, J.; Todeschini, M.; Bresin, E.; Porrati, F.; Gamba, S.; Remuzzi, G.; International Registry of Recurrent and Familial HUS/TTP. Familial Haemolytic Uraemic Syndrome and an MCP Mutation. Lancet 2003, 362, 1542–1547. [Google Scholar] [CrossRef]
- Fremeaux-Bacchi, V.; Kemp, E.J.; Goodship, J.A.; Dragon-Durey, M.-A.; Strain, L.; Loirat, C.; Deng, H.-W.; Goodship, T.H.J. The Development of Atypical Haemolytic-Uraemic Syndrome Is Influenced by Susceptibility Factors in Factor H and Membrane Cofactor Protein: Evidence from Two Independent Cohorts. J. Med. Genet. 2005, 42, 852–856. [Google Scholar] [CrossRef] [Green Version]
- Fremeaux-Bacchi, V.; Dragon-Durey, M.-A.; Blouin, J.; Vigneau, C.; Kuypers, D.; Boudailliez, B.; Loirat, C.; Rondeau, E.; Fridman, W.H. Complement Factor I: A Susceptibility Gene for Atypical Haemolytic Uraemic Syndrome. J. Med. Genet. 2004, 41, e84. [Google Scholar] [CrossRef] [Green Version]
- Frémeaux-Bacchi, V.; Miller, E.C.; Liszewski, M.K.; Strain, L.; Blouin, J.; Brown, A.L.; Moghal, N.; Kaplan, B.S.; Weiss, R.A.; Lhotta, K.; et al. Mutations in Complement C3 Predispose to Development of Atypical Hemolytic Uremic Syndrome. Blood 2008, 112, 4948–4952. [Google Scholar] [CrossRef] [Green Version]
- Goicoechea de Jorge, E.; Harris, C.L.; Esparza-Gordillo, J.; Carreras, L.; Arranz, E.A.; Garrido, C.A.; López-Trascasa, M.; Sánchez-Corral, P.; Morgan, B.P.; Rodríguez de Córdoba, S. Gain-of-Function Mutations in Complement Factor B Are Associated with Atypical Hemolytic Uremic Syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 240–245. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Galbusera, M.; Gastoldi, S.; Macor, P.; Banterla, F.; Bresin, E.; Tripodo, C.; Bettoni, S.; Donadelli, R.; Valoti, E.; et al. Dynamics of Complement Activation in AHUS and How to Monitor Eculizumab Therapy. Blood 2014, 124, 1715–1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, F.; Maga, T.; Meyer, N.C.; Wang, K.; Thomas, C.P.; Nester, C.M.; Smith, R.J.H. Comprehensive Genetic Analysis of Complement and Coagulation Genes in Atypical Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2014, 25, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Józsi, M.; Licht, C.; Strobel, S.; Zipfel, S.L.H.; Richter, H.; Heinen, S.; Zipfel, P.F.; Skerka, C. Factor H Autoantibodies in Atypical Hemolytic Uremic Syndrome Correlate with CFHR1/CFHR3 Deficiency. Blood 2008, 111, 1512–1514. [Google Scholar] [CrossRef] [Green Version]
- Nester, C.M.; Barbour, T.; de Cordoba, S.R.; Dragon-Durey, M.A.; Fremeaux-Bacchi, V.; Goodship, T.H.J.; Kavanagh, D.; Noris, M.; Pickering, M.; Sanchez-Corral, P.; et al. Atypical AHUS: State of the Art. Mol. Immunol. 2015, 67, 31–42. [Google Scholar] [CrossRef]
- Arjona, E.; Huerta, A.; Goicoechea de Jorge, E.; Rodríguez de Córdoba, S. Familial Risk of Developing Atypical Hemolytic-Uremic Syndrome. Blood 2020, 136, 1558–1561. [Google Scholar] [CrossRef]
- Merinero, H.M.; García, S.P.; García-Fernández, J.; Arjona, E.; Tortajada, A.; Rodríguez de Córdoba, S. Complete Functional Characterization of Disease-Associated Genetic Variants in the Complement Factor H Gene. Kidney Int. 2018, 93, 470–481. [Google Scholar] [CrossRef]
- Marinozzi, M.C.; Vergoz, L.; Rybkine, T.; Ngo, S.; Bettoni, S.; Pashov, A.; Cayla, M.; Tabarin, F.; Jablonski, M.; Hue, C.; et al. Complement Factor B Mutations in Atypical Hemolytic Uremic Syndrome-Disease-Relevant or Benign? J. Am. Soc. Nephrol. 2014, 25, 2053–2065. [Google Scholar] [CrossRef] [Green Version]
- Galbusera, M.; Noris, M.; Gastoldi, S.; Bresin, E.; Mele, C.; Breno, M.; Cuccarolo, P.; Alberti, M.; Valoti, E.; Piras, R.; et al. An Ex Vivo Test of Complement Activation on Endothelium for Individualized Eculizumab Therapy in Hemolytic Uremic Syndrome. Am. J. Kidney Dis. 2019, 74, 56–72. [Google Scholar] [CrossRef]
- Rondeau, E.; Scully, M.; Ariceta, G.; Barbour, T.; Cataland, S.; Heyne, N.; Miyakawa, Y.; Ortiz, S.; Swenson, E.; Vallee, M.; et al. The Long-Acting C5 Inhibitor, Ravulizumab, Is Effective and Safe in Adult Patients with Atypical Hemolytic Uremic Syndrome Naïve to Complement Inhibitor Treatment. Kidney Int. 2020, 97, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Ariceta, G.; Dixon, B.P.; Kim, S.H.; Kapur, G.; Mauch, T.; Ortiz, S.; Vallee, M.; Denker, A.E.; Kang, H.G.; Greenbaum, L.A.; et al. The Long-Acting C5 Inhibitor, Ravulizumab, Is Effective and Safe in Pediatric Patients with Atypical Hemolytic Uremic Syndrome Naïve to Complement Inhibitor Treatment. Kidney Int. 2021, 100, 225–237. [Google Scholar] [CrossRef]
- Licht, C.; Greenbaum, L.A.; Muus, P.; Babu, S.; Bedrosian, C.L.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Furman, R.R.; Gaber, O.A.; et al. Efficacy and Safety of Eculizumab in Atypical Hemolytic Uremic Syndrome from 2-Year Extensions of Phase 2 Studies. Kidney Int. 2015, 87, 1061–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grangé, S.; Bekri, S.; Artaud-Macari, E.; Francois, A.; Girault, C.; Poitou, A.-L.; Benhamou, Y.; Vianey-Saban, C.; Benoist, J.-F.; Châtelet, V.; et al. Adult-Onset Renal Thrombotic Microangiopathy and Pulmonary Arterial Hypertension in Cobalamin C Deficiency. Lancet 2015, 386, 1011–1012. [Google Scholar] [CrossRef]
- Socié, G.; Caby-Tosi, M.-P.; Marantz, J.L.; Cole, A.; Bedrosian, C.L.; Gasteyger, C.; Mujeebuddin, A.; Hillmen, P.; Vande Walle, J.; Haller, H. Eculizumab in Paroxysmal Nocturnal Haemoglobinuria and Atypical Haemolytic Uraemic Syndrome: 10-Year Pharmacovigilance Analysis. Br. J. Haematol. 2019, 185, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Pallares, D.E.; Figueroa, J.E.; Densen, P.; Giclas, P.C.; Marshall, G.S. Invasive Haemophilus Influenzae Type b Infection in a Child with Familial Deficiency of the Beta Subunit of the Eighth Component of Complement. J. Pediatr. 1996, 128, 102–103. [Google Scholar] [CrossRef]
- Pettigrew, H.D.; Teuber, S.S.; Gershwin, M.E. Clinical Significance of Complement Deficiencies. Ann. N. Y. Acad. Sci. 2009, 1173, 108–123. [Google Scholar] [CrossRef]
- Bouts, A.; Monnens, L.; Davin, J.-C.; Struijk, G.; Spanjaard, L. Insufficient Protection by Neisseria Meningitidis Vaccination Alone during Eculizumab Therapy. Pediatr. Nephrol. 2011, 26, 1919–1920. [Google Scholar] [CrossRef] [Green Version]
- Lebel, E.; Trahtemberg, U.; Block, C.; Zelig, O.; Elinav, H. Post-Eculizumab Meningococcaemia in Vaccinated Patients. Clin. Microbiol. Infect. 2018, 24, 89–90. [Google Scholar] [CrossRef] [Green Version]
- Platonov, A.E.; Vershinina, I.V.; Kuijper, E.J.; Borrow, R.; Käyhty, H. Long Term Effects of Vaccination of Patients Deficient in a Late Complement Component with a Tetravalent Meningococcal Polysaccharide Vaccine. Vaccine 2003, 21, 4437–4447. [Google Scholar] [CrossRef]
- Gleesing, J.; Chiwane, S.; Rongkavilit, C. Gonococcal Septic Shock Associated with Eculizumab Treatment. Pediatr. Infect. Dis. J. 2012, 31, 543. [Google Scholar] [CrossRef]
- Hublikar, S.; Maher, W.E.; Bazan, J.A. Disseminated Gonococcal Infection and Eculizumab—A “High Risk” Connection? Sex Transm. Dis. 2014, 41, 747–748. [Google Scholar] [CrossRef]
- Ardissino, G.; Possenti, I.; Tel, F.; Testa, S.; Salardi, S.; Ladisa, V. Discontinuation of Eculizumab Treatment in Atypical Hemolytic Uremic Syndrome: An Update. Am. J. Kidney Dis. 2015, 66, 172–173. [Google Scholar] [CrossRef]
- Merrill, S.A.; Brittingham, Z.D.; Yuan, X.; Moliterno, A.R.; Sperati, C.J.; Brodsky, R.A. Eculizumab Cessation in Atypical Hemolytic Uremic Syndrome. Blood 2017, 130, 368–372. [Google Scholar] [CrossRef]
- Fakhouri, F.; Fila, M.; Provôt, F.; Delmas, Y.; Barbet, C.; Châtelet, V.; Rafat, C.; Cailliez, M.; Hogan, J.; Servais, A.; et al. Pathogenic Variants in Complement Genes and Risk of Atypical Hemolytic Uremic Syndrome Relapse after Eculizumab Discontinuation. Clin. J. Am. Soc. Nephrol. 2017, 12, 50–59. [Google Scholar] [CrossRef]
- Fakhouri, F.; Fila, M.; Hummel, A.; Ribes, D.; Sellier-Leclerc, A.-L.; Ville, S.; Pouteil-Noble, C.; Coindre, J.-P.; Le Quintrec, M.; Rondeau, E.; et al. Eculizumab Discontinuation in Children and Adults with Atypical Hemolytic-Uremic Syndrome: A Prospective Multicenter Study. Blood 2021, 137, 2438–2449. [Google Scholar] [CrossRef]
- Sukumar, S.; Brodsky, M.A.; Hussain, S.; Yanek, L.R.; Moliterno, A.R.; Brodsky, R.A.; Cataland, S.R.; Chaturvedi, S. Cardiovascular Disease Is a Leading Cause of Mortality among TTP Survivors in Clinical Remission. Blood Adv. 2021, 6. [Google Scholar] [CrossRef]
- Prevel, R.; Roubaud-Baudron, C.; Tellier, E.; Le Besnerais, M.; Kaplanski, G.; Veyradier, A.; Benhamou, Y.; Coppo, P.; Centre National de Référence des Microangiopathies Thrombotiques. [Endothelial dysfunction in thrombotic thrombocytopenic purpura: Therapeutic perspectives]. Rev. Med. Int. 2021, 42, 202–209. [Google Scholar] [CrossRef]
- Prevel, R.; Roubaud-Baudron, C.; Gourlain, S.; Jamme, M.; Peres, K.; Benhamou, Y.; Galicier, L.; Azoulay, E.; Poullin, P.; Provôt, F.; et al. Prognostic and Long-Term Survival of Immune Thrombotic Thrombocytopenic Purpura in Older Patients. Blood 2019, 134, 2209–2217. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prével, R.; Delmas, Y.; Guillotin, V.; Gruson, D.; Rivière, E. Complement Blockade Is a Promising Therapeutic Approach in a Subset of Critically Ill Adult Patients with Complement-Mediated Hemolytic Uremic Syndromes. J. Clin. Med. 2022, 11, 790. https://doi.org/10.3390/jcm11030790
Prével R, Delmas Y, Guillotin V, Gruson D, Rivière E. Complement Blockade Is a Promising Therapeutic Approach in a Subset of Critically Ill Adult Patients with Complement-Mediated Hemolytic Uremic Syndromes. Journal of Clinical Medicine. 2022; 11(3):790. https://doi.org/10.3390/jcm11030790
Chicago/Turabian StylePrével, Renaud, Yahsou Delmas, Vivien Guillotin, Didier Gruson, and Etienne Rivière. 2022. "Complement Blockade Is a Promising Therapeutic Approach in a Subset of Critically Ill Adult Patients with Complement-Mediated Hemolytic Uremic Syndromes" Journal of Clinical Medicine 11, no. 3: 790. https://doi.org/10.3390/jcm11030790