
Heterogeneity of Genotype–Phenotype in Congenital Hypofibrinogenemia—A Review of Case Reports Associated with Bleeding and Thrombosis


 and
and
Abstract
:1. Fibrinogen’s Structure and Its Function
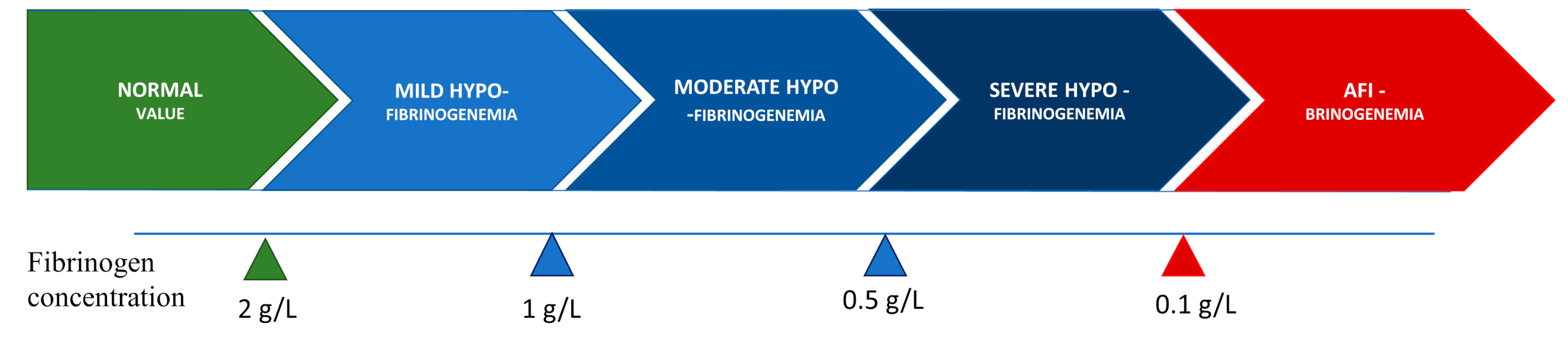
2. Congenital Hypofibrinogenemia—Characterization, Classification, and Clinical Phenotype
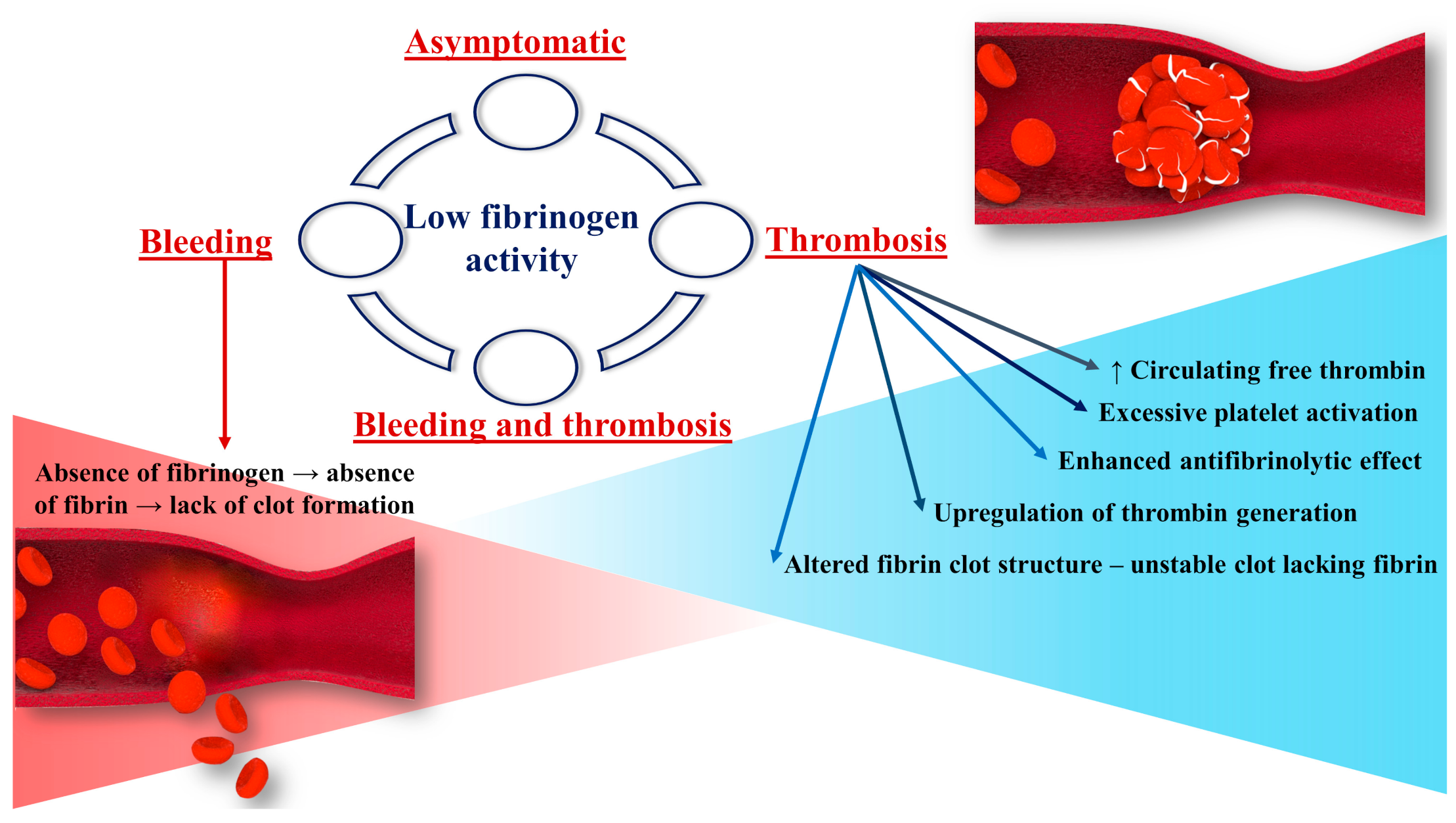
3. Fibrinogen’s Role in the Pathophysiology of Thrombosis and Bleeding in Hypofibrinogenemia
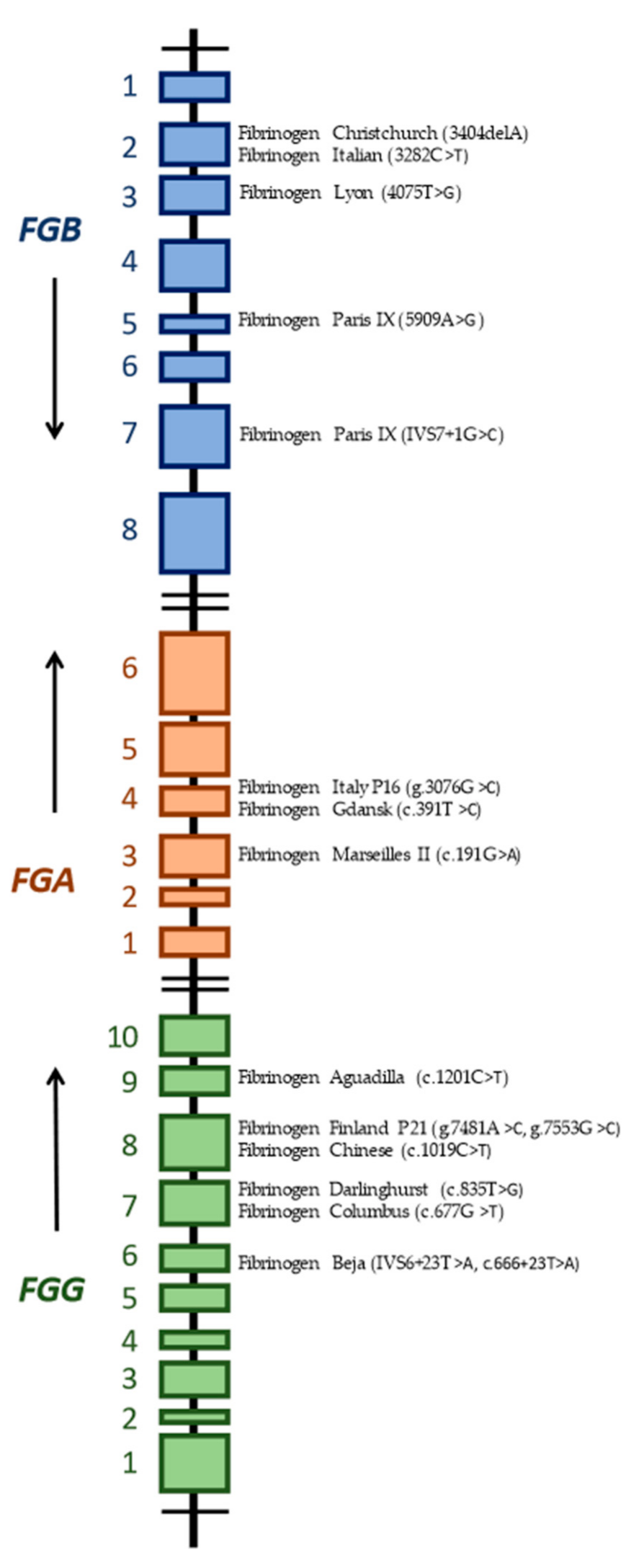
4. Review of Mutations Associated with Hypofibrinogenemia and Both Bleeding and Thrombotic Phenotype
5. Mutations in the FGA Gene
5.1. Fibrinogen MARSEILLES II
5.2. Fibrinogen ITALY P16
5.3. Fibrinogen GDANSK
6. Mutations in the FGB Gene
6.1. Fibrinogen CHRISTCHURCH V
6.2. Fibrinogen ITALIAN
6.3. Fibrinogen LYON
6.4. Fibrinogen PARIS IX
7. Mutations in the FGG Gene
7.1. Fibrinogen BEJA
7.2. Fibrinogen DARLINGHURST
7.3. Fibrinogen COLUMBUS
7.4. Fibrinogen FINLAND P21
7.5. Fibrinogen CHINESE (JIUJIANG)
7.6. Fibrinogen AGUADILLA
8. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Casini, A.; De Moerloose, P. Fibrinogen concentrates in hereditary fibrinogen disorders: Past, present, future. Haemophilia 2019, 26, 25–32. [Google Scholar] [CrossRef]
- Cronje, H.T.; Nienaber-Rousseau, C.; Zandberg, L.; Chikowore, T.; Lange, Z.; van Zyl, T.; Pieters, M. Candidate gene analysis of the fibrinogen phenotype reveales the importance of polygenic co-regulation. Matrix Biol. 2016, 60, 16–26. [Google Scholar] [CrossRef]
- Ridgway, H.J.; Brennan, S.O.; Loreth, R.M.; George, P.M. Fibrinogen Kaiserlautern (γ380 Lys to Asn) a new glycosylated fibrinogen variant with delayed polymerisation. Br. J. Haematol. 1997, 99, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W.; Siebenlist, K.R.; Meh, D.A. The structure and biological features of fibrinogen and fibrin. Ann. N. Y. Acad. Sci. 2001, 93, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Mosesson, M.W. Structure and function of fibrinogen and fibrin. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobrotova, M.; Skornova, I.; Brunclikova, M.; Grendar, M.; Lasabova, Z.; Stasko, J.; et al. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int. J. Hematol. 2020, 111, 795–802. [Google Scholar] [CrossRef]
- Tiscia, G.L.; Margaglione, M. Human Fibrinogen: Molecular and Genetic Aspects of Congenital Disorders. Int. J. Mol. Sci. 2018, 19, 1597. [Google Scholar] [CrossRef] [Green Version]
- Simurda, T.; Asselta, R.; Zolkova, J.; Brunclikova, M.; Dobrotova, M.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Congenital Afibrinogenemia and Hypofibrinogenemia: Laboratory and Genetic Testing in Rare Bleeding Disorders with Life-Threatening Clinical Manifestations and Challenging Management. Diagnostics 2021, 11, 2140. [Google Scholar] [CrossRef]
- Weasel, J.W.; Dempfle, C.E.H. Fibrinogen Structure and Function. Available online: https://oncohemakey.com/fibrinogen-structure-and-function (accessed on 7 September 2021).
- Vilar, R.; Fish, R.J.; Casini, A.; Neerman-Arbez, M. Fibrin(ogen) in human disease: Both friend and foe. Haematologica 2020, 105, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Asselta, R.; Duga, S.; Tenchini, M.L. The molecular basis of quantitative fibrinogen disorders. J. Thromb. Haemost. 2006, 4, 2115–2129. [Google Scholar] [CrossRef]
- Simurda, T.; Caccia, S.; Asselta, R.; Zolkova, J.; Skornova, I.; Snahnicanova, Z.; Loderer, D.; Lasabova, Z.; Kubisz, P. Congenital hypofibrinogenemia associated with as novel heterozygous nonsense mutation in the globular C—Terminal domain of the γ—Chain (p.Glu275Stop). J. Thromb. Thrombolysis 2019, 50, 233–236. [Google Scholar] [CrossRef]
- Simurda, T.; Vilar, R.; Zolkova, J.; Ceznerova, E.; Kolkova, Z.; Loderer, D.; Neerman-Arbez, M.; Casini, A.; Brunclikova, M.; Skornova, I.; et al. A Novel Nonsense Mutation in FGB (c.1421 G>A;p.Trp474Ter) in the Beta Chain of Fibrinogen Causing Hypofibrinogenemia with Bleeding Phenotype. Biomedicines 2020, 8, 605. [Google Scholar] [CrossRef]
- Casini, A.; Undas, A.; Palla, R.; Thachil, J.; de Moerloose, P. Diagnosis and classification of congenital fibrinogen disorders: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2018, 16, 1887–1890. [Google Scholar] [CrossRef]
- Simurda, T.; Brunclikova, M.; Asselta, R.; Caccia, S.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Skornova, I.; Hudecek, J.; Lasabova, Z.; et al. Genetic variants in the FGB and FGG genes mapping in the beta and gamma nodules of the fibrinogen molecule in congenital quantitative fibrinogen disorders associated with a thrombotic phenotype. Int. J. Mol. Sci. 2020, 21, 4616. [Google Scholar] [CrossRef]
- Miesbach, W.; Galanakis, D.; Scharrer, I. treatment of patients with dysfibrinogenemia and history of abortion during pregnancy. Blood Coagul. Fibrinolysis 2009, 20, 366–370. [Google Scholar] [CrossRef]
- Asselta, R.; Platé, M.; Robusto, M.; Borhany, M.; Guella, I.; Soldà, G.; Afrasiabi, A.; Menegatti, M.; Shamsi, T.; Pyvandi, F.; et al. Clinical and molecular characterisation of 21 patients affected by quantitative fibrinogen deficiency. Thromb. Haemost. 2016, 113, 567–576. [Google Scholar] [CrossRef]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and fibrin in hemostasis and thrombosis. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef] [Green Version]
- Beck, E.A.; Vogel, A.; Jackson, D.P. Functional evaluation of an inherited abnormal fibrinogen: Fibrinogen Baltimore. J. Clin. Investig. 1971, 50, 1874–1884. [Google Scholar] [CrossRef]
- Kamijo, T.; Mukai, S.; Taira, C.; Higuchi, Y.; Okumura, N. γD318Y fibrinogen shows no fibrin polymerization due to defective “A-a” and “B-b” interactions, whereas that of γK321E fibrinogen is nearly normal. Thromb. Res. 2019, 182, 150–158. [Google Scholar] [CrossRef]
- Borrell, M.; Gari, M.; Vallve, C.; Tirado, I.; Soria, J.M.; Sala, N.; Munoz, C.; Oliver, A.; Garcia, A.; Fontcuberta, J. Abnormal polymerization and normal binding of plasminogen and t-PA in three new dysfibrinogenemias: Barcelona III and IV (γArg 275→His) and Villajoyosa (γArg 275→Cys). Blood Coagul. Fibrinolysis 1995, 6, 198–206. [Google Scholar] [CrossRef]
- Kotlin, R.; Reicheltova, Z.; Maly, M.; Suttnar, J.; Sobotkova, A.; Salaj, P.; Hirmerova, J.; Riedel, T.; Dyr, J.E. Two cases of congenital dysfibrinogenemia associated with thrombosis—Fibrinogen Prha III and Fibrinogen Plzen. Thromb. Haemost. 2009, 102, 479–486. [Google Scholar] [CrossRef]
- Kaido, T.; Yoda, M.; Kamijo, T.; Taira, C.; Higuchi, Y.; Okumura, N. Comparison of molecular structure and fibrin polymerization between two Bβ—Chain N—terminal region fibrinogen variants, Bβp. G45C and Bβp.R74C. Int. J. Hematol. 2020, 112, 331–340. [Google Scholar] [CrossRef]
- Fuss, C.; Palmaz, J.C.; Sprague, E.A. Fibringen: Structure, function, and surface interactions. J. Vasc. Interv. Radiol. 2001, 12, 677–682. [Google Scholar] [CrossRef]
- Simurda, T.; Casini, A.; Stasko, J.; Hudecek, J.; Skornova, I.; Vilar, R.; Neerman-Arbez, M.; Kubisz, P. Perioperative mangement of a severe congenital hypofibrinogenemia with thrombotic phenotype. Thromb. Res. 2020, 188, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Besser, M.W.; MacDonald, S. Acquired hypofibrinogenemia: Current perspectives. J. Blood Med. 2016, 7, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neerman-Arbez, M.; Casini, A. Clinical Consequences and Molecular Bases of Low Fibrinogen Levels. Int. J. Mol. Sci. 2018, 19, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanss, M.M.L.; Ffrench, P.O.; Morner, J.F.; Chabuet, M.; Biot, F.; De Mazancourt, M. The novel fibrinogen variants found in patients with pulmonary embolism and their families. J. Thromb. Haemost. 2003, 1, 1251–1257. [Google Scholar] [CrossRef] [Green Version]
- Korte, W.; Poon, M.C.; Iorio, A.; Makris, M. Thrombosis in Inherited Fibrinogen Disorders. Transfus. Med. Hemother. 2017, 44, 70–76. [Google Scholar] [CrossRef] [Green Version]
- De Moerloose, P.; Casini, A.; Neerman-Arbez, M. Congenital Fibrinogen Disorders: An Update. Semin. Thromb. Hemost. 2013, 39, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Asselta, R.; Spena, S.; Duga, S.; Tenchini, M.L. Molecular genetics of quantitative fibrinogen disorders. Cardiovasc Hematol. Agents Med. Chem. 2007, 5, 163–173. [Google Scholar] [CrossRef]
- Hamano, A.; Mimuro, J.; Aoshima, M.; Itoh, T.; Kitamura, N.; Nishinarita, S.; Takano, K.; Ishiwata, A.; Kashiwakura, Y.; Niwa, K.; et al. Thrombophilic dysfibrinogen Tokyo V with the amino acid substitution of γAla327Thr: Formation of fragile but fibrinolysis—Resistant fibrin clots and its relevance to arterial thromboembolism. Blood 2004, 103, 3045–3050. [Google Scholar] [CrossRef]
- Duval, C.; Ariens, R.S. Fibrinogen splice variation and cross-linking: Effects on fibrin structure/function and role of fibrinogen γ’as trhombomobulin II. Matrix Biol. 2017, 60, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Soares, A.W.; Maia, M.; Santo, J.E.; Costa, A.P.; Pereira, A.; Catarino, C. Hypofibrinogenemia: A case of spontaneous bleeding and central venous thrombosis in the same lifetime. Eur. J. Case Rep. Intern. Med. 2020, 7, 001424. [Google Scholar] [CrossRef]
- Asselta, R.; Duga, S.; Spena, S.; Peyvandi, F.; Castaman, G.; Malcovati, M.; Mannucci, P.M.; Tenchini, M.L. Missense or splicing mutation? The case of a fibrinogen Bβ—Chain mutation causing severe hypofibrinogenemia. Blood 2004, 103, 3051–3054. [Google Scholar] [CrossRef]
- Hanss, M.; FFrench, P.; Vinciguerra, C.; Bertrands, M.A.; De Mazancourt, P. Four cases of hypofibrinogenemia associated with four novel mutations. J. Thromb. Haemost. 2005, 3, 2347–2349. [Google Scholar] [CrossRef]
- Horellou, M.H.; Chevreaud, C.; Mathieux, V.; Conard, J.; de Mazancourt, P. Fibrinogen Parix IX: A case of asymptomatic hypofibrinogenemia with Bβ Y236C and Bβ IVS7—1C→C mutations. J. Thromb. Haemost. 2006, 4, 1134–1136. [Google Scholar] [CrossRef]
- Amri, Y.; Dabboubi, R.; Mghaieth, F.; Zili, M.; Messaoud, T.; Casini, A.; de Moerloose, P.; Toumi, N.H. Molecular characterization of two hypofibrinogenemic patients associated with a novel FGG IVS6+23T˃A substitution and a previously reported FGB IVS6-10_16delTTTG deletion. Haemophilia 2020, 26, 86–96. [Google Scholar] [CrossRef]
- Davis, R.L.; Mosesson, M.W.; Kerlin, B.A.; Canner, J.A.; Ruymann, F.B.; Brennan, S.O. Fibrinogen Columbus: A novel gamma Gly200Val mutation causing hypofibrinogenemia in a family with associated thrombophilia. Haematologica 2007, 92, 1151–1152. [Google Scholar] [CrossRef] [Green Version]
- Nia, H.; Wu, X.; Bao, B.; Xia, Z.; Tan, D. Cortical venous thrombosis, multiple cortical infarctions, and vaginal bleeding in a Chinese family with hypofibrinogenemia caused by FGG mutation c.1019C˃T: A case report. Neurol. Sci. 2020, 41, 2299–2301. [Google Scholar] [CrossRef]
- Casini, A.; Sokollik, C.; Lukowski, S.W.; Lurz, E.; Rieubland, C.; de Moerlosse, P.; Neerman-Arbez, M. Hypofibrinogenemia and liver disease: A new case of Aguadilla fibrinogen and review of the literature. Haemophilia 2015, 21, 820–827. [Google Scholar] [CrossRef]
- Mital, A.; Undas, A.; Neerman-Arbez, M.; Hellmann, A. Fibrinogen Gdansk: Hypofibrinogenemia associated with a novel missense mutation in FGA (Ser112Pro). Thromb. Res. 2012, 130, e196–e197. [Google Scholar] [CrossRef]
- Brennan, O.S.; Mosesson, M.V.; Lowen, R.; Siebenlits, K.R.; Matsunaga, A. Hypofibrinogenaemia resulting from novel single nucleotide deletion at codon Bβ58/3404delA) associated with thrombotic stroke in infancy. Thromb. Haemost. 2006, 95, 738–739. [Google Scholar]
- Castaman, G.; Lunardi, M.; Rigo, L.; Mastroeni, V.; Bonoldi, E.; Rodeghiero, F. Severe spontaneous arterial thrombotic manifestations in patients with inherited hypo—And afibrinogenemia. Haemophilia 2009, 15, 533–537. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type of CFD | Variants in FGA | Variants in FGB | Variants in FGG | Total Number of Variants |
|---|---|---|---|---|
| Afibrinogenemia | 14 | 6 | 4 | 24 |
| Hypofibrinogenemia | 3 | 5 | 7 | 15 |
| Dysfibrinogenemia | 12 | 2 | 8 | 22 |
| Hypodysfibrinogenemia | 5 | 0 | 3 | 8 |
| Total number | 34 | 13 | 22 | 69 |
| Name/Origin | Mature Protein Variation | Native Protein Variation | cDNA | Gene Status | e–i | Numbers of Studied Patients/Numbers of Patients with Thrombosis | Bleeding Complications | Numbers of Thrombotic Complications and Obstetrical Problems | Localization of Thrombosis | Other Thrombophilic States |
|---|---|---|---|---|---|---|---|---|---|---|
| Fibrinogen Aα Chain Mutations Associated with Hypofibrinogenemia and Bleeding and Thrombosis | ||||||||||
| MARSEILLES II | Aα(45)Cys˃Tyr | p.Cys64Tyr | c.191G˃A | Heterozyg. | e–3 | 2/1 | Spontaneous digestive tract bleeding Metrorrhagia | 1x External iliac VT, common femoral, popliteal | External iliac VT, common femoral, popliteal | Protein S deficiency |
| ITALY P16 | Aα(110)Arg˃Pro | p.Arg129Pro | g.3076G˃C | Heterozyg. | e–4 | 1 | Colon bleeding | Repetitive DVT | DVT | No other thrombophilic state |
| GDANSK | Aα(112)Ser˃Pro | p.Ser131Pro | c.391T˃C | Heterozyg. | e–4 | 4/1 | Easy bruising, bleeding after surgery | 1x Left popliteal and calf veins | Left popliteal and calf veins | No other thrombophilic state |
| Fibrinogen Bβ Chain Mutations Associated with Hypofibrinogenemia and Bleeding and Thrombosis | ||||||||||
| CHRISTCHURCH V | Bβ(58)Lys˃frameshift–stop | p.Lys88Frameshift Stop | 3404delA | Heterozyg. | e–2 | 3/1 | Easy bruising | 1x IS | IS | No other thrombophilic state |
| ITALIAN | Bβ(172)Leu˃Gly | p.Leu202Gly | 3282C˃T | Compound | e–2 | 1 | Bleeding after dental care | 6x Miscar., 1x tibial artery thrombosis | Tibial artery thrombosis | Heterozygous Factor V Leiden |
| LYON | Bβ(118)Met˃Lys | p.Met148Lys | 4075T˃G | Heterozyg. | e–3 | 2/1 | Easy bruising, bleeding after dental care, post—partum bleeding | 3x Miscar. | Miscar. | x |
| PARIS IX | Bβ(236)Tyr˃Cys | p.Tyr266Cys | 5909A˃G IVS7+1G˃C | Compound Compound | e–5 i–7 | 1 | Easy bruising, epistaxis, menorrhagia | 1x Miscar., 1x DiVT | Miscar, DiVT | x |
| Fibrinogen γ Chain Mutations Associated with Hypofibrinogenemia and Bleeding and Thrombosis | ||||||||||
| BEJA | IVS6+23T˃A (c.666+23T˃A) | Homozyg. | i–6 | 1 | Bleeding after dental care | 1x MI | MI | No other thrombophilic state | ||
| DARLINGHURST | γ(253)Trp˃Gly | p.Trp279Gly | c.835T˃G | Homozyg. | e–7 | 2/1 | Bleeding after dental care | CHTED, repetitive miscar. | CHTED, miscra. | No other thrombophilic state |
| COLUMBUS | γ(200)Gly˃Val | p.Gly226Val | c.677G˃T | Heterozyg. | e–7 | 2/2 | x | 1x DVT | DVT | Heterozygous Factor V Leiden, MTHFR C677T mutations |
| FINLAND P21 | γ(277)Thr˃Pro γ(301)Asp˃His | p.Thr303Pro p.Asp327His | g.7481A˃C g.7553G˃C | Compound Compound | e–8 e–8 | 4 | x | x | x | x |
| CHINESE | γ(314)Thr˃IIe | p.Thr340Ile | c.1019C˃T | Heterozyg. | e–8 | 5/1 | Metrorrhagia | 1x CVT | CVT | x |
| AGUADILLA | γ(375)Arg˃Trp | p.Arg401Trp | c.1201C˃T | Heterozyg. | e–9 | 1/1 | x | x | x | x |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunclikova, M.; Simurda, T.; Zolkova, J.; Sterankova, M.; Skornova, I.; Dobrotova, M.; Kolkova, Z.; Loderer, D.; Grendar, M.; Hudecek, J.; et al. Heterogeneity of Genotype–Phenotype in Congenital Hypofibrinogenemia—A Review of Case Reports Associated with Bleeding and Thrombosis. J. Clin. Med. 2022, 11, 1083. https://doi.org/10.3390/jcm11041083
Brunclikova M, Simurda T, Zolkova J, Sterankova M, Skornova I, Dobrotova M, Kolkova Z, Loderer D, Grendar M, Hudecek J, et al. Heterogeneity of Genotype–Phenotype in Congenital Hypofibrinogenemia—A Review of Case Reports Associated with Bleeding and Thrombosis. Journal of Clinical Medicine. 2022; 11(4):1083. https://doi.org/10.3390/jcm11041083
Chicago/Turabian StyleBrunclikova, Monika, Tomas Simurda, Jana Zolkova, Miroslava Sterankova, Ingrid Skornova, Miroslava Dobrotova, Zuzana Kolkova, Dusan Loderer, Marian Grendar, Jan Hudecek, and et al. 2022. "Heterogeneity of Genotype–Phenotype in Congenital Hypofibrinogenemia—A Review of Case Reports Associated with Bleeding and Thrombosis" Journal of Clinical Medicine 11, no. 4: 1083. https://doi.org/10.3390/jcm11041083