
Genome Survey of Stipa breviflora Griseb. Using Next-Generation Sequencing
by
 ,
,
Xiangjun Yun
1,†, 
Jinrui Wu
1,†,
Bo Xu
2,
Shijie Lv
3,
Le Zhang
2,
Wenguang Zhang
3,
Shixian Sun
1,
Guixiang Liu
1,*,
Yazhou Zu
4 and
Bin Liu
4 1
Grassland Research Institute, Chinese Academy of Agriculture Sciences, Hohhot 010019, China
2
Inner Mongolia Forestry Monitoring and Planning Institute, Hohhot 010020, China
3
College of Science, Inner Mongolia Agricultural University, Hohhot 010018, China
4
Nei Mongol BioNew Technology, Hohhot 010018, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Agriculture 2023, 13(12), 2243; https://doi.org/10.3390/agriculture13122243
Submission received: 23 October 2023
/
Revised: 19 November 2023
/
Accepted: 29 November 2023
/
Published: 5 December 2023
(This article belongs to the Section Genotype Evaluation and Breeding)
Abstract
:Due to climate change and global warming, the frequency of sandstorms in northern China is increasing. Stipa breviflora, a dominant species in Eurasian grasslands, can help prevent desertification from becoming more serious. Studies on S. breviflora cover a wide range of fields. To the best of our knowledge, the present study is the first to sequence, assemble, and annotate the S. breviflora genome. In total, 2,781,544 contigs were assembled, and 2,600,873 scaffolds were obtained, resulting in a total length of 649,849,683 bp. The number of scaffolds greater than 1 kb was 70,770. We annotated the assembled genome (>121 kb), conducted a selective sweep analysis, and ultimately succeeded in assembling the Matk gene of S. breviflora. More importantly, our research identified 26 scaffolds that may be responsible for the drought tolerance of S. breviflora Griseb. In summary, the data obtained regarding S. breviflora will be of great significance for future research.
1. Introduction
Belonging to the family Gramineae, Stipa breviflora Griseb. is a dense perennial xerophytic herb. It is widely distributed in the desert area of Yongdeng and Lanzhou and extends along the southeastern edge of the desert steppe to the typical steppe area of northeastern China. The central distribution of Stipa breviflora is the Loess Plateau [1]. The distinguishing geographical distribution of S. breviflora makes it an excellent model for studying the genesis and evolution of grassland species and modern microevolution.
As the climate has become increasingly warmer, more than 90% of natural grasslands in northern China have experienced degradation and desertification, which has led to frequent sandstorms [2]. As a dominant species in Eurasian grasslands, S. breviflora plays an important role in resisting the threat of desertification. As an indicator of the desert steppe, S. breviflora can survive adaptively under drought conditions. S. breviflora has several advantages over other grassland plants, such as drought tolerance, earlier regreening, high productivity, and grazing tolerance. These attributes make S. breviflora one of the most important components of the Eurasian Steppe. Hence, studies on S. breviflora are of great significance not only in theoretical ecology, but also in maintaining the ecological security of China.
S. breviflora is the dominant species in the experimental area, where the natural conditions are harsh and the grassland ecosystem is exceptionally fragile. Over an extended period of unrestricted grazing, this area has been in a state of negative balance between input and output, resulting in severe ecosystem degradation. However, under mild to moderate grazing pressure, such grasslands exhibit relatively stable primary productivity levels. S. breviflora plays a crucial role in shaping the appearance and environmental conditions of the community and gives rise to unique landscape features.
The molecular mechanism of drought tolerance is a concerning issue. The development of next-generation sequencing (NGS) technology has provided researchers with a method through which to address this issue in non-model species, such as S. breviflora. Additionally, the k-mer method has been successfully applied for genome size estimation using NGS technology [3], such as in Chinese jujube [4], Rosa roxburghii Tratt [3], Switchgrass [5], Gracilariopsis lemaneiformis [6], Cucumis sativus [7], and Myrica rubra [8]. Studies on S. breviflora involve a wide range of fields, such as taxonomy, geographical distribution, species morphology, population ecology, community ecology, interspecific relations, and grazing utilization [9,10,11,12,13,14,15]. However, as far as we know, research on the molecular biology of S. breviflora is limited.
The chromosome number (2n = 42) of S. breviflora has been reported [16], and Zhao Lei et al. explored simple sequence repeats (SSRs) as molecular markers [17]. Zhao Qing et al. found that the genetic diversity of the S. breviflora population was related to thermal factors using random amplified polymorphic DNA (RAPD) [18]. An analysis of the S. breviflora genome has not yet been reported. We aimed to accelerate the study of S. breviflora and provide a theoretical basis for the rational utilization and protection of S. breviflora by providing more information about its genome using NGS and by laying a molecular foundation for future study. In this study, we estimated the genome size, GC content, and heterozygosity rate, performed molecular phylogenetic analysis, detected single nucleotide polymorphisms (SNPs), and annotated drought-tolerant loci in S. breviflora scaffolds. These data will be used in our future research.
2. Materials and Methods
2.1. Plant Materials and Sampling Methods
Stipa breviflora plants were collected from the desert steppe of Duhu Sumu Harden Hushu Gacha, Sonid Right Banner, XilinGol League, Inner Mongolia (N42°16′26.2″, E112°47′16.9″), located in the continental monsoon climate region at an altitude of 1100–1150 m. Sonid Right Banner is located in the temperate continental monsoon climate zone, which includes a long and cold winter and a short and cool summer with limited and concentrated rainfall. There is ample sunshine in addition to large temperature variations and numerous windy days. Spring and autumn exhibit significant temperature variations, and the frost-free period is short. The annual average precipitation is 177.2 mm, is unevenly distributed from south to north, and decreases from east to west. Precipitation during the growth stages of grasslands, which amounts to around 160.1 mm, constitutes about 90% of the annual total and is mostly concentrated from June to August. The terrain is gently sloping and is covered by a substantial amount of Tertiary Mesozoic red sandstone, sediment, and gravel layers, with a thinner layer of Quaternary residual deposits on top. The soil is a zonal soil transitioning from desert grassland to desert. The surface is prone to sandification, with organic matter content ranging from 1.0% to 1.8%. The soil type is primarily Haplic Kastanozem (FAO).
Ten individual S. breviflora plants were carefully chosen to represent diverse age structures, with their selection being primarily based on their basal stems, for which the criteria were as follows: seedling, 0.1–0.3 cm, 0.4–1.0 cm, 1.1–2.0 cm, 2.1–3.0 cm, 3.1–4.0 cm, 4.1–5.0 cm, 5.1–7.0 cm, 7.1–9.0 cm, and >9.0 cm. The specimens were transported from the steppe to Beijing at a low temperature. Total DNA was extracted from approximately 100 mg of young leaf tissues using the CTAB method [19], for which we referred to the procedure of Jinlu Li et al. DNA quality and concentrations were assessed using 1% agarose gel electrophoresis with ethidium bromide staining and a Nanodrop K5500 Microvolume Spectrophotometer with a Qubit, respectively.
2.2. Genome Sequencing
The 10 DNA samples were pooled equimolarly before library construction and were designated as ZM1. Library construction and sequencing were performed at the Annoroad facility in Beijing, China. The RAD libraries of the tissues were prepared for paired-end sequencing, with steps including double enzyme restriction, adapter ligation, fragment selection, quantitation, PCR amplification, and quality control [20].
2.2.1. Library Preparation
- (a)
- Fragmentation: A total of 2 μg of genomic DNA of each tissue was digested for 6 h at 37 °C in a 25 μL reaction with MseI and SacI (recognition site: T|TAA and GAGCT|C).
- (b)
- Adapter ligation: After recovery, a single “A” base was added to one end. DNA adapters containing sequences necessary for binding to the Illumina HiSeq 2500 system were ligated to the DNA fragments.
- (c)
- Size selection: Fragments with adapters were selected based on size to ensure consistency in sequencing according to the manufacturer’s protocol.
2.2.2. Library Amplification
PCR amplification: The DNA fragments with attached adapters were amplified through PCR to create enough DNA for sequencing.
2.2.3. Quality Control and Sequencing
The size distribution of the library was assessed. High-throughput sequencing technology that used an Illumina HiSeq 2500 system with PE125bp reads was employed to generate data according to the manufacturer’s protocol. Clean reads were obtained after removing low-quality reads from the libraries, including reads containing contaminated adapters, low-quality nucleotides, and unrecognizable nucleotides.
The steps of data processing were conducted as follows:
- (1)
- A paired read was discarded if at least one read of the pair contained a contaminated adapter.
- (2)
- A paired read was discarded if more than 10% of the bases were uncertain in at least one of the reads.
- (3)
- A paired read was discarded if the proportion of low-quality (Phred quality < 5) bases was over 50% in at least one read (Table 1).
2.3. Genome Size Estimation, Assembly, and GC Content Analysis
After filtering for the genome survey study, 23.087 Gb of clean data were obtained. K-mer analysis was employed to estimate the genome size. The relationship was expressed using the following algorithm: genome size = k-mer num/peak depth. Thus, information on the peak depth and the number of 17-mers was obtained. Genome characteristic estimation (GCE) software was used on the preprocessed reads, where k-mer sizes of 14, 17, 19, and 21 were examined using default parameters, and then we selected the optimal k-mer size according to the N50 length [21]. The Short Oligonucleotide Analysis Package (SOAP) software was used to reconfirm the size. Assembly was performed using SOAPdenovo2 (http://soap.genomics.org.cn, accessed on September 2016)—a genome assembler developed specifically for use with next-generation short-read sequences [22]. First, 125 base reads from fragmented small-insert-size libraries were selected for realignment into contigs using sequence overlap information. Contigs were not extended into regions in which repeat sequences created ambiguous connections. Then, contigs were joined into scaffolds step by step, from the shortest (150 bp) to the longest (10 kb). While these assembled sequences could not be used to construct a high-quality S. breviflora genome, they fulfilled our genome survey requirements. Therefore, they were used for subsequent analyses, such as the calculation of GC content, gene annotation, cluster analysis, and repetitive sequence analysis.
2.4. Gene Annotation and Cluster Analysis
After assembling the S. breviflora genome with a filtering scaffold of <1000 bp for de novo annotation, we annotated genes based primarily on homology to what was reported in the stipe genus genes. From common databases and the NCBI database (https://www.ncbi.nlm.nih.gov, accessed on October 2016), we collected 907 stipe genes (Table S1). BLAST alignment (https://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on October 2016) was performed between the assembled genome (>1 kb) and these genes (Table 2) [23]. In our investigation of the drought tolerance of S. breviflora, we identified 205 drought-tolerant genes that were previously reported in other specimens within the Gramineae family (Table S5). For cluster analysis, we downloaded 134 different MatK gene sequences of chloroplasts from the GenBank database (Table S2). The MAFFT program was used to align them (https://mafft.cbrc.jp/alignment/software/, accessed on January 2017) [24]. For phylogenetic construction, we employed the maximum likelihood method using the Fast Tree program with default parameters (http://www.microbesonline.org/fasttree/, accessed on January 2017) [25].
2.5. Repetitive Sequences and Selective Sweep Analysis
RepeatMasker (http://www.repeatmasker.org/RepeatMasker/, accessed on March 2017) was employed to identify repeats in the assembled genome [26]. The ‘MISA’ mode of SciRoKo was used to identify SSR motifs with the default parameters [27]. At the same time, SSR repeats for mono-, di-, tri-, tetra-, penta-, and hexanucleotides were identified. To detect putative signatures of selection, high-quality SNPs from S. breviflora were identified. Allele counts at SNP positions were used to identify signatures of selection in a sliding 1-kb window with a step size of 500 bp by scanning along S. breviflora chromosomes at all SNP positions. With the numbers of reads corresponding to the most and least abundant alleles (nMAJ and nMIN), we calculated the heterozygosity score in each sliding 1-kb window as follows: Hp = 2∑nMAJ∑nMIN/(∑nMAJ + ∑nMIN)2, where ∑nMAJ and ∑nMIN are the sums of nMAJ and nMIN, respectively, for all SNPs in the window. Individual Hp values were Z-transformed as follows: ZHp = (Hp − μHp)/σHp [28].
3. Results and Discussion
3.1. Genome Size Estimation, Assembly, and GC Content Analysis
We generated about 23.087 Gb of 125 bp paired-end reads from the sample. Before de novo assembly, we analyzed the genome size of S. breviflora using different software-aided GCEs and the KmerFreq_AR model of SOAPec. The results of the GCEs with different k-mers (14, 17, 19, and 21) showed that k-mer 17 was preferable for estimation. The peak of the 17-mer distribution was approximated at 11, and the total K-mer count was 20,624,226,664. According to the formula of genome size = k-mer num/peak depth, the genome size of S. breviflora was approximated to be 1G (Table 1). The assembled genome (606 Mbp) was significantly smaller than the estimated genome size, which was possibly due to the removal of shorter sequences and support for shorter sequences during assembly.
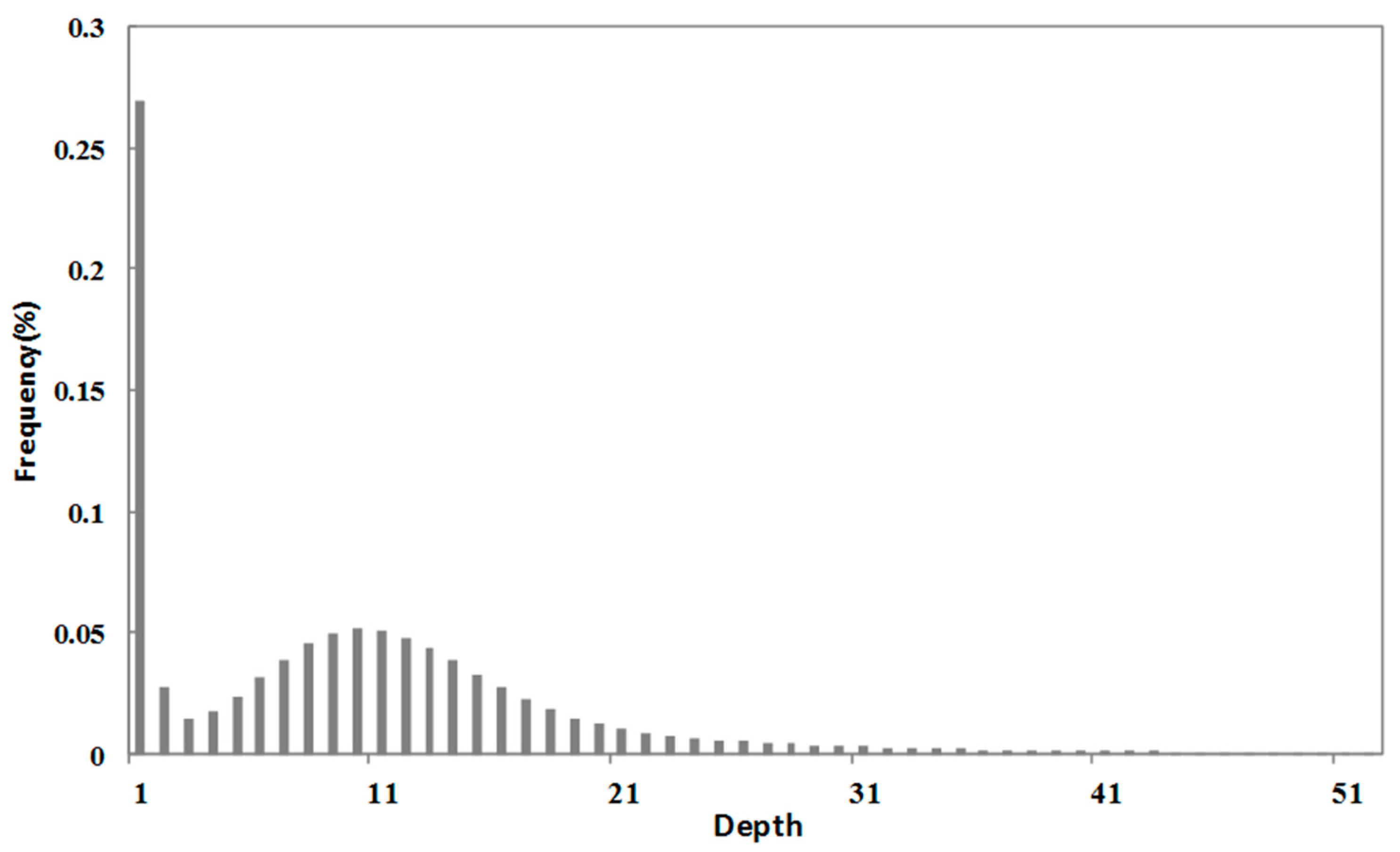
We did not observe a sub-peak in the k-mer analysis; therefore, the heterozygosity rates may have been low. The fat tail shown in Figure 1 indicates that there may be a high level of repeats in S. breviflora.
A total of 23.087 Gb of clean data were used to carry out de novo assembly with the SOAPDenovo software. We contracted k-mer 17 with SOAPDenovo for optimal assembly. At this point, we assembled 2,781,544 contigs, of which 6359 bp was the longest. Then, the total size of the scaffolds was 649,849,683 bp with N50 = 307 bp. The total gap length (Ns) was 7,619,172 bp (Table 3). The average genome coverage was 27.9199%. The average GC content of S. breviflora was about 44.16%, which was higher than those of Arabidopsis thaliana (35.97%), Glycine max (34.21%), Sorghum bicolor (41.56%), and Solanum tuberosum (34.8–36.0%) [29], lower than those of Brachypodium distachyon (46.21%) and Zea mays (46.60%), and similar to that of Oryza sativa (42.41%) [30]. Moreover, the too-high mA content (>65%) and too-low (<25%) GC content may have caused sequence bias on the Illumina sequencing platform, thus seriously affecting the genome assembly [31].
3.2. Gene Annotation and Cluster Analysis
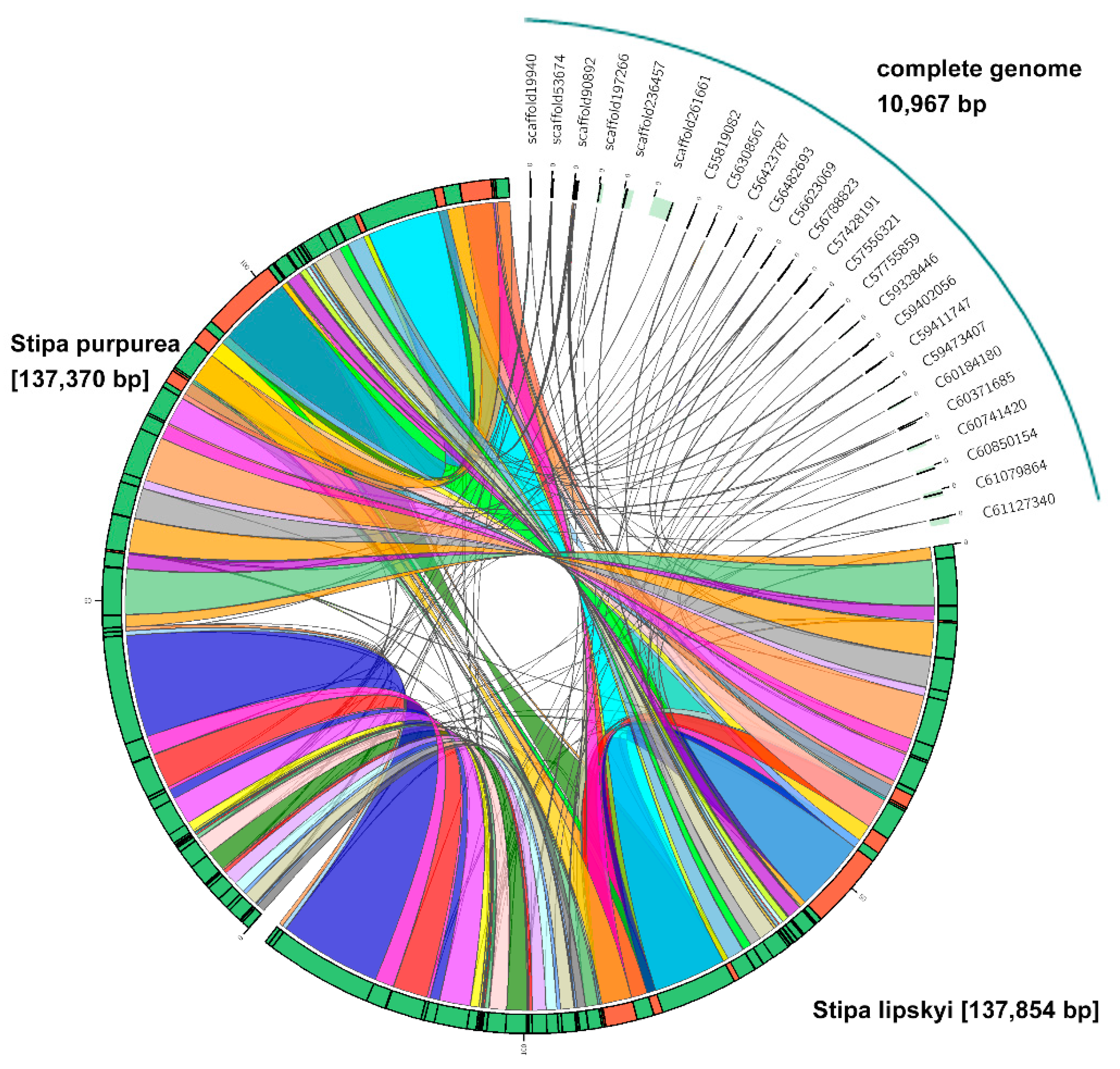
Based on the assembled genome of S. breviflora, 70,770 scaffolds (>1 kb) were used for de novo annotation. Because our data were derived from pooled DNA samples and short assembly scaffolds and were intended for a genome survey of S. breviflora, we opted to align only several specific sets of genes. First, we found that the longest among the 70,770 scaffolds was 10,967 bp, which was a DNA sequence from the chloroplasts. From the NCBI database, we obtained two chloroplast sequences from species of the same genus—Stipa lipskyi and Stipa purpurea. The lengths of those two DNA sequences were 137,854 and 137,370, respectively. C-Sibelia analysis showed that 25 similar sequences were present in these three species (Figure 2). The information on these 25 genes is provided in Table S3. Determining the functions of these 25 genes is a follow-up research direction.
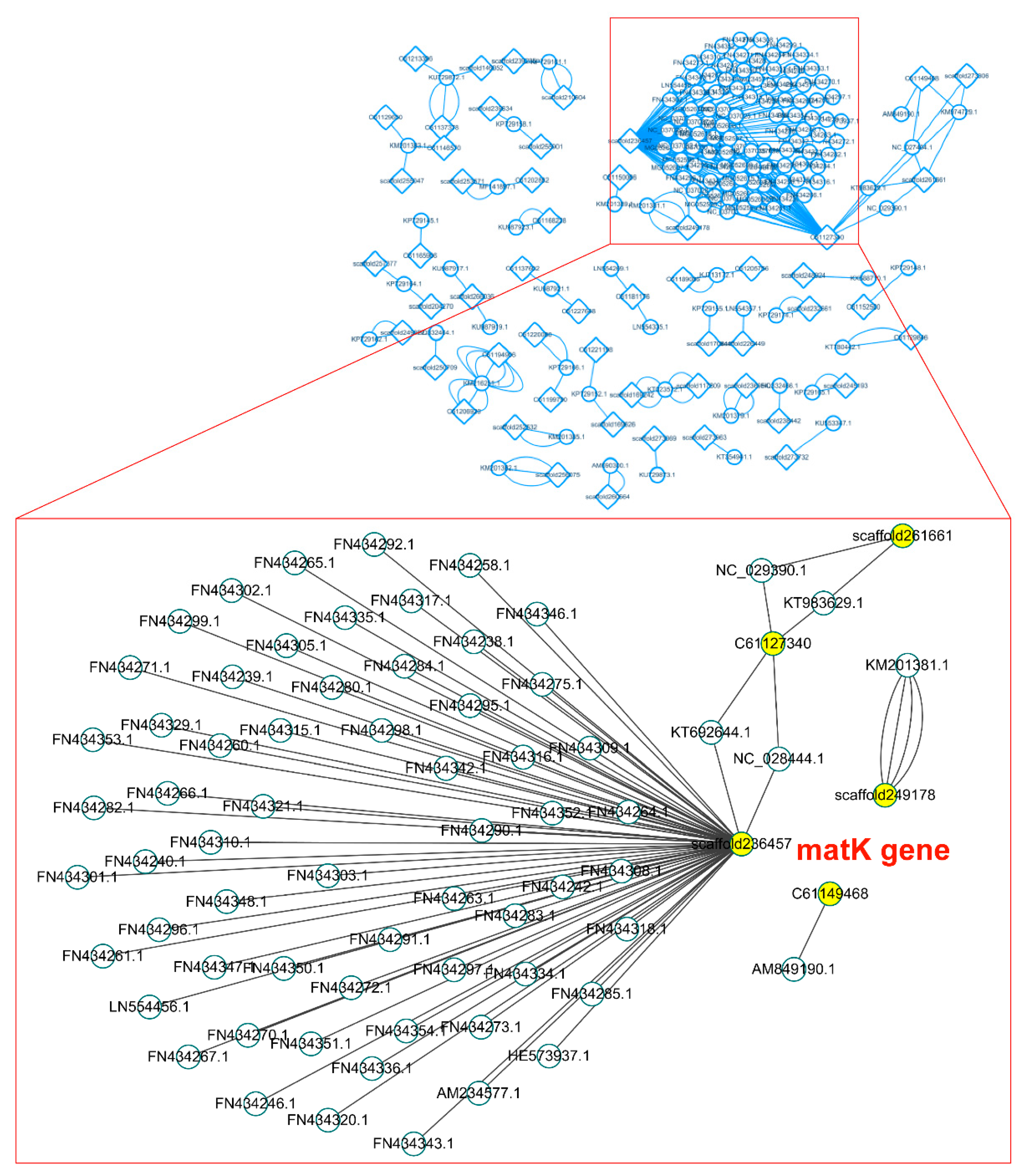
Second, with the 907 genes from common databases (Table S1), we annotated 70,770 scaffolds with a similarity greater than 90%. Scaffold No. 236,457 represented a well-known matK gene (Figure 3).
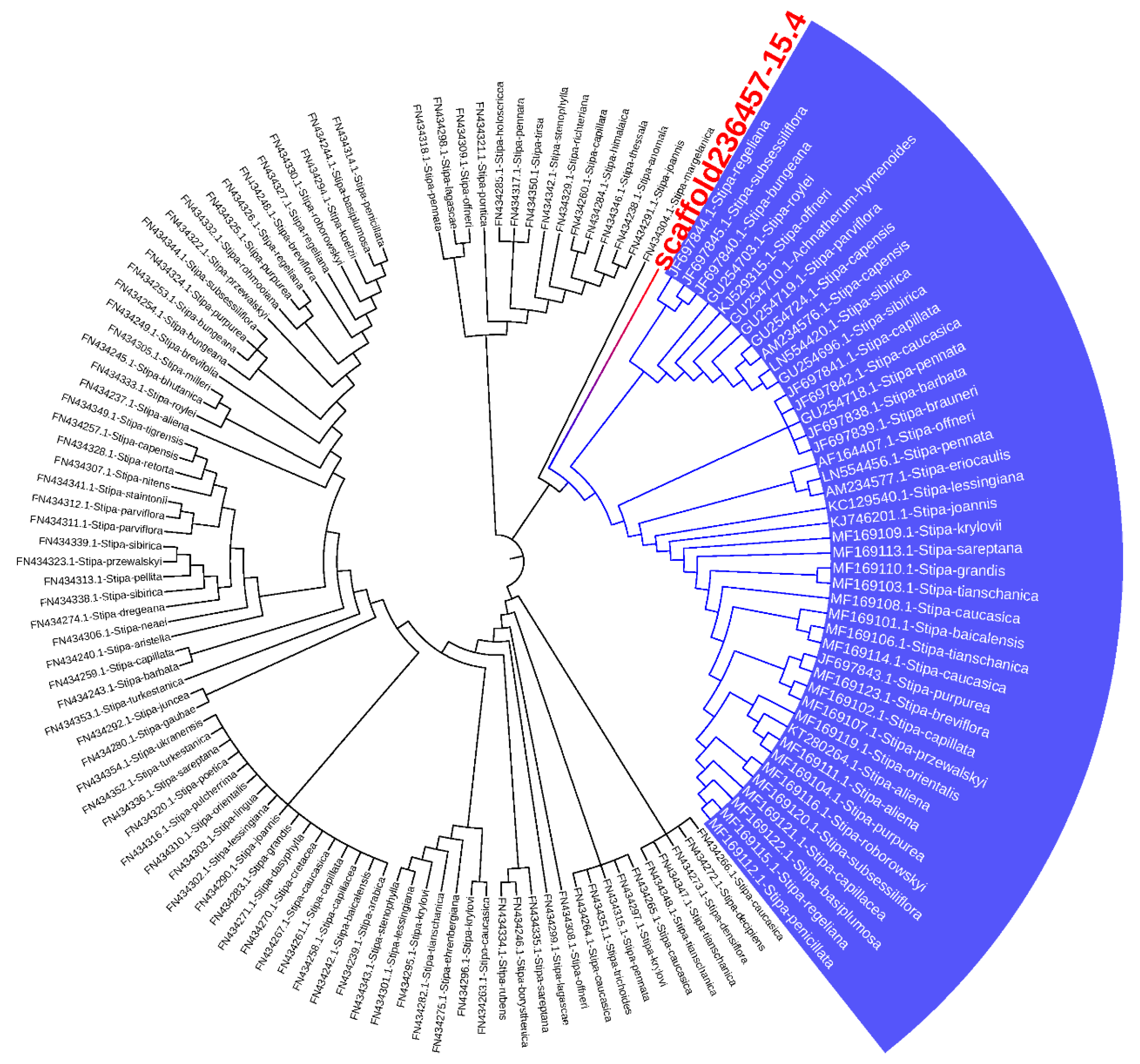
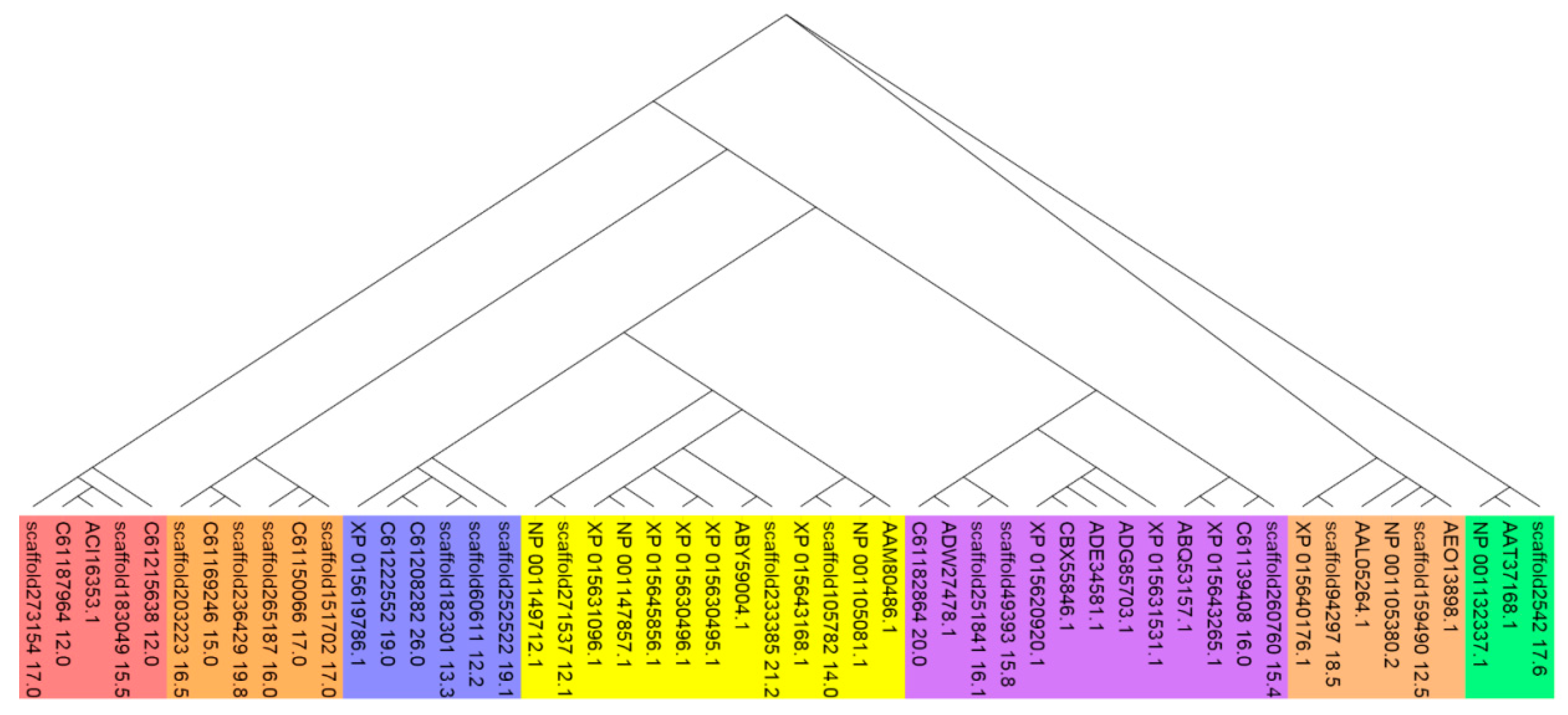
The MatK (MaturaseK) gene is a plant plastidial gene that plays a role in Group II intron splicing [32]. This chloroplast-encoded gene, which is highly conserved in plant systems, was used for the analysis of variants, transition/transversion rates, and molecular phylogeny [33]. The 134 different MatK gene sequences from NCBI and scaffold No. 236,457 were aligned using the MAFFT program. The maximum likelihood method was employed using the Fast Tree program for phylogenetic construction. Cluster analysis showed that two main clusters were separated from each other: One included S. regeliana, S. bugeana, S. subsessilliflora, S. roylei, S. offineri, S. sibirica, and S. capensis, while the second contained S. pennata, S. caucasica, and so on. However, S. breviflora did not belong to either of these two main clusters (Figure 4). Intriguingly, we found that Stipa margelanica was similar to S. breviflora in this respect.
With increasing global temperatures, water scarcity is becoming a major issue for many plants. As one of the most important vegetation types in the desert steppe, S. breviflora has excellent drought resistance. To study the drought tolerance of S. breviflora, we identified 269 reported drought tolerance genes in Gramineae.
For these genes, we annotated the 70,770 scaffolds. We found 34 annotated fragments of 26 scaffolds (Table S4). Then, cluster analysis was performed for the 26 reported genes and 26 scaffolds. UPGMA analysis showed that all 52 fragments were clearly separated from each other (Figure 5), which indicated that the reported genes and assembled ones may have similar functions in which we are interested.
3.3. Repetitive Sequences and Selective Sweep Analysis
Repeated sequences (also known as repetitive elements or repeats) are patterns of nucleic acids (DNA or RNA) that occur in multiple copies throughout the genome. In total, 18,234 repeats were detected in the assembled scaffolds. The percentage of repeats was approximately 25.77%, which was lower than those of Zea mays (64–73%) [34] and Solanum tuberosum (64.2%) [35] but similar to those of Oryza coarctata and Oryza officinalis (25–66%) [36]. The number of repeats was ordered using LINEs (78), DNA elements (29), LTR elements (26), and SINEs (7) (Table 4).
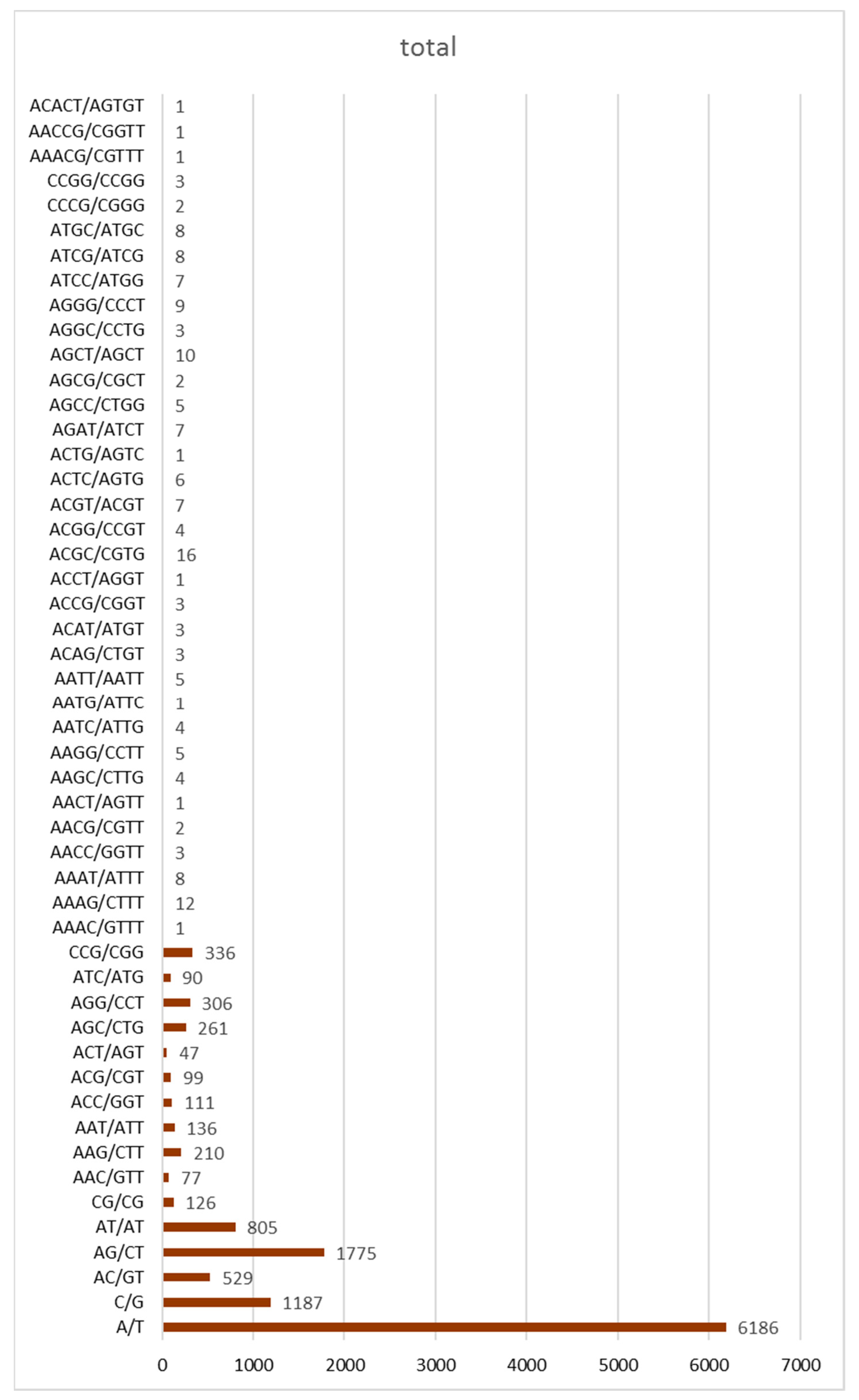
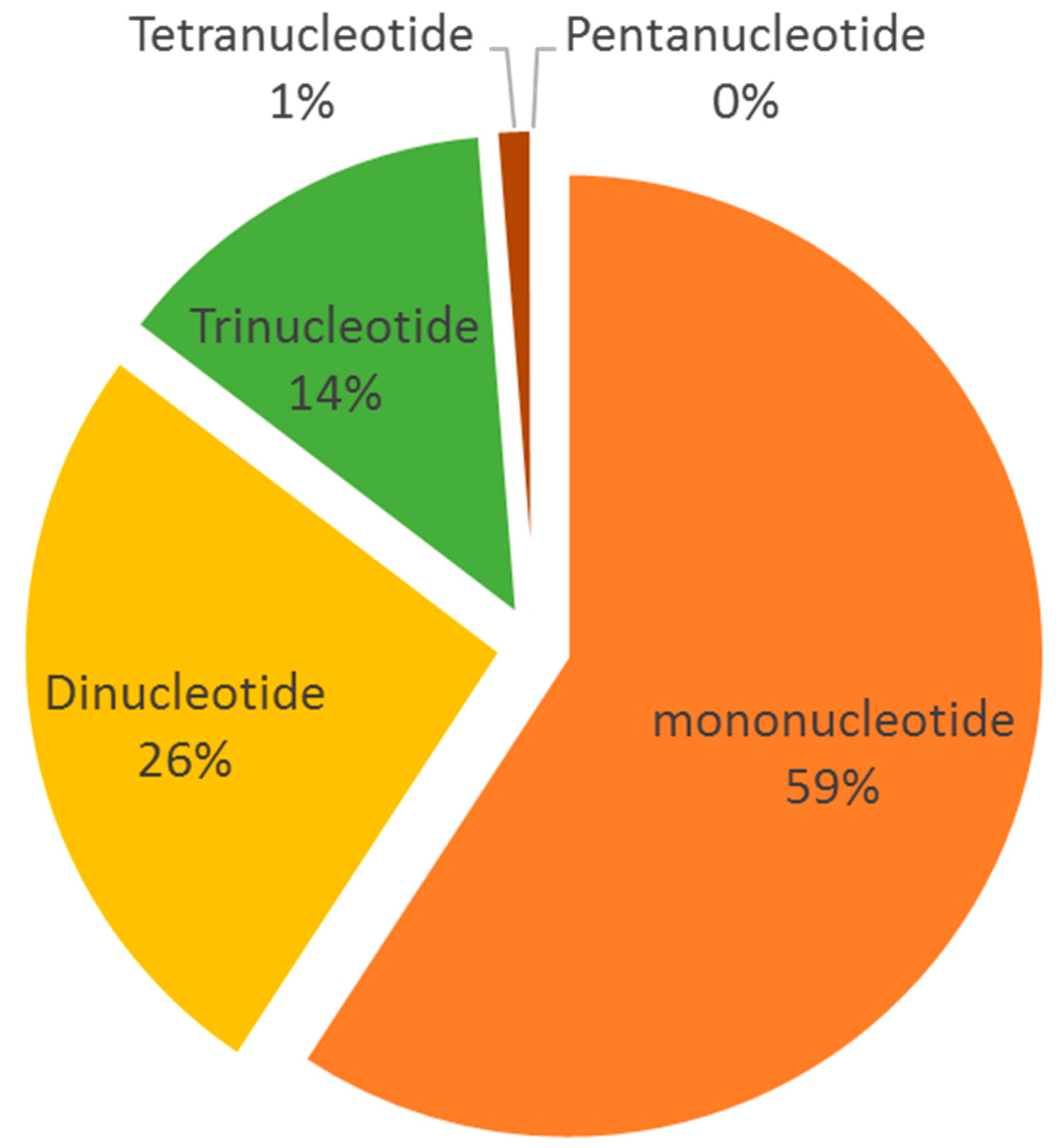
SSR markers are among the most highly polymorphic and reliable tools for genetic map construction and genetic diversity analysis [37]. In total, 12,438 different SSRs, including mono-, di-, tri-, tetra-, and pentanucleotides, were detected (Figure 6). The most abundant types of SSRs, aside from mononucleotides, were dinucleotides, followed by trinucleotides, tetranucleotides, and pentanucleotides. These SSRs can be used for primer design in the future to assess the diversity of S. breviflora, thus providing insights into environmental changes.
AG/CT (1175) and AT/AT (805) were the most numerous in repeats, followed by AC/GT (529), CCG/CGG (336), AGG/CCT (306), AGC/CTG (261), AAG/CTT (210), AAT/ATT (136), and CG/CG (126). The most repeated tetranucleotide motif was ACGC/CGTC (16). Therefore, there was about 1 SSR per 10 kb in the S. breviflora genome. This was in agreement with the results of the k-mer analysis (Figure 7).
Recently, the first draft of the genome of feather grass, Stipa capillata, was reported [38]. This genome features a single-molecule, long-read sequencing dataset, which is distinct from our paired-end sequencing approach. While the genome size and GC content align with our estimates, differences in mono-, di-, and trinucleotide repeats suggest variations in genomic structure between the two species. Additionally, we leveraged FastANI to compare our genome with the reported one, yielding an ANI output of 94.8576, just below the threshold of 95, indicating that S. breviflora and Stipa capillata are indeed distinct species. However, with high similarity, the subsequent utilization of this set of genes can allow perfect assembly of short-flowering needle grass.
Due to their high-density genetic stability, SNPs are most conveniently useful in large-scale and high-throughput genome studies. With the assembled S. breviflora genome sequence as a reference, we realigned all clean reads with the reference to identify heterozygous SNPs.
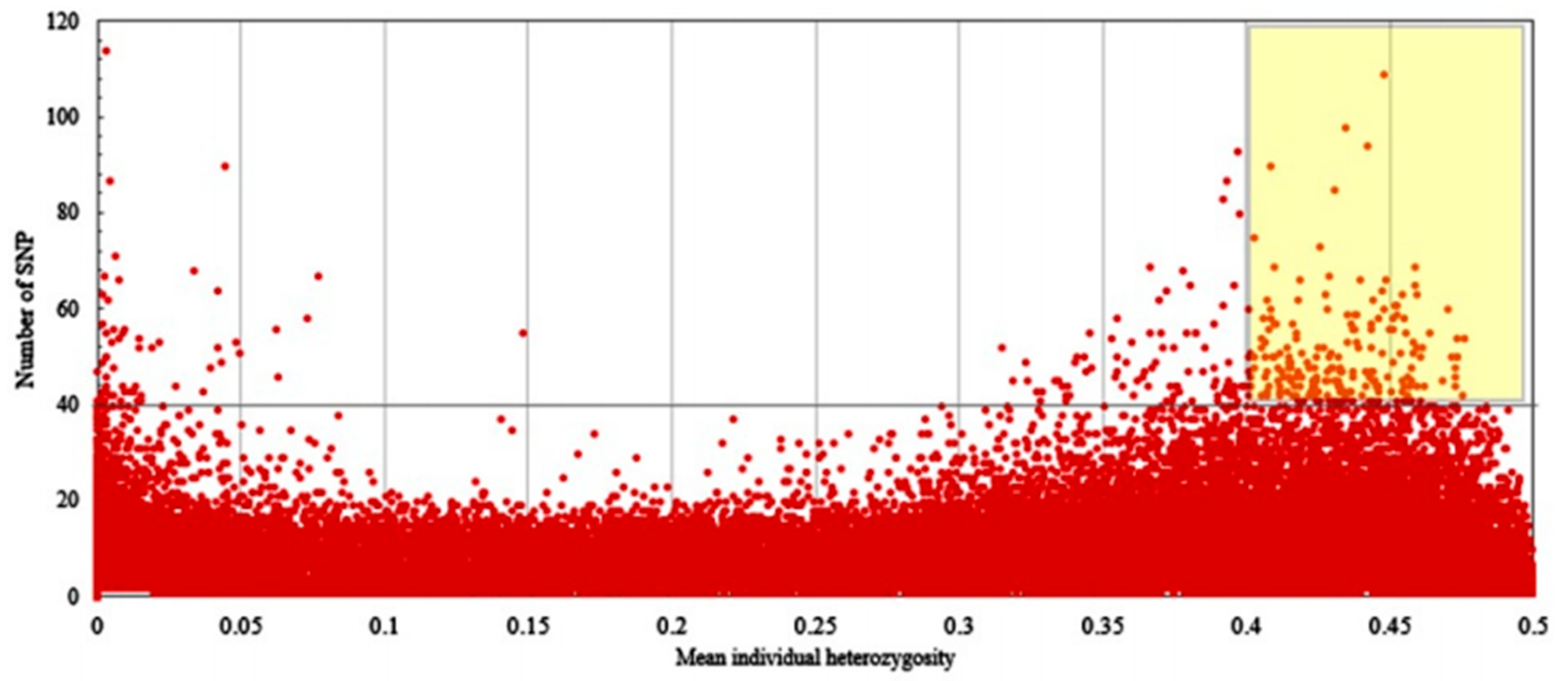
Based on the 100,341,373-locus genome assembly, we identified all positive SNPs and eliminated those with (a) hard filtration with the parameter ‘QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0′, (b) total depth ≥ 10, or (c) allele number = 2. A final set of 79,015 high-quality SNP loci fulfilling these criteria remained and represented regions of the genome that were heterozygous. Heterozygosity (He) refers to the likelihood that an individual at a polymorphic locus carries any two different alleles, i.e., the likelihood of being a heterozygote. The Hp method is an effective genomic selection approach that can be employed in plant breeding for rapid selection and improvement of specific genotypes. Due to genetic drift, it was hard to set a strict threshold that would distinguish true selective sweeps from the regions affected by drift. We were concerned that those regions where the SNP number was greater than 40 and Hp was greater than 0.4 were putative selective sweep areas. As a result, 204 putative sweeps remained (Figure 8). Further functional studies of these loci showed that they were involved in embryo development, ending in seed dormancy (GO:0009793, p = 0.0057) and embryo development (GO:0009790, p = 0.0067). Both were related to the root meristem specification. These results may be justified by the drought conditions of the steppe.
Currently, NGS technology and the k-mer method are being used by researchers to study non-model species. The findings gleaned from this investigation hold potential significance for prospective whole-genome sequencing initiatives involving S. breviflora. In this research, a genome survey of S. breviflora was carried out, and we estimated the genome size of S. breviflora to be about 1 Gb. Flow cytometry is also used to estimate genome size [39]. Thus, flow cytometry could be conducted to estimate the precise genome size of S. breviflora. We assembled these reads and obtained a total of 2,781,544 contigs. Furthermore, we obtained 2,600,873 scaffolds with a total length of 649,849,683 bp. The number of scaffolds (≥1 kb) was 70,770.
With those 70,770 scaffolds, we found that 18,234 scaffolds contained repeat sequences, which comprised 25.76% of the assembled genome. At the same time, the mono-(59%), di- (26%), and tri- (14%) nucleotide repeats comprised nearly 99% of the SSRs, and the sequence motifs AG/CT (1175), CCG/CGG (336), and ACGC/CGTC (16) were the most abundant among the dinucleotide, trinucleotide, and tetranucleotide repeat motifs, respectively. SSRs are among the most useful tools for molecular markers. However, no genome-wide SSR markers have been published. Zhao et al. screened seven primers from the homologous species Stipa purpurea [17]. Limited genomic information on S. breviflora impeded genetic studies. In this study, we shed light on the use of SSR markers. A total of 12,438 SSRs derived from the S. breviflora genome survey could guide the construction of high-density linkage maps. The generated dataset could also contribute to the understanding of the evolution of the S. breviflora genome.
S5 research is growing out of concern for climate change and global food safety. A significant exponential increase has been observed in the rate of transgenic or mutant plants tested for drought resistance in the last 20 years [40]. An assortment of genes in this study with diverse functions were shown to be induced or repressed by drought conditions. Yang et al. [41] classified drought resistance genes into the following three categories: (1) stress-responsive transcriptional regulation (e.g., DREB1, AREB, NF-YB); (2) post-transcriptional RNA or protein modifications, such as phosphorylation/dephosphorylation (e.g., SnRK2, ABI1) and farnesylation (e.g., ERA1); (3) osmoprotectant metabolism or molecular chaperones (e.g., CspB). We divided them, however, into functional genes and regulatory genes according to their different roles in plants. Functional genes can enhance osmotic accumulation, detoxification, antioxidant, and/or bio-molecule synthesis functions. Regulatory genes are transcription factors that can regulate downstream drought tolerance genes. These 26 genes are listed below in Table 5. Cluster analysis indicated their categorization into seven distinct clusters. However, upon closer examination of these genes, we discovered that they perform distinct roles in different species. This suggests that the mechanism of drought tolerance is intricate and multifaceted. The drought tolerance genes found in this study may improve our understanding of the molecular mechanisms of S. breviflora. Stipa breviflora uses various mechanisms to survive and prosper in a drought climate. To gain a better understanding of how these genes work, more research is needed.
4. Conclusions
In this study, we presented the first data on the structure of the S. breviflora genome, which will be useful for future research. We generated approximately 23.087 Gb of clean data to perform a comprehensive genome survey of S. breviflora. Currently, a second-generation sequencing approach is being employed to investigate the genome of S. breviflora, a species of foxtail grass. The objective is to gain initial insights into the research subject in order to pave the way for the subsequent utilization of advanced technologies such as third-generation sequencing for chromosome genome sequencing. This study will also yield reliable short DNA sequences for localized analysis. Through this effort, we aimed to deepen our understanding of its genomic architecture. The assembled data were employed to annotate the genome and conduct a selective sweep analysis. K-mer analysis revealed that the genome of S. breviflora was approximately 1 G in size. Notably, we observed a substantial presence of repetitive elements within the genome. The average GC content was approximately 44.16%. Following the assembly process, we identified the longest scaffold, which spanned 10,967 base pairs and corresponded to a DNA sequence from the chloroplast. Scaffold No. 236,457 represented a well-known matK gene, which, when analyzed, revealed distinct differences between S. breviflora and two clusters of Stipa plants. Additionally, in our research, we identified 26 scaffolds that might be responsible for the drought tolerance of S. breviflora. The SSR search yielded an impressive count of 12,438 different SSRs within the assembled genome. Furthermore, we compiled a set of 79,015 high-quality SNP loci, which were likely indicative of heterozygous regions within the genome. Our selective sweep analysis also pinpointed 204 putative sweeps. This newfound knowledge will accelerate further genetic studies with regard to S. breviflora and related genera.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/agriculture13122243/s1, Table S1: The 907 stipe genes from common databases. Table S2: The 134 different MatK gene sequences of chloroplasts from the GenBank database. Table S3: Twenty-five genes in the assembled genome of Stipa breviflora. Table S4: The 26 genes used to annotate the assembled genome of Stipa breviflora Griseb. Table S5: References for those drought-tolerant genes.
Author Contributions
Conceptualization, X.Y. and G.L.; methodology, J.W., S.L. and G.L.; software, B.X., S.L., L.Z., W.Z. and S.S.; validation, X.Y., J.W., S.L. and G.L.; formal analysis, S.L. and G.L.; investigation, X.Y., J.W., S.S. and G.L.; resources, Y.Z. and B.L.; data curation, X.Y., J.W., Y.Z. and B.L.; writing—original draft preparation, X.Y.; writing—review and editing, X.Y., J.W., B.X., S.L., L.Z., W.Z., S.S. and G.L.; visualization, J.W. and G.L.; supervision, G.L.; project administration, X.Y.; funding acquisition, X.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Science and Technology Innovation Project “Grassland Non-biological Disaster Prevention and Mitigation Team” of the Chinese Academy of Agricultural Sciences (Grant No. CAAS-ASTIP-IGR2015-04), the National Key R&D Program, the Intergovernmental International Cooperation on Science and Technology Innovation, and the Key Technologies for Ecological Restoration of Degraded Grassland between China and Mongolia (2016YFE0116400).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The authors certify that all of the original data used for this research were obtained from public databases. Other data used to support the findings of this study are included within the supplementary information files. All of the raw data from this study are available from the first author or corresponding author upon request.
Conflicts of Interest
No potential conflict of interest are reported by the authors.
References
- Zhang, Q.; Niu, J.M.; Ding, Y.; Kang, S.; Dong, J.J. Current Research Advances and Future Prospects of Biology and Ecology of Stipa breviflora. Chin. J. Grassl. 2010, 32, 93–101. [Google Scholar]
- Sai, S.B. Serious desertification of desert steppe zone and its control in northern Inner Mongolia. J. Arid Land Resour. Environ. 2001, 15, 34–37. [Google Scholar]
- Lu, M.; An, H.M.; Li, L.L. Genome Survey Sequencing for the Characterization of the Genetic Background of Rosa roxburghii Tratt and Leaf Ascorbate Metabolism Genes. PLoS ONE 2016, 11, e0147530. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Zhao, J.; Liu, M.J.; Liu, P.; Dai, L.; Zhao, Z.H. Genome-Wide Characterization of Simple Sequence Repeat (SSR) Loci in Chinese Jujube and Jujube SSR Primer Transferability. PLoS ONE 2015, 10, e0127812. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.K.; Sharma, R.; Cao, P.J.; Jenkins, J.; Bartley, L.E.; Qualls, M.; Grimwood, J.; Schmutz, J.; Rokhsar, D.; Ronald, P.C. A genome-wide survey of switchgrass genome structure and organization. PLoS ONE 2012, 7, e33892. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Hu, Y.Y.; Sui, Z.H.; Fu, F.; Wang, J.G.; Chang, L.P.; Guo, W.H.; Li, B.B. Genome survey sequencing and genetic background characterization of Gracilariopsis lemaneiformis (Rhodophyta) based on next-generation sequencing. PLoS ONE 2013, 8, e69909. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.W.; Li, R.Q.; Zhang, Z.H.; Li, L.; Gu, X.F.; Fan, W.; Lucas, W.J.; Wang, X.W.; Xie, B.Y.; Ni, P.X.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Jia, H.M.; Li, X.W.; Chai, M.L.; Jia, H.J.; Chen, Z.; Wang, G.Y.; Chai, C.Y.; Weg, E.W.; Gao, Z.S. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genom. 2012, 13, 201. [Google Scholar] [CrossRef]
- Zhang, S.; Wei, Z.J.; Lv, S.J.; Liu, H.M.; Wang, T.L.; Sun, S.X. Study on Aboveground Biomass and Vegetation Stability of Main Plant Populations and Community in Stipa breviflora Desert Steppe. Chin. J. Grassl. 2017, 39, 26–32. [Google Scholar]
- Liu, W.T.; Wei, Z.L.; Lv, S.J.; Sun, S.X.; Dai, J.Z.; Yan, B.L. Above-ground biomass in Stipa breviflora desert grassland at different organizational scales. Chin. J. Appl. Environ. Biol. 2015, 21, 912–918. [Google Scholar]
- Sun, S.X.; Wei, Z.J.; Wu, X.H.; Jiang, C.; Guo, L.B. Point pattern and spatial association of primary plant populations in the seasonal regulation of grazing intensity in desert grassland. Acta Ecol. Sin. 2016, 36, 7570–7579. [Google Scholar]
- Fan, R.Y.; Lv, S.J.; Ding, Y.; Li, Q.F. Interactive Effects of Soil Water, Nutrients and Clonal Fragmentation on Root Growth of Xerophilic Plant Stipa breviflora. Agriculture 2022, 12, 2112. [Google Scholar] [CrossRef]
- Wan, T.; Yan, L.; Li, H.; Yi, W.D.; Ma, B.; Liu, M.Y. The morphology of main plant species in the wood-lands of the Helan Mountains. J. Grassl. Forage Sci. 2004, 2, 28–31. [Google Scholar]
- Yan, R.R.; Wei, Z.J.; Yun, X.J.; Chu, W.B.; Xin, X.P. Effects of the grazing systems on diurnal variation of photosynthetic characteristic of major plant species of desert steppe. Acta Agrestia Sinca 2009, 18, 160–167. [Google Scholar]
- Hu, H.L.; Liu, Y.Z.; Li, Y.K.; Lu, D.X.; Gao, M. Use of the N-alkanes to Estimate Intake, Apparent Digestibility and Diet Composition in Sheep Grazing on Stipa breviflora Desert Steppe. J. Integr. Agric. 2014, 13, 1065–1072. [Google Scholar] [CrossRef]
- Yan, G.X.; Zhang, S.Z.; Yun, J.F.; Wang, L.Y.; Fu, X.Q. The geographical distribution and chromosome numbers of 52 forage species. Grassl. China 1991, 2, 53–58. [Google Scholar]
- Zhao, L.; Dang, Z.H.; Zhao, Y.N.; Zhang, Y.; Niu, J.M.; Ren, J.; Zhou, J.M. Optimization of SSR-PCR reaction system and primers screening in Stipa breviflora. J. Inn. Mong. Univ. (Nat. Sci. Ed.) 2016, 47, 58–64. [Google Scholar]
- Zhang, Q.; Niu, J.M.; Dong, J.J. Genetic diversity of Stipa breviflora populations in Inner Mongolia. Acta Ecol. Sin. 2008, 28, 3447–3455. [Google Scholar]
- Doyle, J.J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Yang, Z.L.; Deng, J.X.; Li, D.F.; Sun, T.T.; Xia, L.; Xu, W.W.; Zeng, L.H.; Jiang, H.S.; Yang, X.R. Analysis of Population Structure and Differentially Selected Regions in Guangxi Native Breeds by Restriction Site Associated with DNA Sequencing. G3 Genes Genomes Genet. 2020, 10, 379–386. [Google Scholar] [CrossRef]
- Liu, B.H.; Shi, Y.J.; Yuan, J.Y.; Hu, X.S.; Zhang, H.; Li, N.; Li, Z.Y.; Chen, Y.X.; Mu, D.S.; Fan, W. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome project. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, J.S.; Agarwala, R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics 2007, 23, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Rozewicki, J.; Li, S.L.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum-Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Bridges, S.; Magbanua, Z.V.; Peterson, D.G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008, 36, 2284–2294. [Google Scholar] [CrossRef]
- Kofler, R.; Schlötterer, C.; Lelley, T. SciRoKo: A new tool for whole genome microsatellite search and investigation. Bioinformatics 2007, 23, 1683–1685. [Google Scholar] [CrossRef]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.S.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef]
- Hirakawa, H.; Okada, Y.; Tabuchi, H.; Shirasawa, K.; Watanabe, A.; Tsuruoka, H.; Minami, C.; Nakayama, S.; Sasamoto, S.; Kohara, M. Survey of genome sequences in a wild sweet potato, Ipomoea trifida (H. B. K.) G. Don. DNA Res. 2015, 22, 171–179. [Google Scholar] [CrossRef]
- Shangguan, L.F.; Han, J.; Kayesh, E.; Sun, X.; Zhang, C.Q.; Pervaiz, T.; Wen, X.C.; Fang, J.G. Evaluation of genome sequencing quality in selected plant species using expressed sequence tags. PLoS ONE 2013, 8, e69890. [Google Scholar] [CrossRef]
- Aird, D.; Ross, M.G.; Chen, W.S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Kurz, F.T.; Aon, M.A.; O’Rourke, B.; Armoundas, A.A. Spatio-temporal oscillations of individual mitochondria in cardiac myocytes reveal modulation of synchronized mitochondrial clusters. Proc. Natl. Acad. Sci. USA 2010, 107, 14315–14320. [Google Scholar] [CrossRef] [PubMed]
- Ince, A.G.; Karaca, M.; Onus, A.; Bilgen, M. Chloroplast matK Gene Phylogeny of Some Important Species of Plants. Ziraat Fak. Derg. Akdeniz Univ. 2005, 18, 157–162. [Google Scholar]
- Meyers, B.C.; Tingey, S.V.; Morgante, M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001, 11, 1660–1676. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Pan, S.K.; Cheng, S.F.; Zhang, B.; Mu, D.S.; Ni, P.X.; Zhang, G.Y.; Yang, S.; Li, R.Q.; Wang, J. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [PubMed]
- Zuccolo, A.; Sebastian, A.; Talag, J.; Yu, Y.S.; Kim, H.R.; Collura, K.; Kudrna, D.; Wing, R.A. Transposable element distribution, abundance and role in genome size variation in the genus Oryza. BMC Evol. Biol. 2007, 7, 152. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genic microsatellite markers in plants: Features and applications. Trends Biotechnol. 2005, 23, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Baiakhmetov, E.; Guyomar, C.; Shelest, E.; Nobis, M.; Gudkova, P.D. The first draft genome of feather grasses using SMRT sequencing and its implications in molecular studies of Stipa. Sci. Rep. 2021, 11, 15345. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Blum, A. Genomics for drought resistance—Getting down to earth. Funct. Plant Biol. 2014, 41, 1191–1198. [Google Scholar] [CrossRef]
- Yang, S.; Vanderbeld, B.; Wan, J.; Huang, Y.F. Narrowing Down the Targets: Towards Successful Genetic Engineering of Drought-Tolerant Crops. Mol. Plant 2010, 3, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Székely, G.; Ábrahám, E.; Cséplő, Á.; Rigó, G.; Zsigmond, L.; Csiszár, J.; Ayaydin, F.; Strizhov, N.; Jásik, J.; Schmelzer, E. Duplicated P5CS genes of Arabidopsis play distinct roles in stress regulation and developmental control of proline biosynthesis. Plant J. 2007, 53, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Wang, F.; Si, Z.; Huo, J.X.; Xing, L.; An, Y.Y.; He, S.Z.; Liu, Q.C. A myo-inositol-1-phosphate synthase gene, IbMIPS1, enhances salt and drought tolerance and stem nematode resistance in transgenic sweet potato. Plant Biotechnol. J. 2015, 14, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Niu, X.L.; Yu, J.D.; Yan, J.; Gou, X.J.; Lu, B.R.; Liu, Y.S. Rice choline monooxygenase (OsCMO) protein functions in enhancing glycine betaine biosynthesis in transgenic tobacco but does not accumulate in rice (Oryza sativa L. ssp. japonica). Plant Cell Rep. 2012, 31, 1625–1635. [Google Scholar] [CrossRef]
- Umezawa, T.S.; Sugiyama, N.; Takahashi, F.; Anderson, J.C.; Ishihama, Y.; Peck, S.C.; Shinozaki, K. Genetics and phosphoproteomics reveal a protein phosphorylation network in the abscisic acid signaling pathway in Arabidopsis thaliana. Sci. Signal. 2013, 6, rs8. [Google Scholar] [CrossRef]
- Xiong, L.; Yang, Y. Disease resistance and abiotic stress tolerance in rice are inversely modulated by an abscisic acid-inducible mitogen-activated protein kinase. Plant Cell 2003, 15, 745–759. [Google Scholar] [CrossRef]
- Zhou, S.; Hu, W.; Deng, X.; Ma, Z.B.; Chen, L.H.; Huang, C.; Wang, C.; Wang, J.; He, Y.Z.; Yang, G.X. Overexpression of the wheat aquaporin gene, TaAQP7, enhances drought tolerance in transgenic tobacco. PLoS ONE 2012, 7, e52439. [Google Scholar] [CrossRef]
- Xiang, Y.; Huang, Y.; Xiong, L. Characterization of stress-responsive CIPK genes in rice for stress tolerance improvement. Plant Physiol. 2007, 144, 1416–1428. [Google Scholar] [CrossRef]
- Song, S.Y.; Chen, Y.; Chen, J.; Dai, X.Y.; Zhang, W.H. Physiological mechanisms underlying OsNAC5-dependent tolerance of rice plants to abiotic stress. Planta 2011, 234, 331–345. [Google Scholar] [CrossRef]
- Reis, P.A.B.; Fontes, E.P.B. N-rich protein (NRP)-mediated cell death signaling. Plant Signal. Behav. 2012, 7, 628–632. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, M.; Gao, S.; Zhang, Z.; Zhao, X.P.; Zhao, C.P.; Zhang, F.T.; Chen, X.P. Molecular characterization of novel TaNAC genes in wheat and overexpression of TaNAC2a confers drought tolerance in tobacco. Physiol. Plant. 2011, 144, 210–224. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ying, S.; Zhang, D.F.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. A maize stress-responsive NAC transcription factor, ZmSNAC1, confers enhanced tolerance to dehydration in transgenic Arabidopsis. Plant Cell Rep. 2012, 31, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Bai, X.; Sun, X.L.; Zhu, D.; Liu, B.H.; Ji, W.; Cai, H.; Cao, L.; Wu, J.; Hu, M.R.; et al. Expression of wild soybean WRKY20 in Arabidopsis enhances drought tolerance and regulates ABA signalling. J. Exp. Bot. 2013, 64, 2155–2169. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sakuma, Y.; Todaka, D.; Maruyama, K.; Qin, F.; Mizoi, J.; Kidokoro, S.; Fujita, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Functional analysis of an Arabidopsis heat-shock transcription factor HsfA3 in the transcriptional cascade downstream of the DREB2A stress-regulatory system. Biochem. Biophys. Res. Commun. 2008, 368, 515–521. [Google Scholar] [CrossRef]
- Rolla, A.A.D.; Carvalho, J.D.C.; Fuganti-Pagliarini, R.; Engels, C.; do Rio, A.; Marin, S.R.R.; de Oliveira, M.C.N.; Beneventi, M.A.; Marcelino-Guimaraes, F.C.; Farias, J.R.B. Phenotyping soybean plants transformed with rd29A:AtDREB1A for drought tolerance in the greenhouse and field. Transgen. Res. 2014, 23, 75–87. [Google Scholar] [CrossRef]
- Ying, S.; Zhang, D.F.; Fu, J.; Shi, Y.S.; Song, Y.C.; Wang, T.Y.; Li, Y. Cloning and characterization of a maize bZIP transcription factor, ZmbZIP72, confers drought and salt tolerance in transgenic Arabidopsis. Planta 2012, 235, 253–266. [Google Scholar] [CrossRef]
Figure 1.
Distribution curve of 17 k-mers of Stipa breviflora Griseb.

Figure 2.
C-Sibelia analysis of the assembly sequences and reference chloroplast DNA sequences of Stipa lipskyi and Stipa purpurea. The number of assembly sequences shows that there are 25 similar sequences among the three species.
Figure 2.
C-Sibelia analysis of the assembly sequences and reference chloroplast DNA sequences of Stipa lipskyi and Stipa purpurea. The number of assembly sequences shows that there are 25 similar sequences among the three species.

Figure 3.
The left shows the assembled scaffolds that are homologous to the 907 genes; the right is a magnified section of the left, which shows the Matk gene.
Figure 3.
The left shows the assembled scaffolds that are homologous to the 907 genes; the right is a magnified section of the left, which shows the Matk gene.

Figure 4.
Fast Tree analysis of 134MatK genes from the Stipa genus and scaffold No. 236,457.

Figure 5.
Cluster analysis of drought tolerance genes.

Figure 6.
Distribution of SSRs in the Stipa breviflora Griseb. genome. The X-axis represents the SSR motif type, while the Y-axis shows the number of corresponding motif types.
Figure 6.
Distribution of SSRs in the Stipa breviflora Griseb. genome. The X-axis represents the SSR motif type, while the Y-axis shows the number of corresponding motif types.

Figure 7.
Distribution of 12,438 SSRs in Stipa breviflora Griseb. based on repeats.

Figure 8.
Selective analysis of the Stipa breviflora genome. The box indicates the 204 regions identified.
Figure 8.
Selective analysis of the Stipa breviflora genome. The box indicates the 204 regions identified.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sequencing data and genome size estimation.
| Sample | ZM1 |
|---|---|
| Raw Reads, Number | 155,910,132 |
| Raw Bases, Number | 23,386,519,800 |
| Clean Reads, Number | 153,915,984 |
| Clean Reads, Rate (%) | 98.72 |
| Clean Reads, Number | 23,087,397,600 |
| Raw Q30 Bases, Rate (%) | 93.38 |
| Clean Q30 Bases, Rate (%) | 93.61 |
| Genome size estimated with GCE | 1.09 × 109 |
| Genome size estimated with SOAPec | 1.06 × 109 |
Table 2.
The 907 homologous genes that were sourced from 88 Stipa species within the NCBI database.
| Homologous Genes | Number |
|---|---|
| Chloroplast DNA | 521 |
| Mitochondrial DNA | 20 |
| Others | 366 |
| Total | 907 |
Table 3.
Summary of the sequencing and assembly of the Stipa breviflora Griseb. genome.
| Item | Contigs | Scaffolds |
|---|---|---|
| Total size (bp) | 636,497,947 | 649,849,683 |
| The longest (bp) | 6359 | 10,967 |
| Total number | 2,781,544 | 2,600,873 |
| GapContent_N | 0 | 7,619,172 |
| GC_Content | 44.16% | 44.16% |
Table 4.
Number of repeats in the Stipa breviflora Griseb. genome.
| Number of Elements | Length Occupied | Percentage of Sequence | ||
|---|---|---|---|---|
| SINES: | 7 | 371 bp | 0.00% | |
| ALUS | 1 | 53 bp | 0.00% | |
| MIRs | 4 | 218 bp | 0.00% | |
| LINEs: | 78 | 4767 bp | 0.00% | |
| LINE1 | 34 | 1993 bp | 0.00% | |
| LINE2 | 10 | 627 bp | 0.00% | |
| L3/CR1 | 14 | 864 bp | 0.00% | |
| LTR elements: | 26 | 2250 bp | 0.00% | |
| ERVL | 4 | 226 bp | 0.00% | |
| ERVL-MaLRs | 0 | 0 bp | 0.00% | |
| ERV_classI | 8 | 416 bp | 0.00% | |
| ERV_classII | 1 | 37 bp | 0.00% | |
| DNA elements: | 29 | 1851 bp | 0.00% | |
| hAT-Charlie | 3 | 147 bp | 0.00% | |
| TcMar-Tigger | 4 | 181 bp | 0.00% | |
| Unclassified: | 4 | 490 bp | 0.00% | |
| Total interspersed repeats: | 9729 bp | 0.01% | ||
| Small RNA: | 56 | 3402 bp | 0.00% | |
Table 5.
Twenty-six drought tolerance genes and their functions. The number indicates the homologous fragments of the assembled genome of Stipa breviflora.
Table 5.
Twenty-six drought tolerance genes and their functions. The number indicates the homologous fragments of the assembled genome of Stipa breviflora.
| Drought-Tolerant Genes | |||
|---|---|---|---|
| Categories | Genes | Number | Function |
| Functional genes | P5CS [42] | 2 | Causing proline accumulation to protect membrane protein |
| BADH [43] | 1 | Betaine synthesis and accumulation to maintain osmotic balance | |
| CMO1 [44] | 1 | ||
| MPK3 [45] | 1 | Detoxifying by eliminating ROSs | |
| OsMAPK5 [46] | 2 | Protein kinase to regulate downstream drought-tolerant genes | |
| TaCPK1 | 1 | ||
| AQP7 [47] | 2 | AQP synthesis to improve H2O transportation | |
| CIPK31 [48] | 2 | Reducing water loss | |
| Transcription factor | NAC5 [49] | 1 | NAC family responsible for proline accumulation, osmotic balance, and biological macromolecular synthesis |
| NAC6 [50] | 1 | ||
| OsNAC6 [51] | 2 | ||
| SNAC2 [52] | 1 | ||
| WRKY20 [53] | 1 | WRKY family responsible for reducing water loss and stomatal closure | |
| DREB1 [54,55] | 1 | AP2/ERF family TF responsible for proline accumulation and stomatal closure | |
| bZIP72 [56] | 1 | bZIP family TF responsible for proline and soluble sugar accumulations | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yun, X.; Wu, J.; Xu, B.; Lv, S.; Zhang, L.; Zhang, W.; Sun, S.; Liu, G.; Zu, Y.; Liu, B. Genome Survey of Stipa breviflora Griseb. Using Next-Generation Sequencing. Agriculture 2023, 13, 2243. https://doi.org/10.3390/agriculture13122243
AMA Style
Yun X, Wu J, Xu B, Lv S, Zhang L, Zhang W, Sun S, Liu G, Zu Y, Liu B. Genome Survey of Stipa breviflora Griseb. Using Next-Generation Sequencing. Agriculture. 2023; 13(12):2243. https://doi.org/10.3390/agriculture13122243
Chicago/Turabian StyleYun, Xiangjun, Jinrui Wu, Bo Xu, Shijie Lv, Le Zhang, Wenguang Zhang, Shixian Sun, Guixiang Liu, Yazhou Zu, and Bin Liu. 2023. "Genome Survey of Stipa breviflora Griseb. Using Next-Generation Sequencing" Agriculture 13, no. 12: 2243. https://doi.org/10.3390/agriculture13122243
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.