
Structure and Dynamics of an Archeal Monoglyceride Lipase from Palaeococcus ferrophilus as Revealed by Crystallography and In Silico Analysis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Methods
2.1.1. Expression and Purification of PFL
2.1.2. Crystallization, Data Collection, and Structural Characterization of PFL
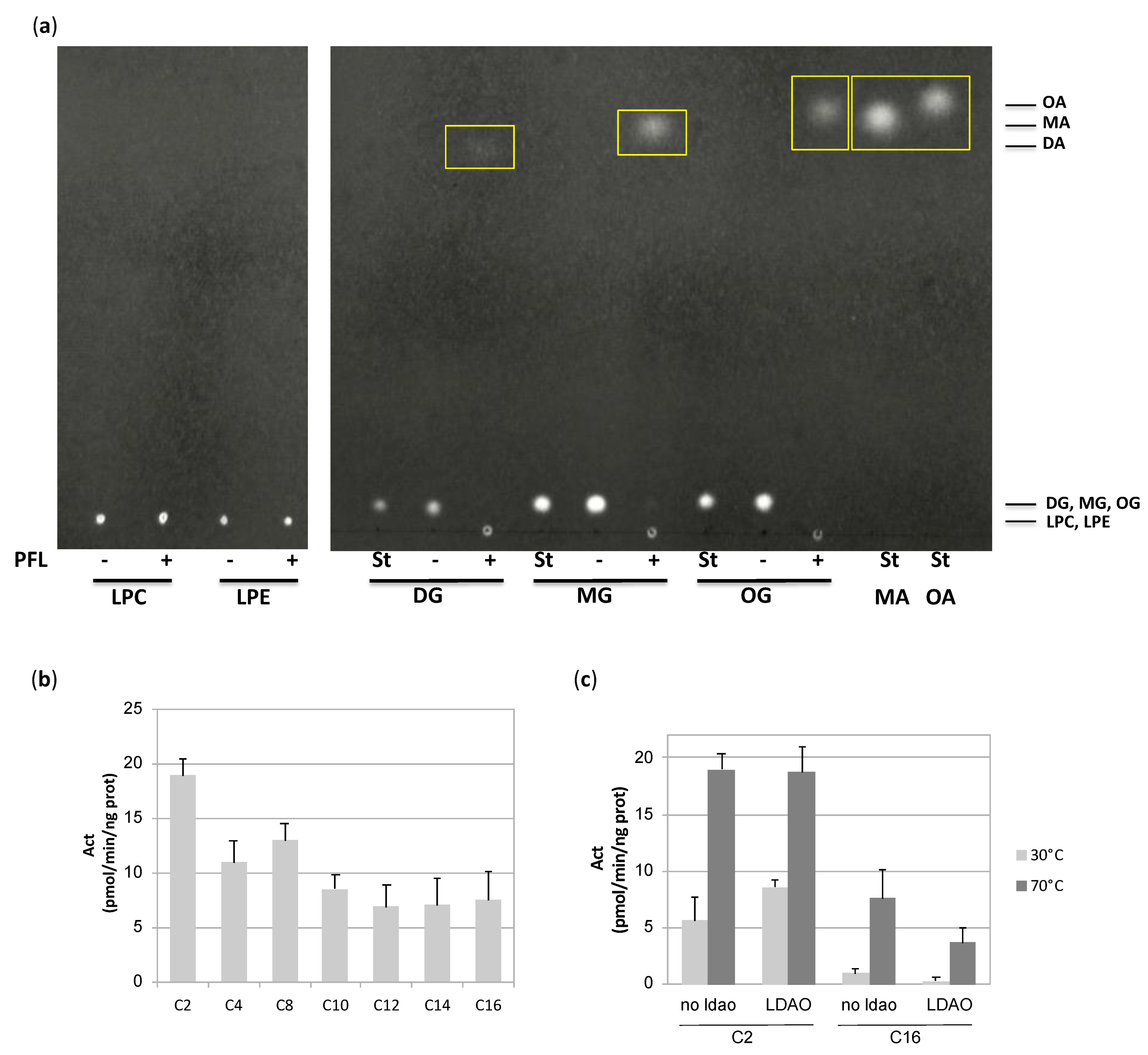
2.1.3. Thin Layer Chromatography Lipase Assay
2.1.4. pNitrophenyl Esters Hydrolase Assay
2.1.5. Thermofluor Assay
2.2. Computational Investigations
2.2.1. Elastic Network Models
2.2.2. Molecular Dynamics Simulations
3. Results
3.1. Experimental Results
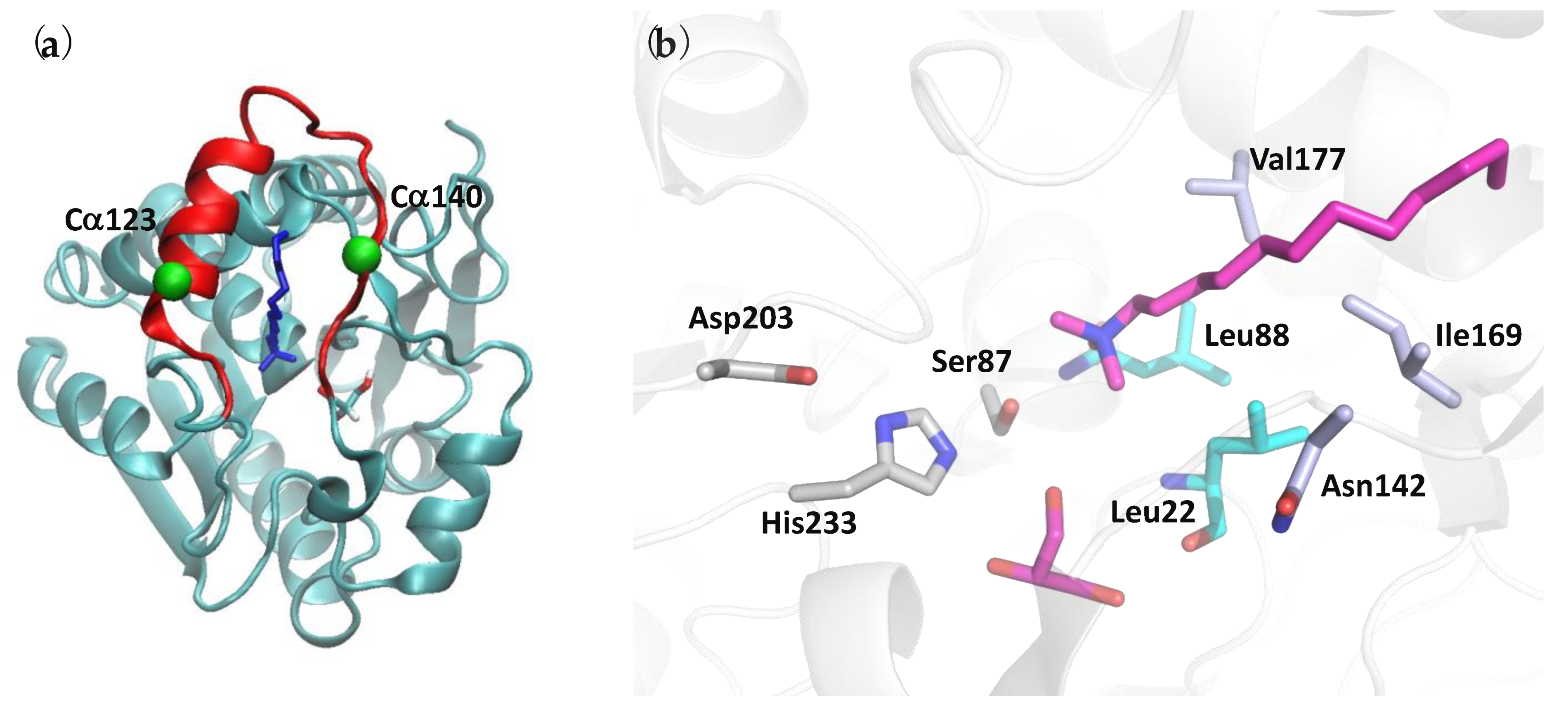
3.1.1. PFL Overall Structure
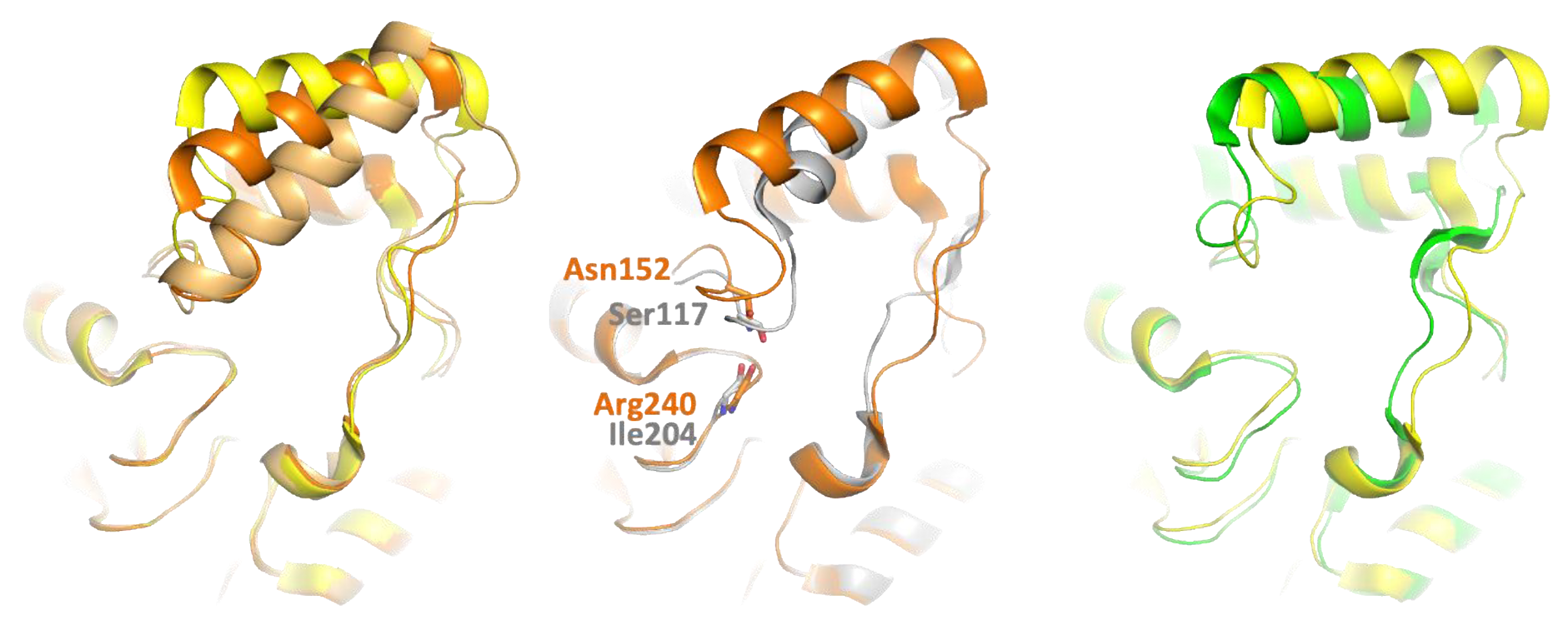
3.1.2. Homology with hMGL and mtMGL
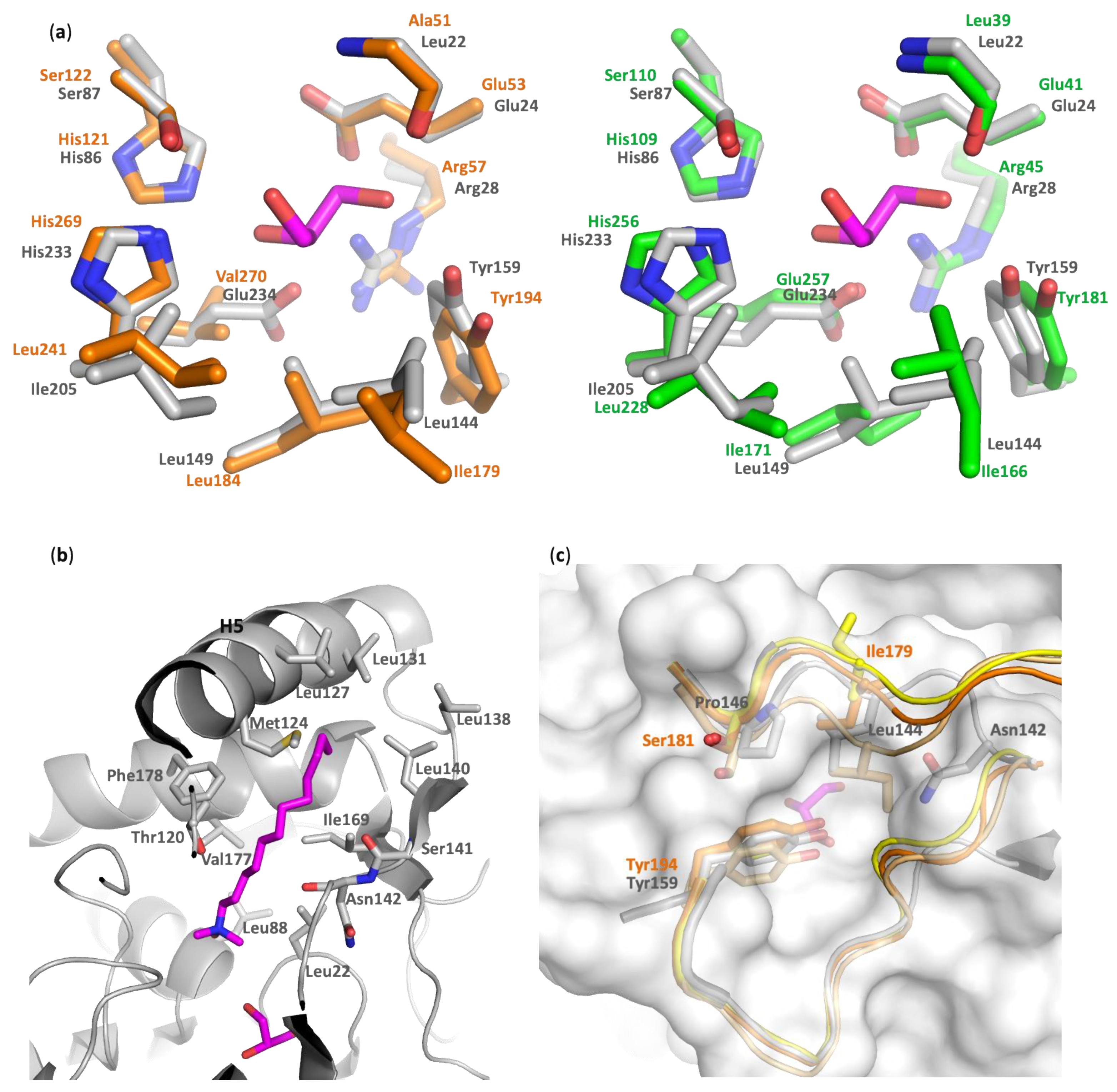
- Hinge and cap conformation. A DALI search retrieved hMGL as the first hit [7,25,26,34]. Other close structural homologs of PFL include mtMGL and Saccharomyces cerevisiae (scMGL) monoglyceride lipases [8,35]. Despite the relatively low sequence identity with these lipases (32%, 37%, and 23% identity with hMGL, mtMGL, and scMGL, respectively) the enzymes retain a striking conservation of structural elements, which are presumed to be of critical importance to support the activity (Figure S1).
- Alcohol-binding pocket. The catalytic site is also well conserved between PFL, hMGL, and mtMGL. In particular, there is a high identity in the alcohol-binding pocket. In the PFL crystal structure, this pocket is bordered by the side chain of Glu24, Arg28, His86, Glu234, Tyr159, Leu149, Ile205, and Leu144, in addition to the Leu22 main chain and the catalytic triad residues Ser87 and His233 (Figure 4a and Figure S1). These residues likely accommodate the substrates by establishing key interactions with its glycerol moiety, which is in agreement with the glycerol or MPD molecules systematically found in the crystal structures of PFL, hMGL, or mtMGL enzymes. These are extremely well conserved in hMGL and mtMGL, with the exception of PFL Glu234, which is involved in a salt bridge with Arg28, which is also present in mtMGL but is replaced by the hydrophobic Val280 in hMGL.
- Acyl-binding pocket site. In the absence of a crystal structure of PFL—or its homologs—in complex with a substrate or a substrate-derived ligand, it is difficult to precisely map the acyl-binding pocket. However, the presence, in the PFL active site, of a co-crystallized detergent molecule with a lauryl chain moiety (Figure S3) allows to delineate this pocket and to identify the residues likely involved in substrate recognition. As expected, the PFL pocket is mainly hydrophobic and bordered on the one side by Asn142, a Thr120 side chain, and a Ser141 main chain and, on the other side, by the side chain of residues Leu22, Leu88, Val177, Phe178, and Ile169 (Figure 4b). Then, the lipid chain projects toward a hydrophobic patch in the lid made of Met124, Leu127, Leu131, Leu138, and Leu140 side chains. Whilst the homology is lower than in the alcohol-binding pocket, several residues of the acyl-binding pocket are also conserved between PFL and hMGL or mtMGL (Figure S1).
3.1.3. PFL Substrate Preference
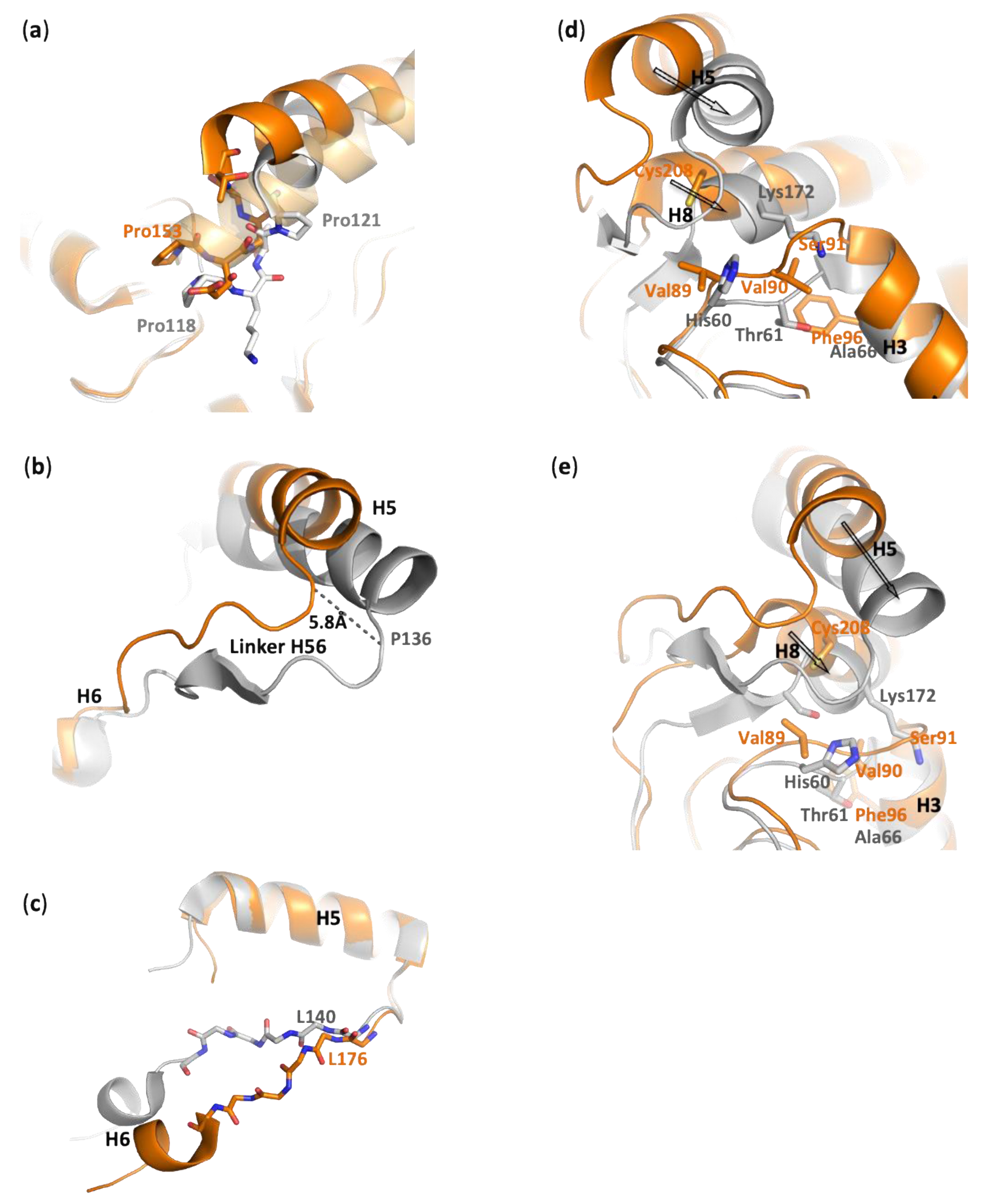
3.1.4. Lid Plasticity
3.2. In Silico Results
3.2.1. Deformability of the Monoglyceride Lipase Structure
3.2.2. Overall and Atomic Fluctuations
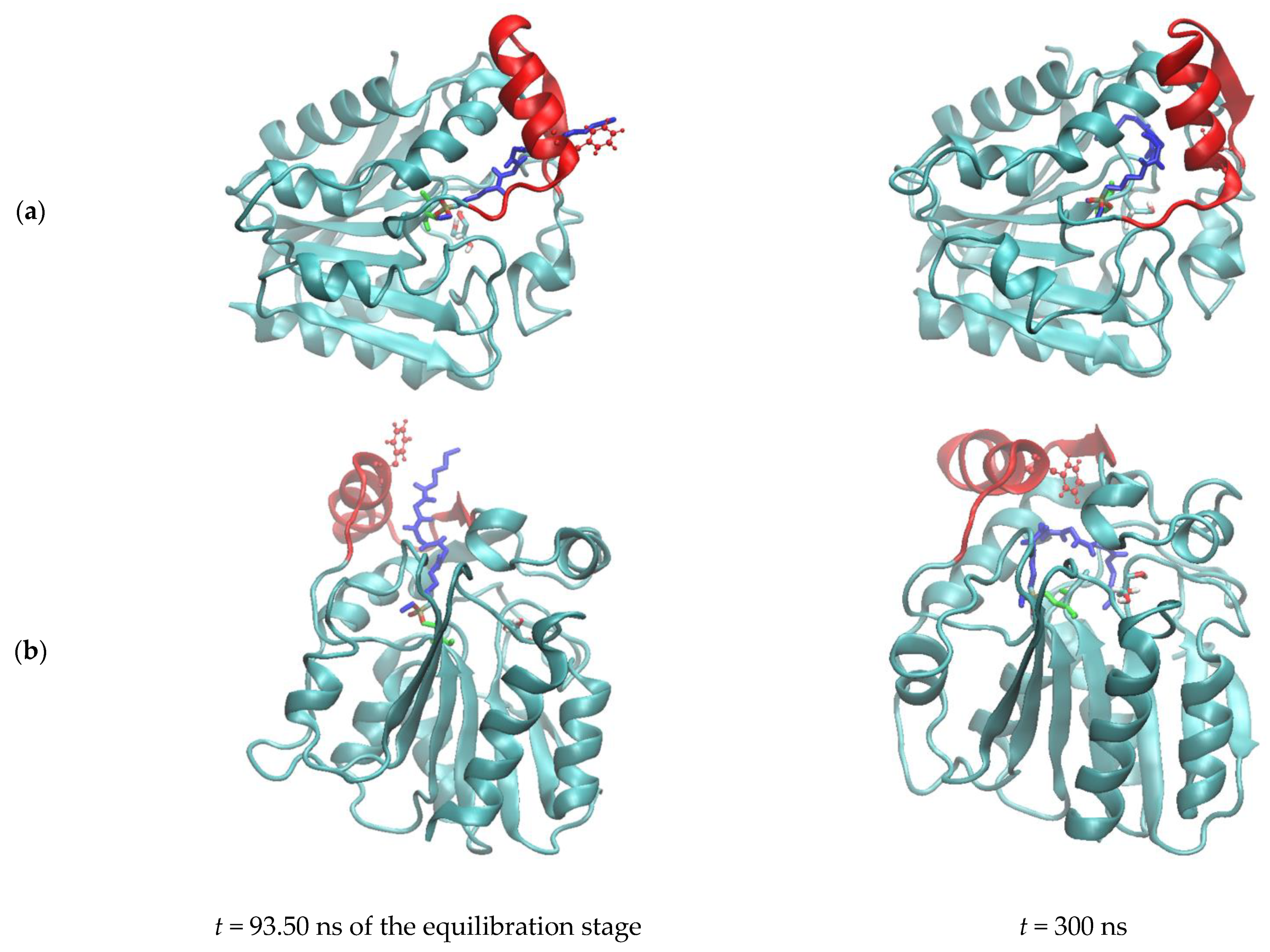
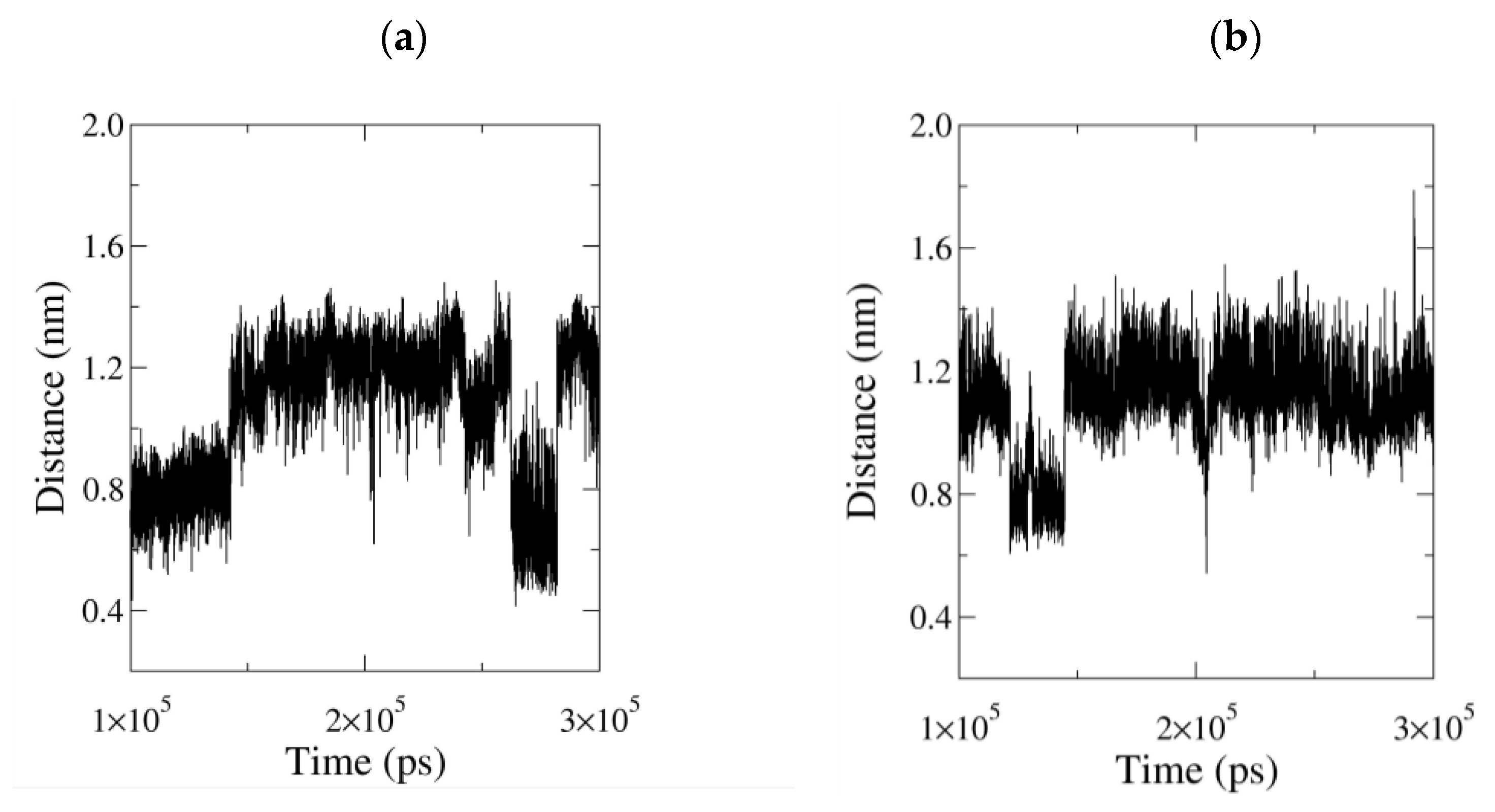
3.2.3. Influence of the Ligand Conformations on the Enzyme Opening
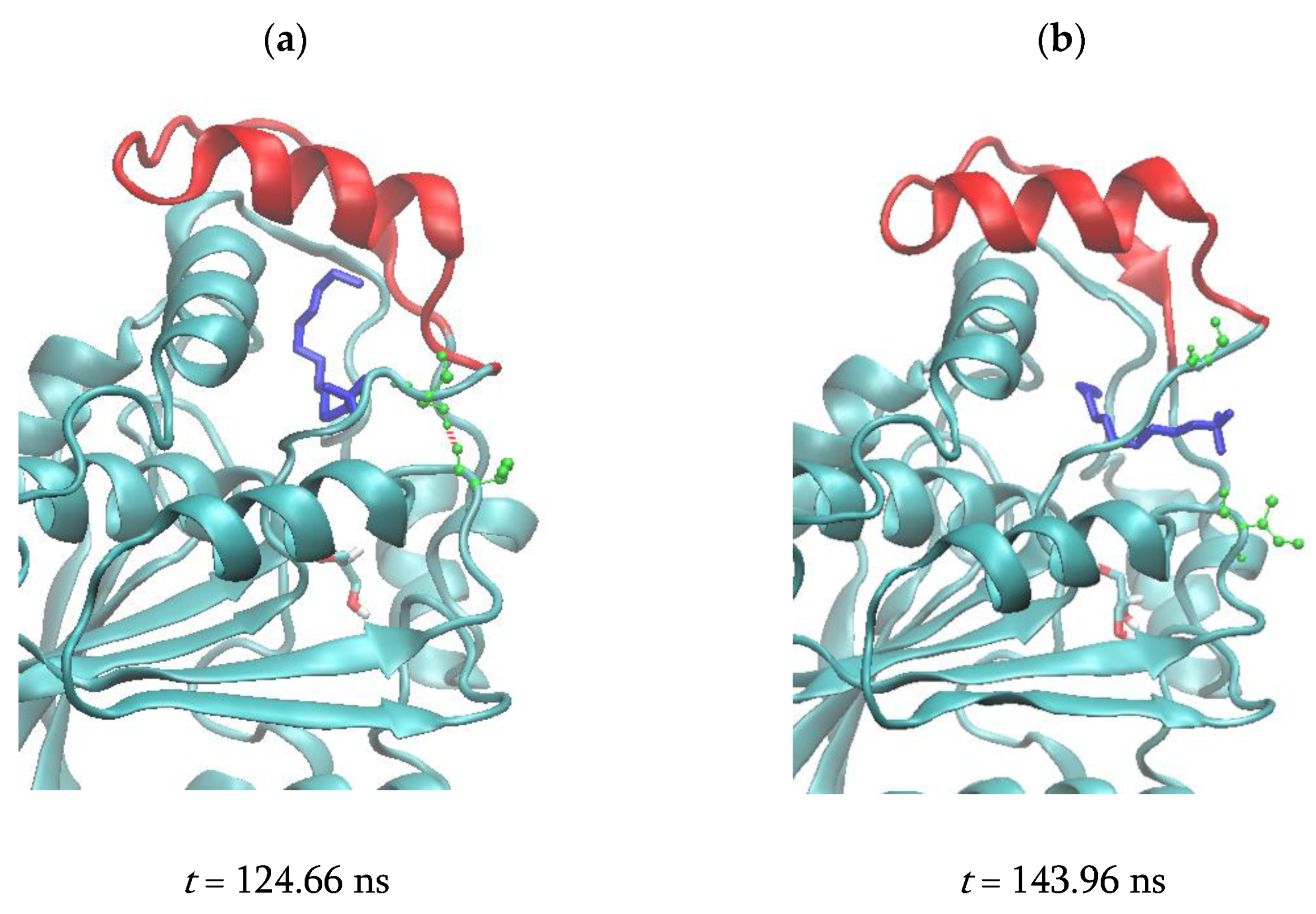
3.2.4. Correlated Lid Motions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Amino Acid |
| ATB | Automated Topology Builder |
| bMGL | Bacillus sp. H257 MGL |
| ENM | Elastic Network Model |
| FF | Force Field |
| hMGL | human monoglyceride lipase |
| LDAO | Lauryldimethylamine oxide |
| MAFP | methyl arachidonyl fluorophosphonate |
| MD | Molecular Dynamics |
| MGL | Monoglyceride lipase |
| mtMGL | Mycobacterium tuberculosis monoglyceride Lipase |
| PCA | Principal Component Analysis |
| PDB | Protein Data Bank |
| PFL | Palaeococcus ferrophilus monoglyceride Lipase |
| RMSD | Root Mean Square Displacement |
| RMSF | Root Mean Square Fluctuation |
| scMGL | Saccharomyces cerevisiae monoglyceride Lipase |
| SPC | Simple Point Charge |
| VMD | Visual Molecular Dynamics |
References
- Priyanka, P.; Tan, Y.; Kinsella, G.K.; Henehan, G.T.; Ryan, B.J. Solvent Stable Microbial Lipases: Current Understanding and Biotechnological Applications. Biotechnol. Lett. 2019, 41, 203–220. [Google Scholar] [CrossRef] [Green Version]
- Hasan, F.; Shah, A.; Hameed, A. Industrial Applications of Microbial Lipases. Enzym. Microb. Technol. 2006, 39, 235–251. [Google Scholar] [CrossRef]
- Borrelli, G.M.; Trono, D. Recombinant Lipases and Phospholipases and Their Use as Biocatalysts for Industrial Applications. Int. J. Mol. Sci. 2015, 16, 20774–20840. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.-M.; Han, S.-S.; Kim, H.-S. Industrial Applications of Enzyme Biocatalysis: Current Status and Future Aspects. Biotechnol. Adv. 2015, 33, 1443–1454. [Google Scholar] [CrossRef]
- Heater, B.S.; Lee, M.M.; Chan, M.K. Direct Production of a Genetically-Encoded Immobilized Biodiesel Catalyst. Sci. Rep. 2018, 8, 12783. [Google Scholar] [CrossRef]
- Grabner, G.F.; Zimmermann, R.; Schicho, R.; Taschler, U. Monoglyceride Lipase as a Drug Target: At the Crossroads of Arachidonic Acid Metabolism and Endocannabinoid Signaling. Pharmacol. Ther. 2017, 175, 35–46. [Google Scholar] [CrossRef]
- Labar, G.; Bauvois, C.; Borel, F.; Ferrer, J.-L.; Wouters, J.; Lambert, D.M. Crystal Structure of the Human Monoacylglycerol Lipase, a Key Actor in Endocannabinoid Signaling. Chembiochem Eur. J. Chem. Biol. 2010, 11, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, P.; Zimmermann, R.; Breinbauer, R.; Pavkov-Keller, T.; Oberer, M. The Crystal Structure of Monoacylglycerol Lipase from M. Tuberculosis Reveals the Basis for Specific Inhibition. Sci. Rep. 2018, 8, 8948. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The Lid Domain in Lipases: Structural and Functional Determinant of Enzymatic Properties. Front. Bioeng. Biotechnol. 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riegler-Berket, L.; Leitmeier, A.; Aschauer, P.; Dreveny, I.; Oberer, M. Identification of Lipases with Activity towards Monoacylglycerol by Criterion of Conserved Cap Architectures. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 679–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; et al. The BioCyc Collection of Microbial Genomes and Metabolic Pathways. Brief. Bioinform. 2019, 20, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.L.; Buchholz, P.C.F.; Pleiss, J. The Modular Structure of α/β-Hydrolases. FEBS J. 2020, 287, 1035–1053. [Google Scholar] [CrossRef]
- Trodler, P.; Schmid, R.D.; Pleiss, J. Modeling of Solvent-Dependent Conformational Transitions in Burkholderia Cepacia Lipase. BMC Struct. Biol. 2009, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehm, S.; Trodler, P.; Pleiss, J. Solvent-Induced Lid Opening in Lipases: A Molecular Dynamics Study. Protein Sci. Publ. Protein Soc. 2010, 19, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Tian, R.; Ni, Z.; Zhang, Z.; Chen, H.; Guo, Q.; Saier, M.H.; Vastermark, A. Conformational Transition Pathway in the Inhibitor Binding Process of Human Monoacylglycerol Lipase. Protein J. 2014, 33, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, L.; Arencibia, J.M.; Bono, L.; Armirotti, A.; Girotto, S.; De Vivo, M. Lid Domain Plasticity and Lipid Flexibility Modulate Enzyme Specificity in Human Monoacylglycerol Lipase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 441–451. [Google Scholar] [CrossRef]
- Ali, S.; Khan, F.I.; Chen, W.; Rahaman, A.; Wang, Y. Open and Closed States of Mrlip1 DAG Lipase Revealed by Molecular Dynamics Simulation. Mol. Simul. 2018, 44, 1520–1528. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular Structure Determination Using X-Rays, Neutrons and Electrons: Recent Developments in Phenix. Acta Crystallogr. Sect. Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.8; Schrödinger, LLC: New York, NY, USA, 2020. [Google Scholar]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural Summaries of PDB Entries. Protein Sci. Publ. Protein Soc. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Afonine, P.V.; Moriarty, N.W.; Mustyakimov, M.; Sobolev, O.V.; Terwilliger, T.C.; Turk, D.; Urzhumtsev, A.; Adams, P.D. FEM: Feature-Enhanced Map. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 646–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder Maps: Improving OMIT Maps by Excluding Bulk Solvent. Acta Crystallogr. Sect. Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, T.; Augé, F.; Houtmann, J.; Rak, A.; Vallée, F.; Mikol, V.; Berne, P.F.; Michot, N.; Cheuret, D.; Hoornaert, C.; et al. Structural Basis for Human Monoglyceride Lipase Inhibition. J. Mol. Biol. 2010, 396, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Schalk-Hihi, C.; Schubert, C.; Alexander, R.; Bayoumy, S.; Clemente, J.C.; Deckman, I.; DesJarlais, R.L.; Dzordzorme, K.C.; Flores, C.M.; Grasberger, B.; et al. Crystal Structure of a Soluble Form of Human Monoglyceride Lipase in Complex with an Inhibitor at 1.35 Å Resolution. Protein Sci. Publ. Protein Soc. 2011, 20, 670–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. IMODS: Internal Coordinates Normal Mode Analysis Server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Orellana, L.; Rueda, M.; Ferrer-Costa, C.; Lopez-Blanco, J.R.; Chacón, P.; Orozco, M. Approaching Elastic Network Models to Molecular Dynamics Flexibility. J. Chem. Theory Comput. 2010, 6, 2910–2923. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and Testing of the GROMOS Force-Field Versions 54A7 and 54B7. Eur. Biophys. J. EBJ 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14. [Google Scholar] [CrossRef]
- Rengachari, S.; Bezerra, G.A.; Riegler-Berket, L.; Gruber, C.C.; Sturm, C.; Taschler, U.; Boeszoermenyi, A.; Dreveny, I.; Zimmermann, R.; Gruber, K.; et al. The Structure of Monoacylglycerol Lipase from Bacillus Sp. H257 Reveals Unexpected Conservation of the Cap Architecture between Bacterial and Human Enzymes. Biochim. Biophys. Acta 2012, 1821, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. DALI and the Persistence of Protein Shape. Protein Sci. Publ. Protein Soc. 2020, 29, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Aschauer, P.; Rengachari, S.; Lichtenegger, J.; Schittmayer, M.; Das, K.M.P.; Mayer, N.; Breinbauer, R.; Birner-Gruenberger, R.; Gruber, C.C.; Zimmermann, R.; et al. Crystal Structure of the Saccharomyces Cerevisiae Monoglyceride Lipase Yju3p. Biochim. Biophys. Acta 2016, 1861, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rengachari, S.; Aschauer, P.; Schittmayer, M.; Mayer, N.; Gruber, K.; Breinbauer, R.; Birner-Gruenberger, R.; Dreveny, I.; Oberer, M. Conformational Plasticity and Ligand Binding of Bacterial Monoacylglycerol Lipase. J. Biol. Chem. 2013, 288, 31093–31104. [Google Scholar] [CrossRef] [Green Version]
- Griebel, G.; Pichat, P.; Beeské, S.; Leroy, T.; Redon, N.; Jacquet, A.; Françon, D.; Bert, L.; Even, L.; Lopez-Grancha, M.; et al. Selective Blockade of the Hydrolysis of the Endocannabinoid 2-Arachidonoylglycerol Impairs Learning and Memory Performance While Producing Antinociceptive Activity in Rodents. Sci. Rep. 2015, 5, 7642. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Roder, H.; Dickson, A.; Karanicolas, J. Isothermal Analysis of ThermoFluor Data Can Readily Provide Quantitative Binding Affinities. Sci. Rep. 2019, 9, 2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasr, M.L.; Shi, X.; Bowman, A.L.; Johnson, M.; Zvonok, N.; Janero, D.R.; Vemuri, V.K.; Wales, T.E.; Engen, J.R.; Makriyannis, A. Membrane Phospholipid Bilayer as a Determinant of Monoacylglycerol Lipase Kinetic Profile and Conformational Repertoire. Protein Sci. Publ. Protein Soc. 2013, 22, 774–787. [Google Scholar] [CrossRef]
- Varejão, N.; De-Andrade, R.A.; Almeida, R.V.; Anobom, C.D.; Foguel, D.; Reverter, D. Structural Mechanism for the Temperature-Dependent Activation of the Hyperthermophilic Pf2001 Esterase. Struct. Lond. Engl. 1993 2018, 26, 199–208.e3. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.A.M.; Loccisano, A.E.; Firestine, S.M.; Evanseck, J.D. Chapter 13 Principal Components Analysis: A Review of its Application on Molecular Dynamics Data. In Annual Reports in Computational Chemistry; Spellmeyer, D.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 233–261. ISBN 1574-1400. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glycerol | Ligand | No. of SPC Water Molecules | Final Box Size (nm) | |
|---|---|---|---|---|
| PFL | - | - | 14,383 | 7.83904 |
| PFL-G | X | - | 14,472 | 7.84390 |
| PFL-GL | X | LDAO | 14,372 | 7.83805 |
| PFL-GM | X | MAFP | 14,379 | 7.83855 |
| PFL-70 | - | - | 14,383 | 7.93524 |
| / | MAFP | |
|---|---|---|
| / | n.d. | 89.9 ± 2.1 |
| LDAO 0.1% | 76.4 ± 1.8 | 83.5 ± 1.3 |
| Cα123–Cα140 Distance (nm) | Cα124–Cα142 Distance (nm) | |
|---|---|---|
| PFL | 1.061 ± 0.071 | 0.722 ± 0.063 |
| PFL-G | 0.764 ± 0.120 | 0.779 ± 0.112 |
| PFL-GL | 1.014 ± 0.061 | 1.048 ± 0.144 |
| PFL-GM | 0.922 ± 0.076 | 1.006 ± 0.061 |
| PFL-70 | 1.240 ± 0.117 | 1.147 ± 0.126 |
| Crystal structure | 1.493 | 1.076 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labar, G.; Brandt, N.; Flaba, A.; Wouters, J.; Leherte, L. Structure and Dynamics of an Archeal Monoglyceride Lipase from Palaeococcus ferrophilus as Revealed by Crystallography and In Silico Analysis. Biomolecules 2021, 11, 533. https://doi.org/10.3390/biom11040533
Labar G, Brandt N, Flaba A, Wouters J, Leherte L. Structure and Dynamics of an Archeal Monoglyceride Lipase from Palaeococcus ferrophilus as Revealed by Crystallography and In Silico Analysis. Biomolecules. 2021; 11(4):533. https://doi.org/10.3390/biom11040533
Chicago/Turabian StyleLabar, Geoffray, Nathalie Brandt, Amaury Flaba, Johan Wouters, and Laurence Leherte. 2021. "Structure and Dynamics of an Archeal Monoglyceride Lipase from Palaeococcus ferrophilus as Revealed by Crystallography and In Silico Analysis" Biomolecules 11, no. 4: 533. https://doi.org/10.3390/biom11040533