
A Multigene Phylogeny of Native American Hawkweeds (Hieracium Subgen. Chionoracium, Cichorieae, Asteraceae): Origin, Speciation Patterns, and Migration Routes
 ,
,
Abstract
:1. Introduction
2. Results
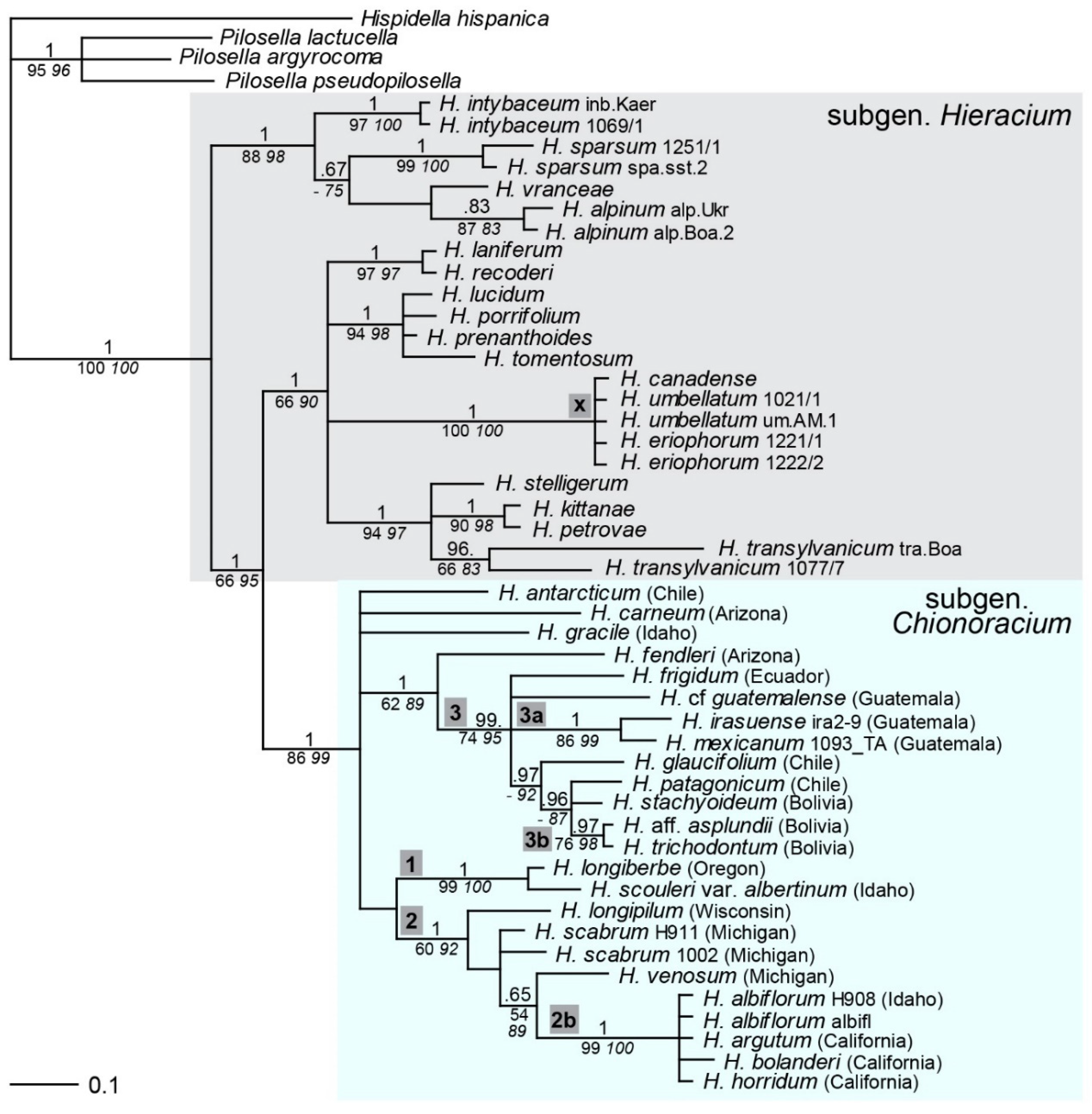
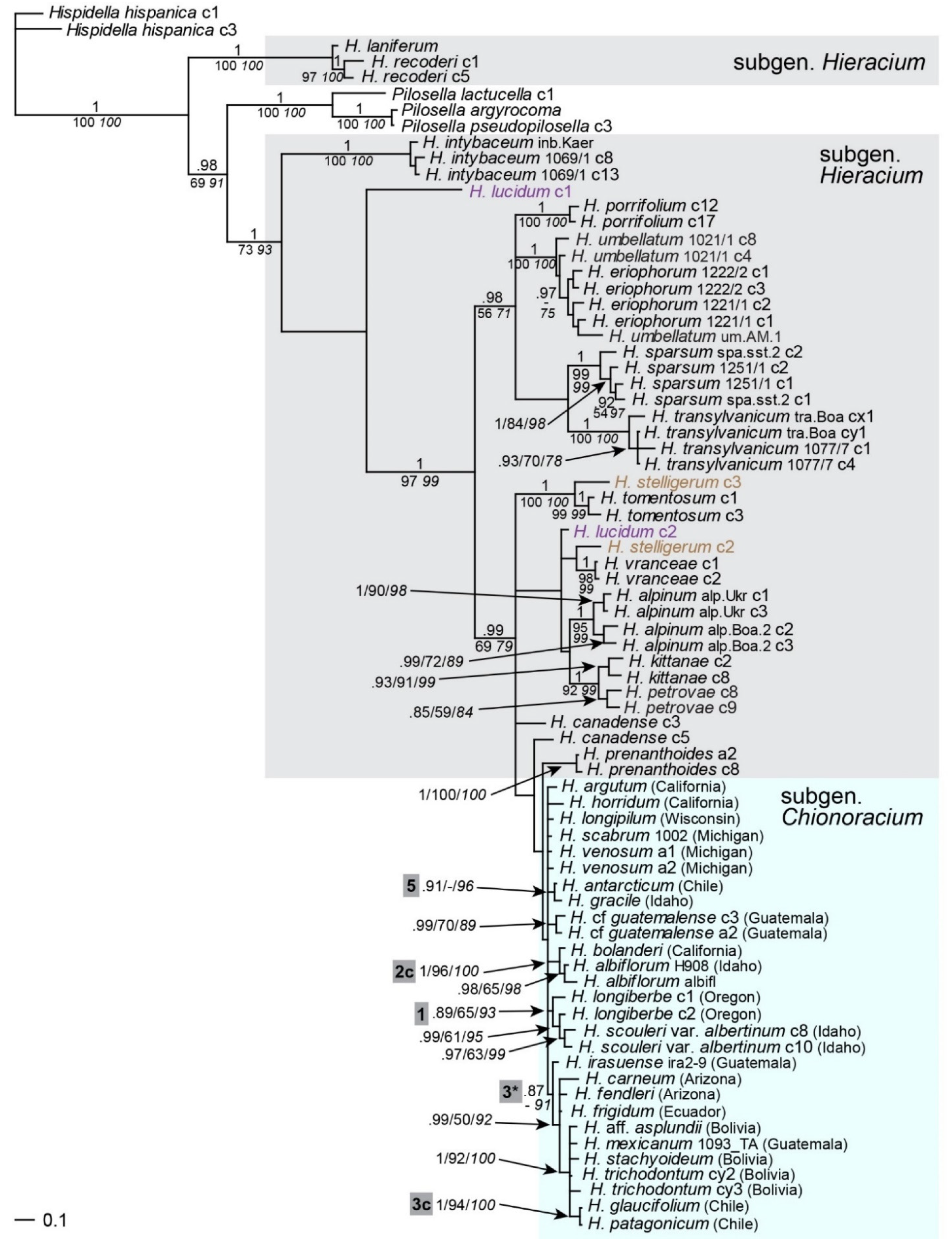
2.1. Phylogenetic Inference Based on ptDNA
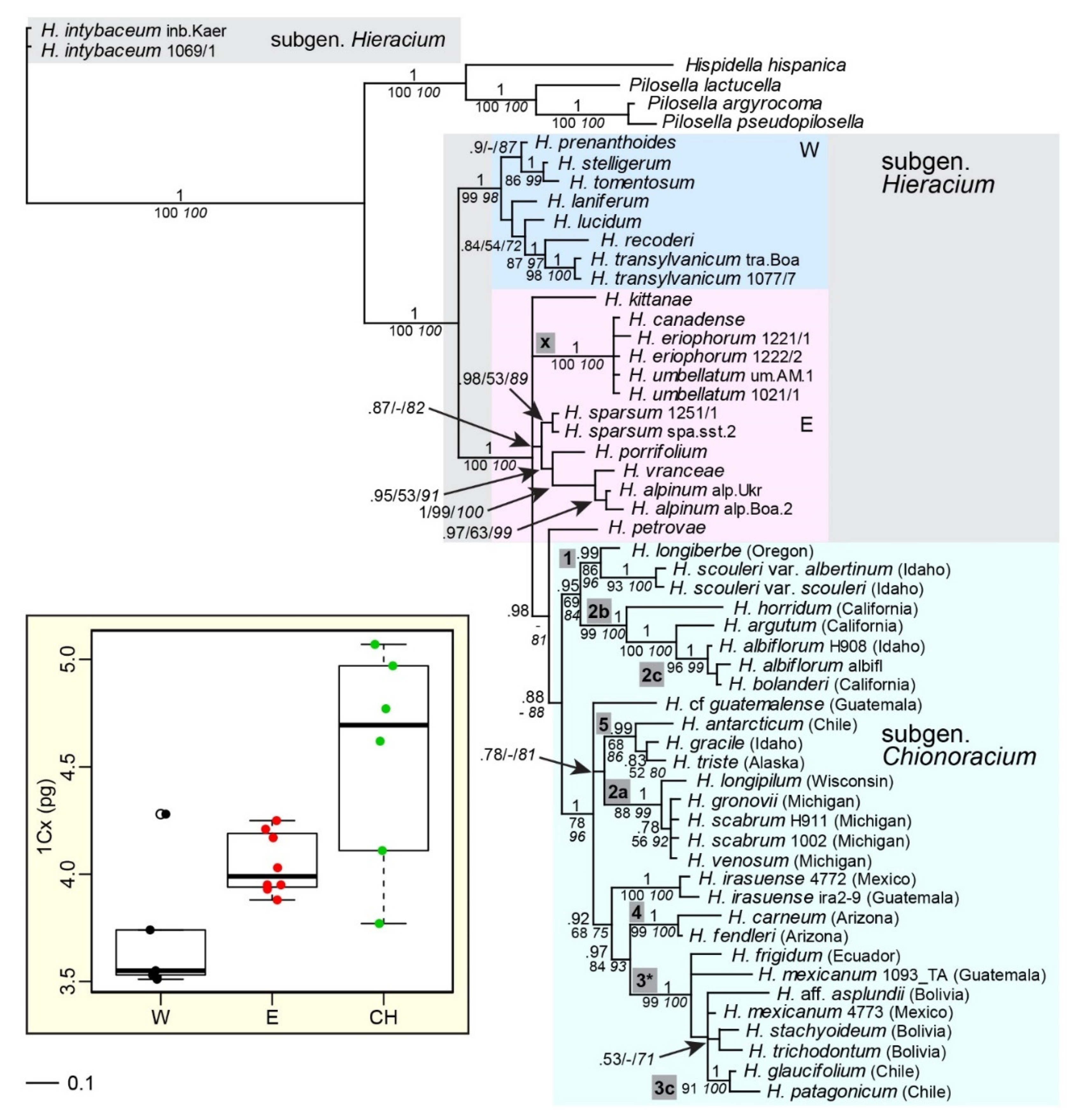
2.2. Phylogenetic Inference Based on Multi-Copy nrDNA (ITS, ETS)
2.3. Phylogenetic Inference Based on Multi-Copy nrDNA (5S-NTS)
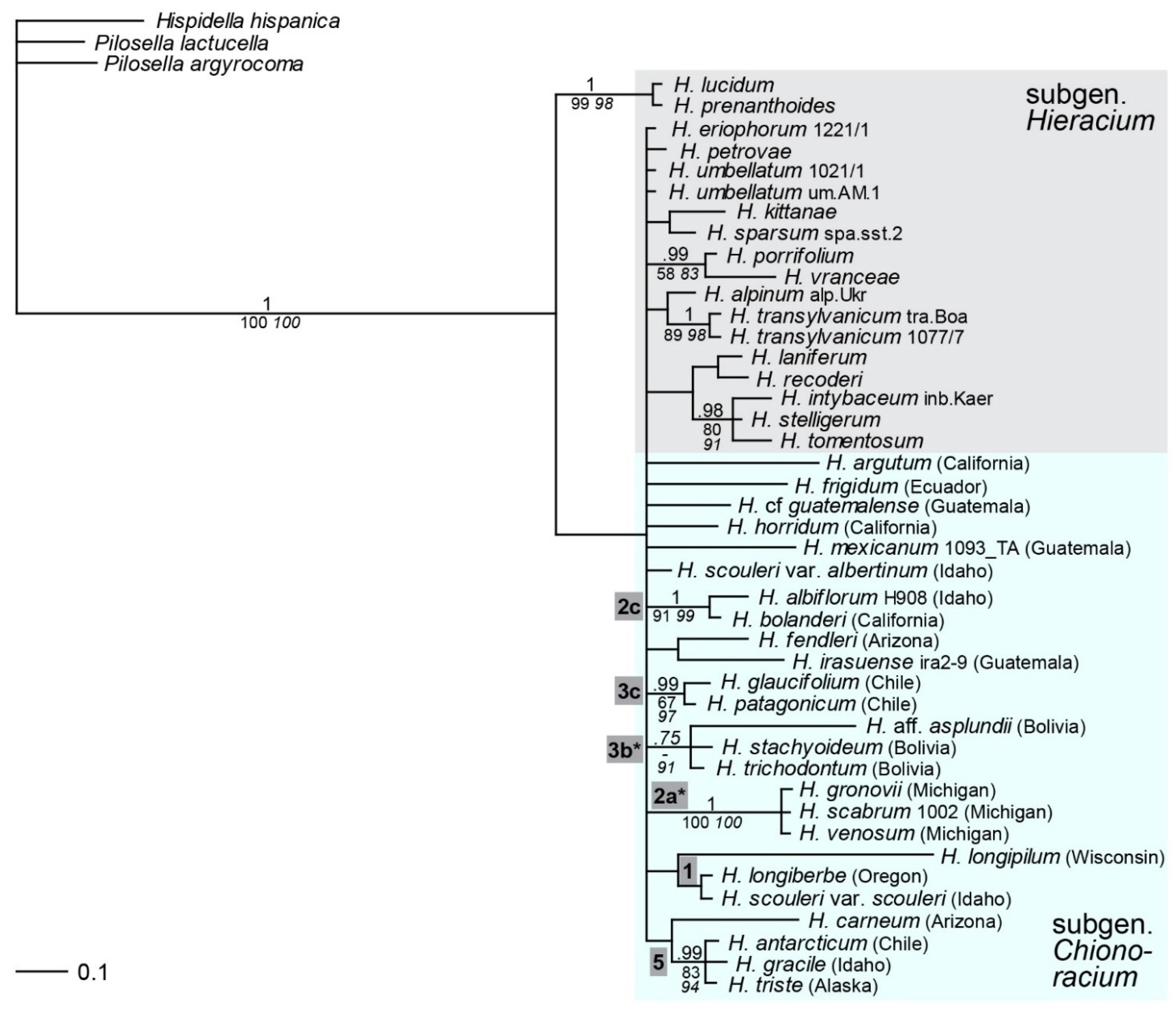
2.4. Phylogenetic Inference Based on the Low-Copy Nuclear Marker Gsh1
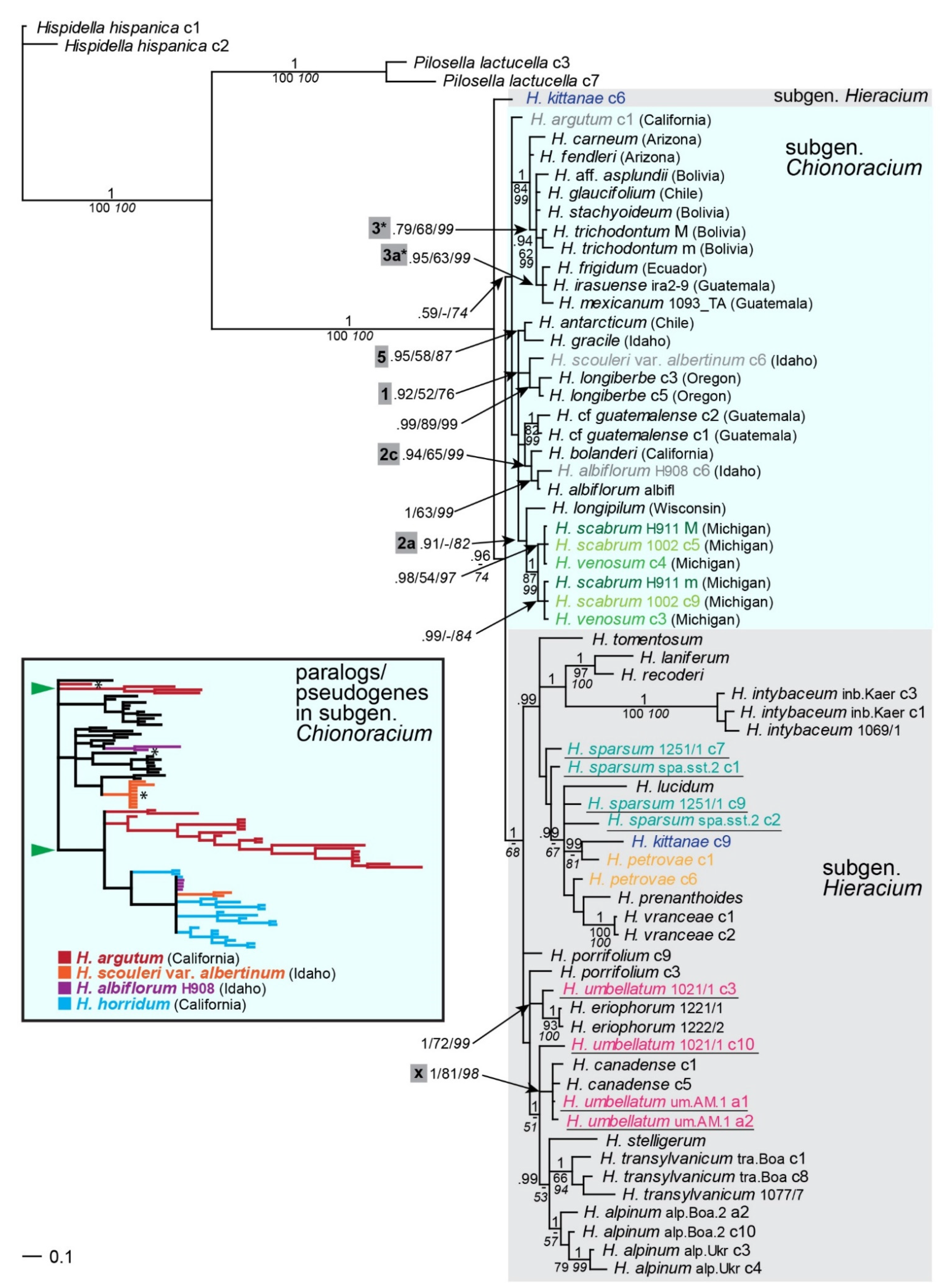
2.5. Paralogs and Pseudogenes of the Low-Copy Nuclear Marker Gsh1
2.6. Phylogenetic Inference Based on the Low-Copy Nuclear Marker Sqs
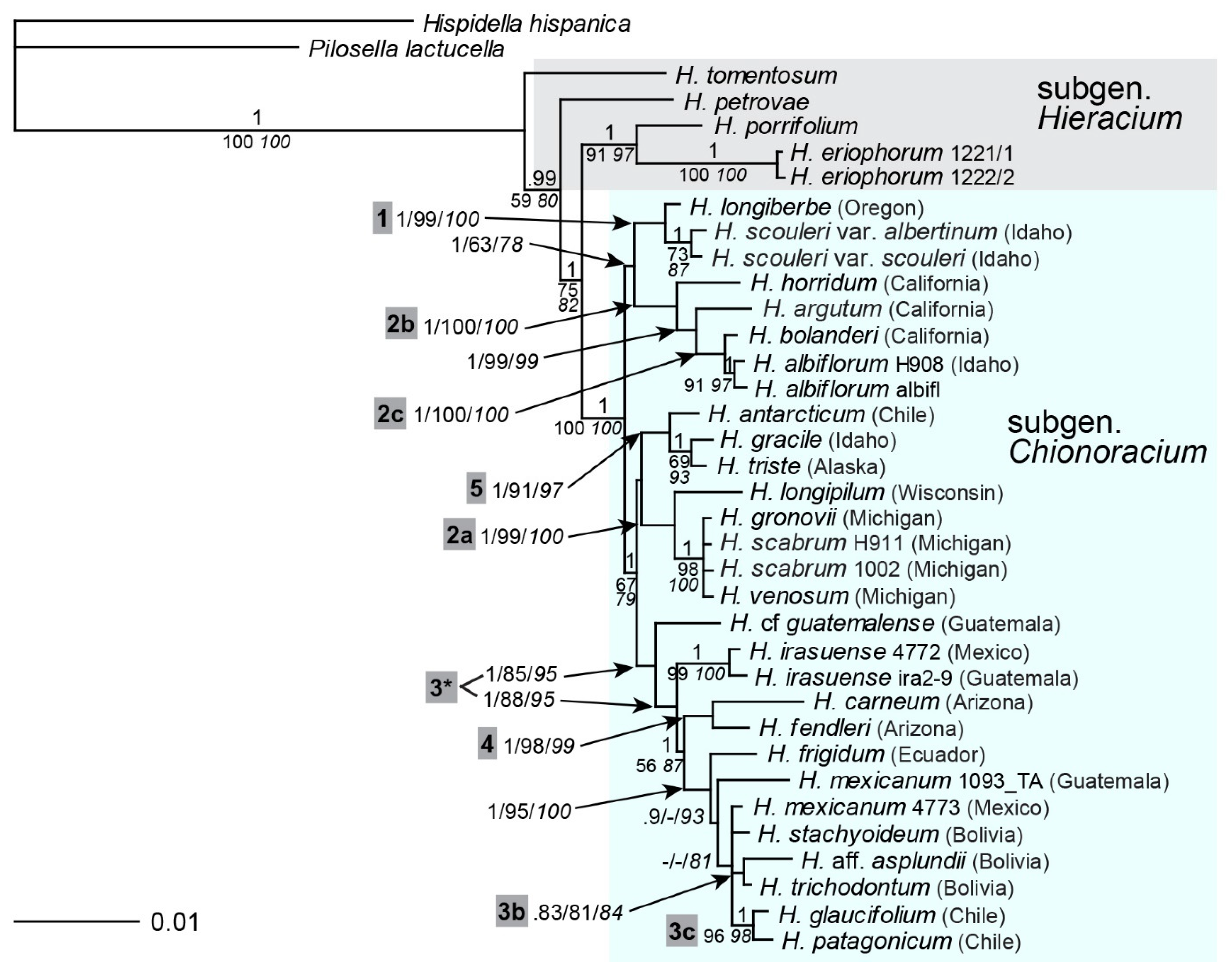
2.7. Multigene Phylogeny of Subgen. Chionoracium
2.8. Sectional Classification in Comparison with Phylogenetic Reconstruction
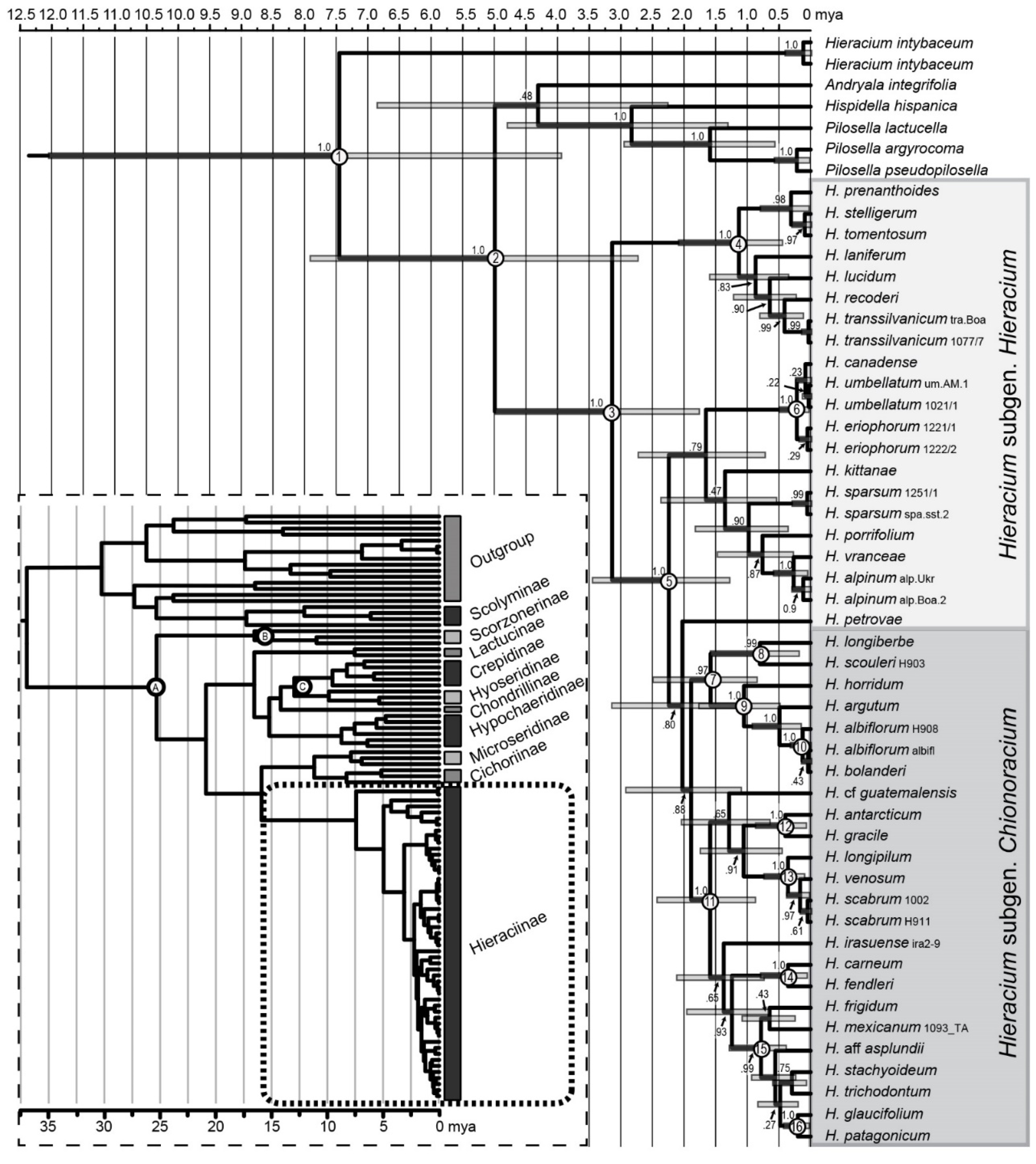
2.9. Dating of Chionoracium Lineages
3. Discussion
3.1. Features of the Molecular Markers
3.2. Paralogs and Pseudogenization of Gsh1 in Some Samples of Chionoracium
3.3. Maternal Lineages of Chionoracium
3.4. Evidence from Nuclear Markers and Genome Size
3.5. Synthesis of Phylogenetic Analyses
3.6. Morphological Classification in the Light of Phylogenetic Reconstruction
3.7. Dating of Hieraciinae and Chionoracium Nodes
3.8. Colonization of the New World
3.9. A Case of Long Distance Dispersal
3.10. Establishment of Species and Reproductive Assurance
4. Materials and Methods
4.1. Plant Material
4.2. Genome Size Determination
4.3. Molecular Procedures
4.4. Phylogenetic Analyses
4.5. Treatment of Paralogs/Pseudogenes
4.6. Molecular Dating
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Vouchers and GenBank Accession Numbers
References
- Schuhwerk, F. Some thoughts on the taxonomy of Hieracium. Ber. Bayer. Bot. Ges. 2002, 72, 193–198. [Google Scholar]
- Torrey, J.; Gray, A. A Flora of North America 2(3); Wiley and Putnam: New York, NY, USA, 1843. [Google Scholar]
- Fries, E. Epicrisis Generis Hieraciorum; Edquist & Berglund: Uppsala, Sweden, 1862. [Google Scholar]
- Peter, A. Hieracium. In Die Natürlichen Pflanzenfamilien 4(5); Engler, A., Prantl, K., Eds.; Wilhelm Engelmann: Leipzig, Germany, 1894; pp. 375–387. [Google Scholar]
- Schultz, C.H. Ueber die Hieracien Amerika’s. Bonplandia 1861, 9, 172–175. [Google Scholar]
- Arvet-Touvet, C.J.-M. Spicilegium Rariorum vel Novorum Hieraciorum, Praecipue Americanorum et Europaeorum; Impr. et Lithographie Veuve Rigaudin: Grenoble, France, 1881. [Google Scholar]
- Zahn, K.H. Hieracium Subgenus Stenotheca. In Das Pflanzenreich IV.280 (Heft 79); Engler, A., Ed.; Wilhelm Engelmann: Leipzig, Germany, 1922; pp. 1075–1142. [Google Scholar]
- Babcock, E.B.; Stebbins, G.L., Jr. The Genus Youngia; Carnegie Inst. of Washington Publ.: Washington, DC, USA, 1937; p. 484. [Google Scholar]
- Jeffrey, C. Notes on Compositae: I. The Cichorieae in east tropical Africa. Kew Bull. 1966, 18, 427–486. [Google Scholar] [CrossRef]
- Tutin, T.G. Tolpis Adanson. In Flora Europaea 4; Tutin, T.G., Heywood, V.H., Burges, N.A., Moore, D.M., Valentine, D.H., Walters, S.M., Webb, D.A., Eds.; Cambridge University Press: Cambridge, UK, 1976; p. 306. [Google Scholar]
- Blackmore, S.; Jarvis, C.E. Palynology of the genus Tolpis Adanson (Compositae: Lactuceae). Pollen Spores 1986, 28, 111–122. [Google Scholar]
- Sleumer, H. Die Hieracien Argentiniens unter Berücksichtigung der Nachbarländer. Bot. Jahrb. Syst. 1956, 77, 85–148. [Google Scholar]
- Sell, P.D. An introduction to the study of the British hieracia, 1. History and classification. Watsonia 1987, 16, 365–371. [Google Scholar]
- Chrtek, J., Jr.; Mráz, P.; Severa, M. Chromosome numbers in selected species of Hieracium s.str. (Hieracium subgen. Hieracium) in the Western Carpathians. Preslia 2004, 76, 119–139. [Google Scholar]
- Hand, M.L.; Vít, P.; Krahulcová, A.; Johnson, S.D.; Oelkers, K.; Siddons, H.; Chrtek, J., Jr.; Fehrer, J.; Koltunow, A.M.G. Evolution of apomixis loci in Pilosella and Hieracium (Asteraceae) inferred from the conservation of apomixis-linked markers in natural and experimental populations. Heredity 2015, 114, 17–26. [Google Scholar] [CrossRef]
- Mráz, P.; Zdvořák, P. Reproductive pathways in Hieracium s.s. (Asteraceae): Strict sexuality in diploids and apomixis in polyploids. Ann. Bot. 2019, 123, 391–403. [Google Scholar] [CrossRef]
- Guppy, G.A. Species relationships of Hieracium (Asteraceae) in British Columbia. Canad. J. Bot. 1978, 56, 3008–3019. [Google Scholar] [CrossRef]
- Strother, J.L. Hieracium. In Flora of North America. North of Mexico 19; Flora of North America Editorial Committee, Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 278–294. [Google Scholar]
- Bräutigam, S.; Greuter, W. A new treatment of Pilosella for the Euro-Mediterranean flora [Notulae ad floram euro-mediterraneam pertinentes 24]. Willdenowia 2007, 37, 123–137. [Google Scholar] [CrossRef]
- Morgan-Richards, M.; Trewick, S.A.; Chapman, H.M.; Krahulcová, A. Interspecific hybridization among Hieracium species in New Zealand: Evidence from flow cytometry. Heredity 2004, 93, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.M.; Fehrer, J.; Bräutigam, S.; Grosskopf, G. A new invasive hawkweed, Hieracium glomeratum (Lactuceae, Asteraceae), in the Pacific Northwest. Can. J. Bot. 2006, 84, 133–142. [Google Scholar] [CrossRef]
- Krahulec, F.; Krahulcová, A. Ploidy levels and reproductive behaviour in invasive Hieracium pilosella in Patagonia. NeoBiota 2011, 11, 25–31. [Google Scholar] [CrossRef]
- Fehrer, J.; Krahulcová, A.; Krahulec, F.; Chrtek, J., Jr.; Rosenbaumová, R.; Bräutigam, S. Evolutionary aspects in Hieracium subgenus Pilosella. In Apomixis: Evolution, Mechanisms and Perspectives (Regnum Vegetabile 147); Hörandl, E., Grossniklaus, U., van Dijk, P., Sharbel, T., Eds.; Koeltz: Königstein, Germany, 2007; pp. 359–390. [Google Scholar]
- Koltunow, A.M.G.; Johnson, S.D.; Rodrigues, J.C.M.; Okada, T.; Hu, Y.; Tsuchiya, T.; Wilson, S.; Fletcher, P.; Ito, K.; Suzuki, G.; et al. Sexual reproduction is the default mode in apomictic Hieracium subgenus Pilosella, in which two dominant loci function to enable apomixis. Plant J. 2011, 66, 890–902. [Google Scholar] [CrossRef]
- Fehrer, J.; Gemeinholzer, B.; Chrtek, J., Jr.; Bräutigam, S. Incongruent plastid and nuclear DNA phylogenies reveal ancient intergeneric hybridization in Pilosella hawkweeds (Hieracium, Cichorieae, Asteraceae). Mol. Phylogen. Evol. 2007, 42, 347–361. [Google Scholar] [CrossRef]
- Fehrer, J.; Krak, K.; Chrtek, J., Jr. Intra-individual polymorphism in diploid and apomictic polyploid hawkweeds (Hieracium, Lactuceae, Asteraceae): Disentangling phylogenetic signal, reticulation, and noise. BMC Evol. Biol. 2009, 9, 239. [Google Scholar] [CrossRef]
- Majeský, Ľ.; Krahulec, F.; Vašut, R.J. How apomictic taxa are treated in current taxonomy: A review. Taxon 2017, 66, 1017–1040. [Google Scholar] [CrossRef]
- Beaman, J.H.; De Jong, D.C.D.; Stoutamire, W.P. Chromosome studies in the alpine and subalpine floras of Mexico and Guatemala. Amer. J. Bot. 1962, 49, 41–50. [Google Scholar] [CrossRef]
- Beaman, J.H. Revision of Hieracium (Asteraceae) in Mexico and Central America. Syst. Bot. Monogr. 1990, 29, 1–77. [Google Scholar] [CrossRef]
- Schuhwerk, F. Published Chromosome Counts in Hieracium. 1996. Available online: http://www.botanischestaatssammlung.de/projects/chrzlit.html (accessed on 5 June 2021).
- Urtubey, E. Hieracium reitzianum (Asteraceae, Cichorieae), a new species from Brazil. Novon 2019, 27, 140–143. [Google Scholar] [CrossRef]
- Nägeli, C.; Peter, N. Die Hieracien Mittel-Europas. Monographische Bearbeitung der Piloselloiden Mit Besonderer Berücksichtigung der Mitteleuropäischen Sippen; R. Oldenburg: München, Germany, 1885. [Google Scholar]
- Gaskin, J.F.; Wilson, L.M. Phylogenetic relationships among native and naturalized Hieracium (Asteraceae) in Canada and the United States based on plastid DNA sequences. Syst. Bot. 2007, 32, 478–485. [Google Scholar] [CrossRef]
- Krak, K.; Álvarez, I.; Caklová, P.; Costa, A.; Chrtek, J.; Fehrer, J. Development of novel low-copy nuclear markers for Hieraciinae (Asteraceae) and their perspective for other tribes. Amer. J. Bot. 2012, 99, e74–e77. [Google Scholar] [CrossRef] [PubMed]
- Malme, G.O.A.N. Hieracia brasiliensia. Herbarii Regnelliani. Ark. Bot. 1931, 23, 1–10. [Google Scholar]
- Hind, D.J.N. An Annotated Preliminary Checklist of the Compositae of Bolivia (Version 2). 2011. Available online: https://www.kew.org/sites/default/files/2019-01/Bolivian%20compositae%20checklist.pdf (accessed on 18 August 2022).
- Ariza Espinar, L.; Cerana, M.M. Asteraceae: Subfamilia Cichorioidea. Tribus Lactuceae: Genero Hieracium. In Flora Argentina; Zuloaga, F.O., Anton, A., Eds.; Estudio Sigma: Buenos Aires, Argentina, 2015; Volume 7, pp. 15–42. [Google Scholar]
- Long, E.O.; Dawid, I.B. Repeated genes in eukaryotes. Annu. Rev. Biochem. 1980, 49, 727–764. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N. Concerted evolution of multigene families. In Evolution of Genes and Proteins; Nei, M., Koehn, R.K., Eds.; Sinauer: Sunderland, MA, USA, 1983; pp. 38–61. [Google Scholar]
- Álvarez, I.; Wendel, J.F. Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogen. Evol. 2003, 29, 417–434. [Google Scholar] [CrossRef]
- Sang, T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit. Rev. Biochem. Mol. Biol. 2002, 37, 121–147. [Google Scholar] [CrossRef]
- Nieto Feliner, G.; Rosselló, J.A. Better the devil you know? Guidelines for insightful utilization of nrDNA ITS in species-level evolutionary studies in plants. Mol. Phylogen. Evol. 2007, 44, 911–919. [Google Scholar] [CrossRef]
- Fehrer, J.; Slavíková, R.; Paštová, L.; Josefiová, J.; Mráz, P.; Chrtek, J.; Bertrand, Y.J.K. Molecular evolution and organization of ribosomal DNA in the hawkweed tribe Hieraciinae (Cichorieae, Asteraceae). Front. Plant Sci. 2021, 12, 647375. [Google Scholar] [CrossRef]
- Krak, K.; Caklová, P.; Chrtek, J.; Fehrer, J. Reconstruction of phylogenetic relationships in a highly reticulate group with deep coalescence and recent speciation (Hieracium, Asteraceae). Heredity 2013, 110, 138–151. [Google Scholar] [CrossRef]
- Ferreira, M.Z.; Zahradníček, J.; Kadlecová, J.; Menezes de Sequeira, M.; Chrtek, J., Jr.; Fehrer, J. Tracing the evolutionary history of the little-known Mediterranean-Macaronesian genus Andryala (Asteraceae) by multigene sequencing. Taxon 2015, 62, 535–551. [Google Scholar] [CrossRef]
- Mráz, P.; Filipaş, L.; Bărbos, M.I.; Kadlecová, J.; Paštová, L.; Belyayev, A.; Fehrer, J. An unexpected new diploid Hieracium from Europe: Integrative taxonomic approach with a phylogeny of diploid Hieracium taxa. Taxon 2019, 68, 1258–1277. [Google Scholar] [CrossRef]
- Chrtek, J.; Mráz, P.; Belyayev, A.; Paštová, L.; Mrázová, V.; Caklová, P.; Josefiová, J.; Zagorski, D.; Hartmann, M.; Jandová, M.; et al. Evolutionary history and genetic diversity of apomictic allopolyploids in Hieracium s.str.: Morphological versus genomic features. Amer. J. Bot. 2020, 107, 66–90. [Google Scholar] [CrossRef] [PubMed]
- Chrtek, J., Jr.; Zahradníček, J.; Krak, K.; Fehrer, J. Genome size in Hieracium subgenus Hieracium (Asteraceae) is strongly correlated with major phylogenetic groups. Ann. Bot. 2009, 104, 161–178. [Google Scholar] [CrossRef]
- Appels, R.; Honeycutt, R.L. rDNA evolution over a billion years. In DNA Systematics: Plants II Plant DNA; Dutta, S.K., Ed.; CRC Press: Boca Raton, FL, USA, 1986; pp. 81–125. [Google Scholar]
- Standley, P.C.; Steyermark, J.A. Studies of Central American plants–IV. Field Mus. Nat. Hist. Bot. Ser. 1944, 23, 31–109. [Google Scholar]
- Olmstead, R.G.; Palmer, J.D. Chloroplast DNA systematics: A review of methods and data analysis. Amer. J. Bot. 1994, 81, 1205–1224. [Google Scholar] [CrossRef]
- Cronn, R.C.; Zhao, X.; Paterson, A.H.; Wendel, J.F. Polymorphism and concerted evolution in a tandemly repeated gene family: 5S ribosomal DNA in diploid and allopolyploid cottons. J. Mol. Evol. 1996, 42, 685–705. [Google Scholar] [CrossRef]
- Kaplan, Z.; Jarolímová, V.; Fehrer, J. Revision of chromosome numbers of Potamogetonaceae: A new basis for taxonomic and evolutionary implications. Preslia 2013, 85, 421–482. [Google Scholar]
- Mahelka, V.; Kopecký, D.; Baum, B.R. Contrasting patterns of evolution of 45S and 5S rDNA families uncover new aspects in the genome constitution of the agronomically important grass Thinopyrum intermedium (Triticeae). Mol. Biol. Evol. 2013, 30, 2065–2086. [Google Scholar] [CrossRef] [Green Version]
- Whittemore, A.T.; Schaal, B.A. Interspecific gene flow in sympatric oaks. Proc. Natl. Acad. Sci. USA 1991, 88, 2540–2544. [Google Scholar] [CrossRef]
- Acosta, M.C.; Premoli, A.C. Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogen. Evol. 2010, 54, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wang, G.; Ma, Q.; Liang, K.; Yang, Z. Multilocus data reveal deep phylogenetic relationships and intercontinental biogeography of the Eurasian-North American genus Corylus (Betulaceae). Mol. Phylogen. Evol. 2020, 142, 106658. [Google Scholar] [CrossRef] [PubMed]
- Small, R.L.; Cronn, R.C.; Wendel, J.F. Use of nuclear genes for phylogeny reconstruction in plants. Aust. Syst. Bot. 2004, 17, 145–170. [Google Scholar] [CrossRef]
- Smith, S.A.; Donoghue, M.J. Rates of molecular evolution are linked to life history in flowering plants. Science 2008, 322, 86–89. [Google Scholar] [CrossRef]
- Lanfear, R.; Ho, S.Y.W.; Davies, T.J.; Moles, A.T.; Aarssen, L.; Swenson, N.G.; Warman, L.; Zanne, A.E.; Allen, A.P. Taller plants have lower rates of molecular evolution. Nat. Commun. 2013, 4, 1879. [Google Scholar] [CrossRef]
- Tremetsberger, K.; Gemeinholzer, B.; Zetzsche, H.; Blackmore, S.; Kilian, N.; Talavera, S. Divergence time estimation in Cichorieae (Asteraceae) using a fossil-calibrated relaxed molecular clock. Org. Divers. Evol. 2013, 13, 1–13. [Google Scholar] [CrossRef]
- Barba-Montoya, J.; dos Reis, M.; Schneider, H.; Donoghue, P.C.J.; Yang, Z. Constraining uncertainty in the timescale of angiosperm evolution and the veracity of a Cretaceous Terrestrial Revolution. New Phytol. 2018, 218, 819–834. [Google Scholar] [CrossRef]
- Yang, Z.; Rannala, B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 2006, 23, 212–226. [Google Scholar] [CrossRef]
- Carruthers, T.; Muñoz-Rodríguez, P.; Wood, J.R.I.; Scotland, R.W. The temporal dynamics of evolutionary diversification in Ipomoea. Mol. Phylogen. Evol. 2020, 146, 106768. [Google Scholar] [CrossRef]
- Coates, A.G.; Obando, J.A. The geologic evolution of the Central American isthmus. In Evolution and Environment in Tropical America; Jackson, J.B.C., Budd, A.F., Coates, A.G., Eds.; University of Chicago Press: Chicago, IL, USA, 1996; pp. 21–56. [Google Scholar]
- Gregory-Wodzicki, K.M. Uplift history of the Central and Northern Andes: A review. GSA Bull. 2000, 112, 1091–1105. [Google Scholar] [CrossRef]
- Bershaw, J.; Garzione, C.N.; Higgins, P.; MacFadden, B.J.; Anaya, F.; Alvarenga, H. Spatial-temporal changes in Andean plateau climate and elevation from stable isotopes of mammal teeth. Earth Planet. Sci. Lett. 2010, 289, 530–538. [Google Scholar] [CrossRef]
- Richardson, J.E.; Pennington, R.T.; Pennington, T.D.; Hollingsworth, P.M. Rapid diversification of a species-rich genus of neotropical rain forest trees. Science 2001, 293, 2242–2245. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Pennington, R.T.; Antonelli, A. Neotropical plant evolution: Assembling the big picture. Bot. J. Linn. Soc. 2013, 171, 1–18. [Google Scholar] [CrossRef]
- Colston, T.J.; Grazziotin, F.G.; Shepard, D.B.; Vitt, L.J.; Colli, G.R.; Henderson, R.W.; Hedges, S.B.; Bonatto, S.; Zaher, H.; Noonan, B.P.; et al. Molecular systematics and historical biogeography of tree boas (Corallus spp.). Mol. Phylogen. Evol. 2013, 66, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, F.S.; Ojanguren-Affilastro, A.A.; Ramírez, M.J.; Ochoa, J.A.; Mattoni, C.I.; Prendini, L. Andean uplift drives diversification of the bothriurid scorpion genus Brachistosternus. J. Biogeogr. 2016, 43, 1942–1954. [Google Scholar] [CrossRef]
- Jabaily, R.S.; Sytsma, K.J. Historical biogeography and life history evolution of Andean Puya (Bromeliaceae). Bot. J. Linn. Soc. 2012, 171, 201–224. [Google Scholar] [CrossRef]
- Drew, B.T.; Sytsma, K.J. Phylogenetics, biogeography and evolution of dioecy in South American Lepechinia (Lamaiaceae). Bot. J. Linn. Soc. 2012, 171, 171–190. [Google Scholar] [CrossRef]
- Milne, R.I. Phylogeny and biogeography of Rhododendron subsection Pontica, a group with a tertiary relict distribution. Mol. Phylogen. Evol. 2004, 33, 389–401. [Google Scholar] [CrossRef]
- Denk, T.; Grímsson, F.; Zetter, R. Episodic migration of oaks to Iceland: Evidence for a North Atlantic “land bridge” in the latest Miocene. Amer. J. Bot. 2010, 97, 276–287. [Google Scholar] [CrossRef]
- Marincovich, L., Jr.; Gladenkov, A.Y. New evidence for the age of Bering Strait. Quat. Sci. Rev. 2001, 20, 329–335. [Google Scholar] [CrossRef]
- Abbott, R.J.; Smith, L.C.; Milne, R.I.; Crawford, R.M.M.; Wolff, K.; Balfour, J. Molecular analysis of plant migration and refugia inthe Arctic. Science 2000, 289, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Ickert-Bond, S.M.; Murray, D.F.; DeChaine, E. Contrasting patterns of plant distribution in Beringia. Alsk. Park Sci. 2009, 8, 26–32. [Google Scholar]
- Les, D.H.; Crawford, D.J.; Kimball, R.T.; Moody, M.L.; Landolt, E. Biogeography of discontinuously distributed hydrophytes: A molecular appraisal of intercontinental disjunctions. Int. J. Plant Sci. 2003, 164, 917–932. [Google Scholar] [CrossRef]
- Blattner, F.R. Multiple intercontinental dispersals shaped the distribution area of Hordeum (Poaceae). New Phytol. 2006, 169, 603–614. [Google Scholar] [CrossRef]
- Villaverde, T.; Escudero, M.; Martín-Bravo, S.; Jiménez-Mejías, P.; Sanmartín, I.; Vargas, P.; Luceño, M. Bipolar distributions in vascular plants: A review. Amer. J. Bot. 2017, 104, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Fehrer, J.; Iida, S.; Kaplan, Z. Cryptic species of pondweeds (Potamogetonaceae) at an intercontinental scale revealed by molecular phylogenetic analyses. Taxon 2022, 71, 531–551. [Google Scholar] [CrossRef]
- Kirby, J.S.; Stattersfield, A.J.; Butchart, S.H.M.; Evans, M.I.; Grimmett, R.F.A.; Jones, V.R.; O’Sullivan, J.; Tucker, G.M.; Newton, I. Key conservation issues for migratory land- and waterbird species on the world’s major flyways. Bird Conserv. Int. 2008, 18, S49–S73. [Google Scholar] [CrossRef]
- Stern, D.L. The genetic causes of convergent evolution. Nat. Rev. Genet. 2013, 14, 751–764. [Google Scholar] [CrossRef]
- Guppy, G.A. The Systematics of Indigenous Species of Hieracium (Asteraceae) in British Columbia. Ph.D. Thesis, The University of British Columbia, Vancouver, BC, Canada, 1975. [Google Scholar] [CrossRef]
- Bennett, M.D.; Leitch, I.J.; Hanson, L. DNA amounts in two samples of angiosperm weeds. Ann. Bot. 1998, 82, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Knight, C.A.; Molinari, N.A.; Petrov, D.A. The large genome constraint hypothesis: Evolution, ecology and phenotype. Ann. Bot. 2005, 95, 177–190. [Google Scholar] [CrossRef]
- Mráz, P. Mentor effects in the genus Hieracium s.str. (Compositae, Lactuceae). Folia Geobot. 2003, 38, 345–350. [Google Scholar] [CrossRef]
- Lysák, M.A.; Doležel, J. Estimation of nuclear DNA content in Sesleria (Poaceae). Caryologia 1998, 51, 123–132. [Google Scholar] [CrossRef]
- Otto, F. DAPI staining of fixed cells for high-resolution flow cytometry of nuclear DNA. In Methods in Cell Biology: Flow Cytometry; Crissman, H.A., Darzynkiewicz, Z., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 105–110. [Google Scholar]
- Štorchová, H.; Hrdličková, R.; Chrtek, J., Jr.; Tetera, M.; Fitze, D.; Fehrer, J. An improved method of DNA isolation from plants collected in the field and conserved in saturated NaCl/CTAB solution. Taxon 2000, 49, 79–84. [Google Scholar] [CrossRef]
- Zagorski, D.; Hartmann, M.; Bertrand, Y.J.K.; Paštová, L.; Slavíková, R.; Josefiová, J.; Fehrer, J. Characterization and dynamics of repeatomes in closely related species of Hieracium (Asteraceae) and their synthetic and apomictic hybrids. Front. Plant Sci. 2020, 11, 591053. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit, a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Borchsenius, F. FastGap 1.2. Department of Biosciences, Aarhus University, Denmark. 2009. Available online: http://www.aubot.dk/FastGap_home.htm (accessed on 17 August 2022).
- Simmons, M.P.; Ochoterena, H. Gaps as characters in sequence-based phylogenetic analyses. Syst. Biol. 2000, 49, 369–381. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4; Sinauer: Sunderland MA, USA, 2002. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Modeltest: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Kilian, N.; Sennikov, A.; Wang, Z.-H.; Gemeinholzer, B.; Zhang, J.-W. Sub-Paratethyan origin and Middle to Late Miocene principal diversification of the Lactucinae (Compositae: Cichorieae) inferred from molecular phylogenetics, divergence-dating and biogeographic analysis. Taxon 2017, 66, 675–703. [Google Scholar] [CrossRef]
- Barba-Montoya, J.; dos Reis, M.; Yang, Z. Comparison of different strategies for using fossil calibrations to generate the time prior in Bayesian molecular clock dating. Mol. Phylogen. Evol. 2017, 114, 386–400. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. Beast 2: A software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Douglas, J.; Zhang, R.; Bouckaert, R. Adaptive dating and fast proposals: Revisiting the phylogenetic relaxed clock model. PLoS Comput. Biol. 2021, 17, e1008322. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Drummond, J.J. bModelTest: Bayesian phylogenetic site model averaging and model comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Russel, P.M.; Brewer, B.J.; Klaere, S.; Bouckaert, R.R. Model selection and parameter inference in phylogenetics using nested sampling. Syst. Biol. 2018, 68, 219–233. [Google Scholar] [CrossRef]
- Kass, R.E.; Raftery, A.E. Bayes factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Clades | Lineage | ptDNA | ITS | ETS | ITS+ ETS | 5S-NTS | gsh1 | sqs | Comb. |
|---|---|---|---|---|---|---|---|---|---|
| H. longiberbe, H. scouleri | 1 | +++ | + | – | +++ | – | + | + | +++ |
| H. longipilum, H. scabrum, H. venosum, H. horridum, H. argutum, H. albiflorum, H. bolanderi | 2 | ++ | – | – | – | – | – | – | – |
| H. longipilum, H. scabrum, H. venosum (+ H. gronovii, ETS and 5S-NTS only) | 2a/2a* | – | +++ | ++ | +++ | +++ | + | – | +++ |
| H. horridum, H. argutum, H. albiflorum, H. bolanderi | 2b | +++ | +++ | – | +++ | – | – | – | +++ |
| H. albiflorum, H. bolanderi | 2c | – | +++ | – | +++ | +++ | + | +++ | +++ |
| H. mexicanum, H. cf. guatemalense, H. irasuense, H. trichodontum, H. stachyoideum, H. glaucifolium, H. patagonicum, H. aff. asplundii, H. frigidum | 3/3* | +++ | ++ | +++ | +++ | – | ++ | + | +++ |
| H. mexicanum, H. irasuense | 3a | +++ | – | – | – | – | – | – | – |
| H. trichodontum, H. aff. asplundii | 3b/3b* | +++ | – | – | – | – | – | – | ++ |
| H. glaucifolium, H. patagonicum | 3c | – | +++ | ++ | +++ | ++ | n.d. | +++ | +++ |
| H. carneum, H. fendleri | 4 | – | +++ | +++ | +++ | – | – | – | +++ |
| H. antarcticum, H. gracile (+ H. triste, ETS and 5S-NTS only) | 5 | – | +++ | + | ++ | +++ | ++ | + | +++ |
| H. canadense, H. umbellatum (+ H. eriophorum) | x | +++ | +++ | +++ | +++ | n.d. | +++ | – | n.d. |
| Accession, Clone | Sequence Features | ||||
|---|---|---|---|---|---|
| AG | TG | Stop | Poly-T | Exon Missing | |
| H. horridum c1–5, 7, 12–15, 17–18, 20 | + | ||||
| H. horridum c6, 8, 9, 11 | + | + | |||
| H. horridum c16, 19 | + | + | + | ||
| H. argutum c1, 10, 16, 22 | + | ||||
| H. argutum c2, 3, 6, 8–9, 13–18, 23 | + | + | |||
| H. argutum c4, 7, 20–21 | + | ||||
| H. argutum c5, 11–12 | + | ||||
| H. argutum c19 | + | + | |||
| H. albiflorum H908 c1 | (+) | ||||
| H. albiflorum H908 c2, 4–5 | + | ||||
| H. albiflorum H908 c3, 6 | |||||
| H. scouleri var. albertinum c1–2, 4–10 | |||||
| H. scouleri var. albertinum c3 | + | + | |||
| H. scouleri var. albertinum c11 | + | ||||
| Node Number | Description | Age Estimates Mya (Range) |
|---|---|---|
| 1 | Hieraciinae | 7.45 (3.95–12.07) |
| 2 | Hieraciinae without H. intybaceum | 4.99 (2.74–7.94) |
| 3 | Hieracium subgenera Hieracium and Chionoracium | 3.14 (1.78–5.01) |
| 4 | H. subgen. Hieracium, W European clade | 1.13 (0.46–2.11) |
| 5 | H. subgen. Hieracium, E European clade & Chionoracium | 2.24 (1.29–3.47) |
| 6 | Divergence in the H. umbellatum clade (incl. H. canadense) | 0.22 (0.04–0.52) |
| 7 | wNA species, lineages 1, 2b & 2c | 1.58 (1.19–3.16) |
| 8 | H. longiberbe/H. scouleri (wNA, lineage 1) | 0.82 (0.20–1.61) |
| 9 | H. horridum/H. argutum/H. albiflorum/H. bolanderi (wNA, lineage 2b) | 1.05 (0.50–1.79) |
| 10 | H. albiflorum/H. bolanderi (wNA, lineage 2c) | 0.04 (0.0–0.17) |
| 11 | all other species of H. subgen. Chionoracium | 1.59 (0.89–2.45) |
| 12 | H. antarcticum/H. gracile (sSA, nNA/wNA, lineage 5) | 0.40 (0.08–0.90) |
| 13 | H. longipilum/H. venosum/H. scabrum (eNA, lineage 2a) | 0.36 (0.03–0.41) |
| 14 | H. carneum/H. fendleri (wNA, lineage 4) | 0.36 (0.07–0.82) |
| 15 | all SA species (except H. antarcticum) and H. mexicanum (CA) | 0.79 (0.40–1.31) |
| 16 | H. glaucifolium/H. patagonicum (sSA, lineage 3c) | 0.20 (0.04–0.45) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehrer, J.; Bertrand, Y.J.K.; Hartmann, M.; Caklová, P.; Josefiová, J.; Bräutigam, S.; Chrtek, J. A Multigene Phylogeny of Native American Hawkweeds (Hieracium Subgen. Chionoracium, Cichorieae, Asteraceae): Origin, Speciation Patterns, and Migration Routes. Plants 2022, 11, 2584. https://doi.org/10.3390/plants11192584
Fehrer J, Bertrand YJK, Hartmann M, Caklová P, Josefiová J, Bräutigam S, Chrtek J. A Multigene Phylogeny of Native American Hawkweeds (Hieracium Subgen. Chionoracium, Cichorieae, Asteraceae): Origin, Speciation Patterns, and Migration Routes. Plants. 2022; 11(19):2584. https://doi.org/10.3390/plants11192584
Chicago/Turabian StyleFehrer, Judith, Yann J. K. Bertrand, Matthias Hartmann, Petra Caklová, Jiřina Josefiová, Siegfried Bräutigam, and Jindřich Chrtek. 2022. "A Multigene Phylogeny of Native American Hawkweeds (Hieracium Subgen. Chionoracium, Cichorieae, Asteraceae): Origin, Speciation Patterns, and Migration Routes" Plants 11, no. 19: 2584. https://doi.org/10.3390/plants11192584