
Insulin Resistance and High Blood Pressure: Mechanistic Insight on the Role of the Kidney
 ,
,
Abstract
:1. Introduction
2. Insulin Resistance: General Concepts
3. Insulin and the Kidney
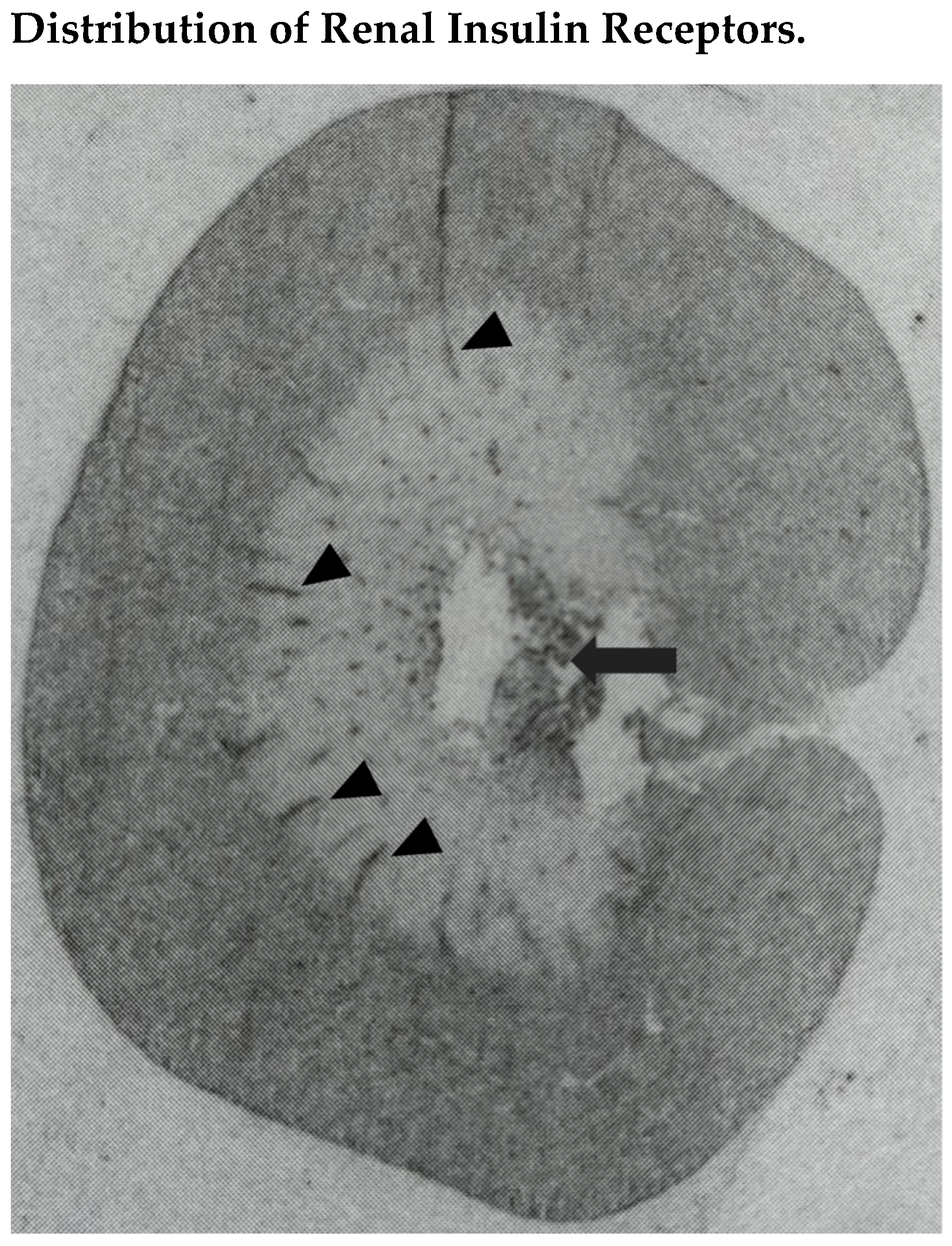
3.1. Renal Insulin Receptors
3.2. Renal Effects of Insulin
3.3. Renal Effects of Insulin in Diabetes
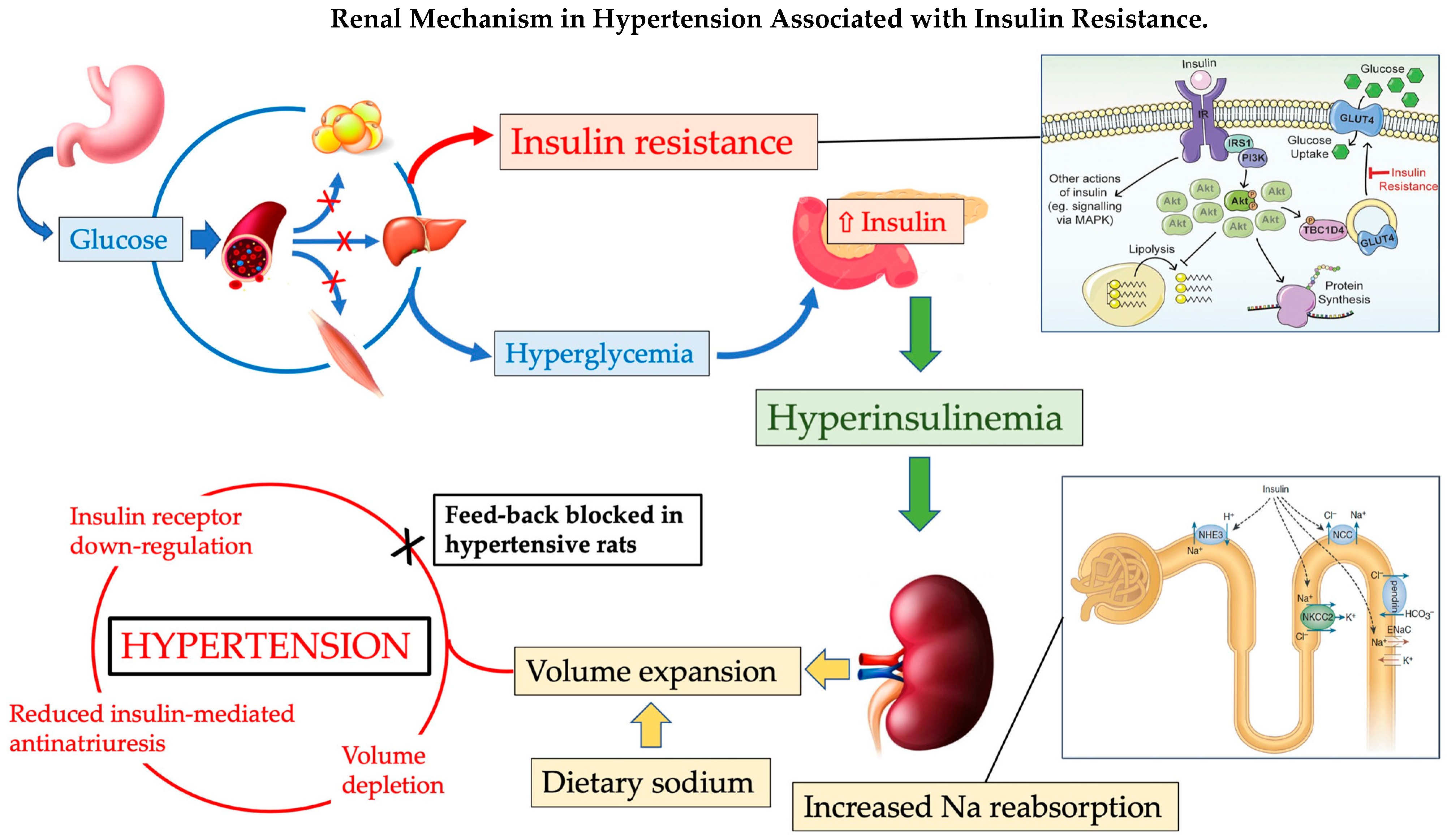
4. Insulin Resistance in Hypertension
4.1. Association of Insulin Resistance with Essential Hypertension
4.2. Mechanisms Linking Insulin Resistance with Hypertension: The Role of Sodium
4.3. Molecular Mechanisms of Insulin Resistance in Hypertension
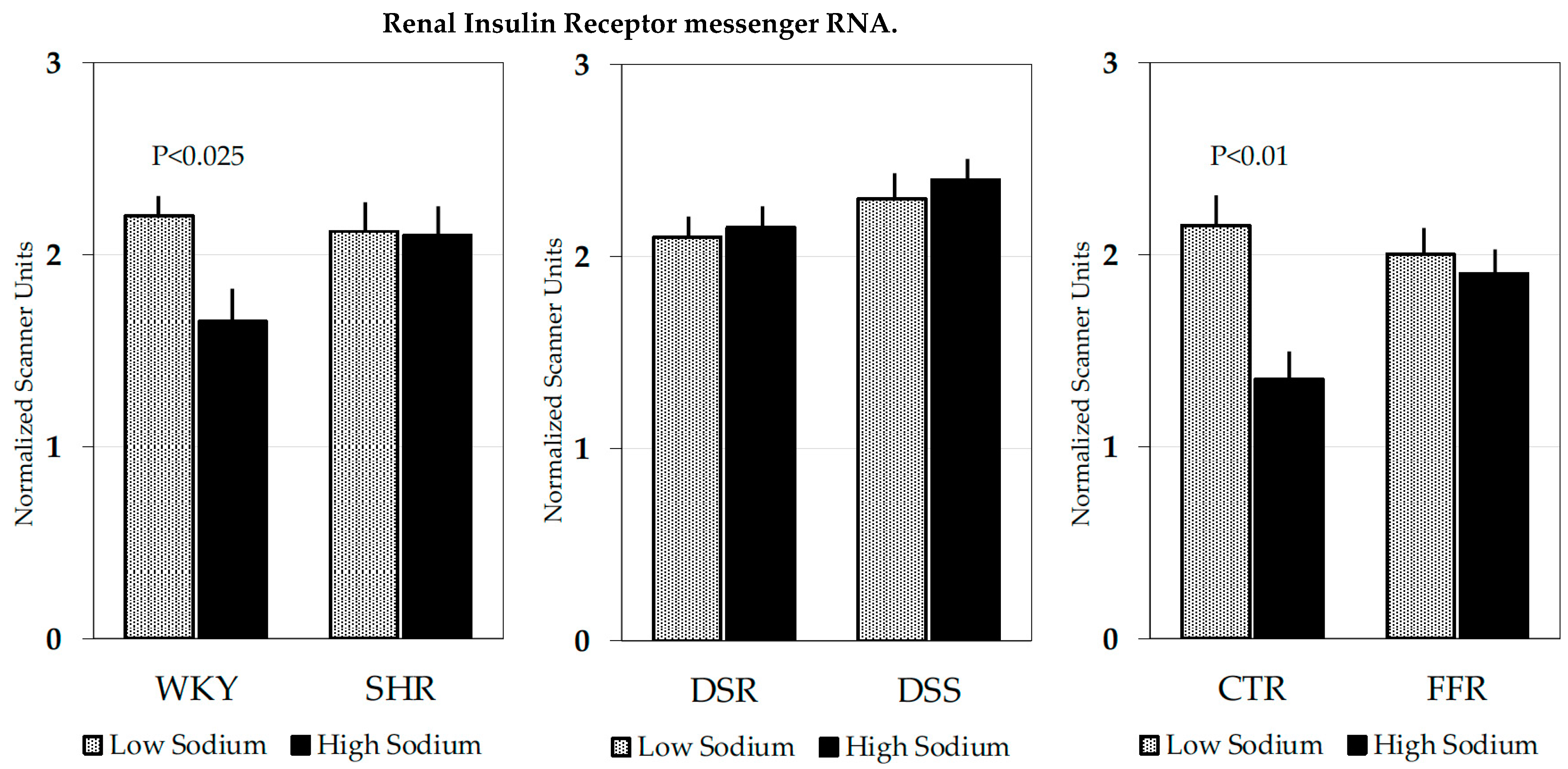
4.3.1. Spontaneously Hypertensive Rats
4.3.2. Dahl Hypertensive Rats
4.3.3. Fructose-Fed Rats
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Modan, M.; Halkin, H.; Almog, S.; Lusky, A.; Eshkol, A.; Shefi, M.; Shitrit, A.; Fuchs, Z. Hyperinsulinemia: A link between hypertension obesity and glucose intolerance. J. Clin. Investig. 1985, 75, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Buzzigoli, G.; Bonadonna, R.; Giorico, M.A.; Oleggini, M.; Graziadei, L.; Pedrinelli, R.; Brandi, L.; Bevilacqua, S. Insulin resistance in essential hypertension. N. Engl. J. Med. 1987, 317, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, N.M. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Arch. Intern. Med. 1989, 149, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef]
- Hill, M.A.; Yang, Y.; Zhang, L.; Sun, Z.; Jia, G.; Parrish, A.R.; Sowers, J.R. Insulin resistance, cardiovascular stiffening and cardiovascular disease. Metabolism 2021, 119, 154766. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Melis, A.; Tedde, R. Insulin hypersecretion: A potential role in essential but not secondary hypertension. Metabolism 1992, 41, 1261–1266. [Google Scholar] [CrossRef]
- Facchini, F.; Chen, Y.-D.I.; Clinkingbeard, C.; Jeppesen, J.; Reaven, G.M. Insulin resistance, hyperinsulinemia, and dyslipidemia in nonobese individuals with a family history of hypertension. Am. J. Hypertens. 1992, 5, 694–699. [Google Scholar] [CrossRef]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.; Wang, Z.; Hall, M.E. Obesity, kidney dysfunction and hypertension: Mechanistic links. Nat. Rev. Nephrol. 2019, 15, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Poirier, P.; Despres, J.P. Cardiovascular and metabolic heterogeneity of obesity: Clinical challenges and implications for management. Circulation 2018, 137, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.W. Obesity, adipose tissue and vascular dysfunction. Circ. Res. 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Whaley-Connell, A.; Sowers, J.R. Obesity and kidney disease: From population to basic science and the search for new therapeutic targets. Kidney Int. 2017, 92, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signal pathways and substrate flux. J. Clin. Invest. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Shulman, G.I. Mechanisms of insulin action and insulin resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Di Pino, A.; DeFronzo, R.A. Insulin resistance and atherosclerosis: Implications for insulin-sensitizing agents. Endocr. Rev. 2019, 40, 1447–1467. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Bartoli, E. Mechanisms of insulin resistance leading to hypertension: What we can learn from experimental rat models. J. Investig. Med. 1997, 45, 238–251. [Google Scholar] [PubMed]
- Abdul-Ghani, M.; DeFronzo, R.A. Insulin resistance and hyperinsulinemia: The egg and the chicken. J. Clin. Endocrinol. Metab. 2021, 106, 1897–1899. [Google Scholar] [CrossRef]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell. Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. Insulin action at a molecular level-100 years of progress. Mol. Metab. 2021, 52, 101304. [Google Scholar] [CrossRef]
- Whittaker, J. Structure and Function of the Insulin Receptor; Post, T.W., Ed.; Uptodate: Waltham, MA, USA, 2020. [Google Scholar]
- Gutmann, T.; Kim, K.H.; Grzybek, M.; Walz, T.; Coskun, U. Visualization of ligand-induced transmembrane signaling in the full-length human insulin receptor. J. Cell. Biol. 2018, 217, 1643–1649. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin receptor isoforms in physiology and disease: An updated review. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Shoelson, S.E.; Keutmann, H.; Kahn, C.R. A cascade of tyrosine autophosphorylation in the beta-subunit activates the phosphotransferase of the insulin receptor. J. Biol. Chem. 1988, 263, 2969–2980. [Google Scholar] [CrossRef]
- Sun, X.J.; Rothenberg, P.; Kahn, C.R.; Backer, J.M.; Araki, E.; Wilden, P.A.; Cahill, D.A.; Goldstein, B.J.; White, M.F. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991, 352, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Sadagurski, M.; Dong, X.C.; Myers Jr., M.G.; White, M.F. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol. Metab. 2013, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Low Wang, C.C.; Goalstone, M.L.; Draznin, B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes 2004, 53, 2735–2740. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Kahn, C.R. From mice to man: Insight into the insulin resistance syndromes. Annu. Rev. Physiol. 2006, 68, 123–158. [Google Scholar] [CrossRef]
- Da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of hyperinsulinemia and insulin resistance in hypertension: Metabolic syndrome revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef]
- Tiwari, S.; Riazi, S.; Ecelbarger, C.A. Insulin’s impact on renal sodium transport and blood pressure in health, obesity, and diabetes. Am. J. Physiol. Renal Physiol. 2007, 293, F974–F984. [Google Scholar] [CrossRef]
- Butlen, D.; Vadrot, S.; Roseau, S.; Morel, F. Insulin receptors along the rat nephron: [125I] insulin binding in microdissected glomeruli and tubules. Pflügers Arch. 1988, 412, 604–612. [Google Scholar] [CrossRef]
- Sechi, L.A.; De Carli, S.; Bartoli, E. In situ characterization of renal insulin receptors in the rat. J. Recept. Res. 1994, 14, 347–356. [Google Scholar] [CrossRef]
- Bisbis, S.; Derouet, M.; Simon, J. Characterization of insulin receptors in chicken kidneys: Effect of nutritional status. Gen. Comp. Endocrinol. 1994, 96, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Griffin, C.A.; Grady, E.F.; Grunfeld, C.; Kalinyak, J.E.; Schambelan, M. Tissue-specific regulation of insulin receptor mRNA levels in rats with STZ-induced diabetes mellitus. Diabetes 1992, 41, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Halagappa, V.K.; Riazi, S.; Hu, X.; Ecelbarger, C.A. Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J. Am. Soc. Nephrol. 2007, 18, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Tedde, R.; Sechi, L.A.; Marigliano, A.; Scano, L.; Pala, A. In-vitro action of insulin on erythrocyte sodium transport mechanisms: Its possible role in the pathogenesis of essential hypertension. Clin. Exp. Hypertens. 1988, 10, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Baum, M. Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J. Clin. Investig. 1987, 79, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Klisic, J.; Hu, M.C.; Nief, V.; Reyes, L.; Fuster, D.; Moe, O.W.; Ambuhl, P.M. Insulin activated Na+/H+ exchanger 3: Biphasic response and glucocorticoid dependence. Am. J. Physiol. Renal Physiol. 2002, 283, F532–F539. [Google Scholar] [CrossRef]
- Ferraille, E.; Carranza, M.L.; Rousselot, M.; Favre, H. Insulin enhances sodium sensitivity of Na-K-ATPase in isolated rat proximal convoluted tubule. Am. J. Physiol. Renal Fluid Electrolyte Physiol. 1994, 267, F55–F62. [Google Scholar] [CrossRef]
- Ito, O.; Kondo, Y.; Takahashi, N.; Kudo, K.; Igarashi, Y.; Omata, K.; Imai, Y.; Abe, K. Insulin stimulates NaCl transport in isolated perfused MTAL of Henle’s loop of rabbit kidney. Am. J. Physiol. Renal Fluid Electrolyte Physiol. 1994, 267, F265–F270. [Google Scholar] [CrossRef]
- Sechi, L.A.; Marigliano, A.; Tedde, R. Evaluation of insulin-induced changes in the renal response to furosemide in normal subjects. Miner. Electrolyte Metab. 1991, 17, 383–389. [Google Scholar]
- Blazer-Yost, B.L.; Esterman, M.A.; Vlahos, C.J. Insulin-stimulated trafficking of ENaC in renal cells requires PI3-kinase activity. Am. J. Physiol. Cell Physiol. 2003, 284, C1645–C1653. [Google Scholar] [CrossRef]
- Ferraille, E.; Rousselot, M.; Rajerison, R.; Favre, H. Effect of insulin on Na+, K+ -ATPase in rat collecting duct. J. Physiol. 1995, 488, 171–180. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Cooke, C.R.; Andres, R.; Faloona, G.R.; Davis, P.J. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J. Clin. Investig. 1975, 55, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Skott, P.; Hother-Nielsen, O.; Bruun, N.E.; Giese, J.; Nielsen, M.D.; Beck-Nielsen, H.; Parving, H.H. Effect of insulin on kidney function and sodium excretion in healthy subjects. Diabetologia 1989, 32, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Bolinder, J.; Alvestrand, A. Effects of insulin on renal hemodynamics and the proximal and distal tubular sodium handling in healthy subjects. Diabetologia 1992, 35, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Griffin, C.A.; Grady, E.F.; Kalinyak, J.E.; Schambelan, M. Insulin receptor concentration and gene expression are modulated by sodium intake in the rat kidney. J. Hypertens. 1992, 29, S212–S213. [Google Scholar]
- Sechi, L.A.; Griffin, C.A.; Schambelan, M. Effect of dietary sodium chloride on insulin receptor number and mRNA levels in the kidney of normal rats. Am. J. Physiol. Renal Fluid Electrolyte Physiol. 1994, 266, F31–F38. [Google Scholar] [CrossRef]
- Tiwari, S.; Sharma, N.; Gill, P.S.; Igarashi, P.; Kahn, R.C.; Wade, J.B.; Ecelbarger, C.E. Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 6469–6474. [Google Scholar] [CrossRef]
- Pavlov, T.S.; Ilatovskaya, D.V.; Levchenko, V.; Li, L.; Ecelbarger, C.M.; Staruschenko, A. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J. 2013, 27, 2723–2732. [Google Scholar] [CrossRef]
- Li, L.; Garikepati, M.; Tsukerman, S.; Kohan, D.; Wade, J.B.; Tiwari, S.; Ecelbarger, C.E. Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. Am. J. Physiol. Renal Physiol. 2013, 304, F279–F288. [Google Scholar] [CrossRef]
- Sechi, L.A.; Valentin, J.P.; Griffin, C.A.; Lee, E.; Bartoli, E.; Humphreys, M.H.; Schambelan, M. Receptors for atrial natriuretic peptide are decreased in the kidney of rats with streptozotocin-induced diabetes mellitus. J. Clin. Investig. 1995, 95, 2451–2457. [Google Scholar] [CrossRef]
- Siddiqui, M.R.; Moorthy, K.; Taha, A.; Hussain, M.E.; Baquer, N.Z. Low doses of vanadate and Trigonella synergistically regulate Na+/K+-ATPase activity and GLUT4 translocation in alloxan-diabetic rats. Mol. Cell. Biochem. 2006, 285, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Knepper, M.A.; Verbalis, J.G.; Ecelbarger, C.A. Increased renal ENaC subunit and sodium transporter abundances in streptozotocin-induced type 1 diabetes. Am. J. Physiol. Renal Physiol. 2003, 285, F1125–F1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Sands, J.M.; Klein, J.D. Changes in renal medullary transport proteins during uncontrolled diabetes mellitus in rats. Am. J. Physiol. Renal Physiol. 2003, 285, F303–F309. [Google Scholar] [CrossRef] [PubMed]
- Bickel, C.A.; Knepper, M.A.; Verbalis, J.G.; Ecelbarger, C.A. Dysregulation of renal salt and water transport proteins in diabetic Zucker rats. Kidney Int. 2002, 61, 2099–2110. [Google Scholar] [CrossRef] [PubMed]
- Riazi, S.; Khan, O.; Tiwari, S.; Hu, X.; Ecelbarger, C.A. Rosiglitazone regulates ENaC and Na-K-2Cl cotransporters [NKCC(2)] abundance in the obese Zucker rat. Am. J. Nephrol. 2006, 26, 245–257. [Google Scholar] [CrossRef]
- Sechi, L.A.; Griffin, C.A.; Zingaro, L.; Valentin, J.P.; Bartoli, E.; Schambelan, M. Effects of angiotensin II on insulin receptor binding and mRNA levels in normal and diabetic rats. Diabetologia 1997, 40, 770–777. [Google Scholar] [CrossRef]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and progression of kidney disease in type 2 diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Hou, Y.C.; Zheng, C.M.; Yen, T.H.; Lu, K.C. Molecular mechanisms of SGLT2 inhibitor on cardiorenal protection. Int. J. Mol. Sci. 2020, 21, 7833. [Google Scholar] [CrossRef]
- Hosokawa, Y.; Ogawa, W. SGLT2 inhibitors for genetic and acquired insulin resistance: Considerations for clinical use. J. Diabetes Investig. 2020, 11, 1431–1433. [Google Scholar] [CrossRef]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Fiorentino, T.V.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514. [Google Scholar] [CrossRef]
- Latva-Rasku, A.; Honka, M.-J.; Kullberg, J.; Mononen, N.; Lehtimaki, T.; Saltevo, J.; Kirjavainen, A.K.; Saunavaara, V.; Iozzo, P.; Johansson, L.; et al. The SGLT2 inhibitor dapagliflozin reduces liver fat but does not affect insulin sensitivity: A randomized, double-blind, placebo-controlled study with 8-week treatment in type 2 diabetes patients. Diabetes Care 2019, 42, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Pereira-Moreira, R.; Seghieri, M.; Rebelos, E.; Souza, A.L.; Chueire, V.B.; Arvia, C.; Muscelli, E. Insulin enhances renal glucose excretion: Relation to insulin sensitivity and sodium-glucose transport. BMJ Open Diabetes Res. Care 2020, 8, e001178. [Google Scholar] [CrossRef] [PubMed]
- Nizar, J.M.; Shepard, B.D.; Vo, V.T.; Bhalla, V. Renal tubule insulin receptor modestly promotes elevated blood pressure and markedly stimulates glucose reabsorption. JCI Insight 2018, 3, e95107. [Google Scholar] [CrossRef] [PubMed]
- Welborn, T.A.; Breckenridge, A.; Rubinstein, A.H.; Dollery, C.T.; Fraser, T.R. Serum insulin in essential hypertension and in peripheral vascular disease. Lancet 1966, 1, 1136–1137. [Google Scholar] [CrossRef]
- Shen, D.-C.; Shieh, S.M.; Fuh, M.; Wu, D.-A.; Chen, Y.-D.I.; Reaven, G.M. Resistance to insulin stimulated glucose uptake in patients with hypertension. J. Clin. Endocrinol. Metab. 1988, 66, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Alberti, K.G.; Grundy, S.M.; Zimmer, P.Z. The metabolic syndrome. Lancet 2010, 375, 181–183. [Google Scholar] [CrossRef]
- Saad, M.F.; Lillioja, S.; Nyomba, B.L.; Castillo, C.; Ferraro, R.; DeGregorio, M.; Ravussin, E.; Knowler, W.C.; Bennett, P.H.; Havard, V.V.; et al. Racial difference in the relation between blood pressure and insulin resistance. New Engl. J. Med. 1991, 324, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Natali, A.; Capaldo, B.; Lehtovirta, M.; Jacob, S.; Yki Järvinen, H.; for the European Group for the Study of Insulin resistance (EGIR). Insulin resistance, hyperinsulinemia, and blood pressure. Role of age and obesity. Hypertension 1992, 30, 1144–1149. [Google Scholar] [CrossRef]
- Marigliano, A.; Tedde, R.; Sechi, L.A.; Pala, A.; Pisanu, G.; Pacifico, A. Insulinemia and blood pressure: Relationships in patients with primary and secondary hypertension, and with or without glucose metabolism impairment. Am. J. Hypertens. 1990, 3, 521–526. [Google Scholar] [CrossRef]
- Shamiss, A.; Carroll, J.; Rosenthall, T. Insulin resistance in secondary hypertension. Am. J. Hypertens. 1992, 5, 26–28. [Google Scholar] [CrossRef]
- Allemann, Y.; Horber, F.F.; Colombo, M.; Ferrari, P.; Shaw, S.; Jaeger, P.; Weidman, P. Insulin sensitivity and body fat distribution in normotensive offspring of hypertensive parents. Lancet 1993, 341, 327–331. [Google Scholar] [CrossRef]
- Beatyy, O.L.; Harper, R.; Sheridan, B.; Atkinson, A.B.; Bell, P.M. Insulin resistance in offspring of hypertensive parents. BMJ 1993, 307, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarfors, E.T.; Lithell, H.O.; Selinus, I. Risk factors for the development of hypertension: A 10-year longitudinal study in middle-aged men. J. Hypertens. 1991, 9, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lissner, L.; Bengtsson, C.; Lapidus, L.; Kristjansson, K.; Wedel, H. Fasting insulin in relation to subsequent blood pressure changes and hypertension in women. Hypertension 1992, 20, 797–801. [Google Scholar] [CrossRef]
- Taittonen, L.; Uhari, M.; Nuutinen, M.; Turtinen, J.; Pokka, T.; Akerblom, H.K. Insulin and blood pressure among healthy children. Am. J. Hypertens. 1996, 9, 193–199. [Google Scholar] [CrossRef]
- Meigs, J.B. Invited commentary: Insulin resistance syndrome? Syndrome X? A syndrome at all? Factor analysis reveals patterns in the fabric of correlated metabolic risk factors. Am. J. Epidemiol. 2000, 152, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Zavaroni, I.; Mazza, S.; Dall’Aglio, E.; Gasparini, P.; Passeri, M.; Reaven, G.M. Prevalence of hyperinsulinemia in patients with high blood pressure. J. Intern. Med. 1992, 231, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.L.; Liess, C.; Poljak, A.; Xu, A.; Zhang, J.; Thoma, C.; Trenell, M.; Milner, B.; Jenkins, A.B.; Chisholm, D.J.; et al. Phenotypic characterization of insulin-resistant and insulin-sensitive obesity. J. Clin. Endocrinol. Metab. 2015, 100, 4082–4091. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Bakris, G.L. The antinatriuretic effect of insulin: And unappreciated mechanism for hypertension associated with insulin resistance. Am. J. Nephrol. 2007, 27, 44–54. [Google Scholar] [CrossRef]
- Shi, Z.; Cassaglia, P.A.; Pelletier, N.E.; Brooks, V.L. Sex differences in the sympathoexcitatory response to insulin in obese rats: Role of neuropeptide Y. J. Physiol. 2019, 597, 1757–1775. [Google Scholar] [CrossRef]
- He, F.J.; Tan, M.; Ma, Y.; MacGregor, G.A. Salt reduction to prevent hypertension and cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2020, 75, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Balafa, O.; Kalaitzidis, R.G. Salt sensitivity and hypertension. J. Hum. Hypertens. 2021, 35, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Ertuglu, L.A.; Elijovic, F.; Laffer, C.L.; Kirabo, A. Salt-sensitivity of blood pressure and insulin resistance. Front. Physiol. 2021, 12, 793924. [Google Scholar] [CrossRef] [PubMed]
- Tedde, R.; Sechi, L.A.; Marigliano, A.; Pala, A.; Scano, L. Antihypertensive effect of insulin reduction in diabetic-hypertensive patients. Am. J. Hypertens. 1989, 2, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Rocchini, A.P.; Katch, V.; Kveselis, D.; Moorehead, C.; Martin, M.; Lampman, R.; Gregory, M. Insulin and renal sodium retention in obese adolescents. Hypertension 1989, 14, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Natali, A.; Quinones, G.A.; Santoro, D.; Percori, N.; Taddei, S.; Salvetti, A.; Ferrannini, E. Relationship between insulin release, antinatriuresis and hypokalemia after glucose ingestion in normal and hypertensive men. Clin. Sci. 1993, 85, 327–335. [Google Scholar] [CrossRef]
- Muscelli, E.; Natali, A.; Bianchi, S.; Bigazzi, R.; Galvan, A.Q.; Sironi, A.M.; Frascerra, S.; Ciociaro, D.; Ferrannini, E. Effect of insulin on renal sodium and uric acid handling in essential hypertension. Am. J. Hypertens. 1996, 9, 746–752. [Google Scholar] [CrossRef]
- Nosadini, R.; Sambataro, M.; Thomaseth, K.; Pacini, G.; Cipollina, M.R.; Brocco, E.; Solini, A.; Carraro, A.; Velussi, M.; Frigato, F. Role of hyperglycemia and insulin resistance in determining sodium retention in non-insulin-dependent diabetes. Kidney Int. 1993, 44, 139–146. [Google Scholar] [CrossRef]
- Sechi, L.A. Mechanisms of insulin resistance in rat models of hypertension and their relationships with salt sensitivity. J. Hypertens. 1999, 17, 1229–1237. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Bartoli, E. Molecular mechanisms of insulin resistance in arterial hypertension. Blood Pressure 1996, 5 (Suppl. 1), 47–54. [Google Scholar]
- Reaven, G.M.; Chang, H. Relationship between blood pressure, plasma insulin and triglyceride concentration, and insulin action in SHR and WKY rats. Am. J. Hypertens. 1991, 4, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nessa, N.; Toba, H.; Kobara, M.; Nakata, T. Angelica acutiloba exerts antihypertensive effect and improves insulin resistance in spontaneously hypertensive rats fed with high-fat diet. Pharmacology 2022, 107, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, D.A.; Zhu, S.; Wyss, J.M.; Oparil, S. Diurnal blood pressure variation and dietary salt in spontaneously hypertensive rats. Hypertension 1994, 24, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaven, G.M.; Chang, H.; Hoffman, B.B.; Azhar, S. Resistance to insulin-stimulated glucose uptake in adipocytes isolated from spontaneously hypertensive rats. Diabetes 1989, 38, 1155–1160. [Google Scholar] [CrossRef]
- Paternostro, G.; Clarke, K.; Heath, J.; Seymour, A.M.; Radda, G.K. Decreased GLUT-4 mRNA content and insulin-sensitive deoxyglucose uptake show insulin resistance in the hypertensive rat heart. Cardiovasc. Res. 1995, 30, 205–2011. [Google Scholar] [CrossRef]
- Kahan, C.R.; Saad, M.J.A. Alterations in insulin receptor and substrate phosphorylation in hypertensive rats. J. Am. Soc. Nephrol. 1992, 3 (Suppl. 1), S69–S77. [Google Scholar] [CrossRef]
- Farrace, S.; Frontoni, S.; Gambardella, S.; Menzinger, G.; Rossetti, L. Glycogen synthase activity in two rat models of hypertension. Am. J. Hypertens. 1995, 8, 949–953. [Google Scholar] [CrossRef]
- Xing, W.; Yan, W.; Liu, P.; Ji, L.; Li, Y.; Sun, L.; Tao, L.; Zhang, H.; Gao, F. A novel mechanism for vascular insulin resistance in normotensive young SHRs: Hypoadiponectinemia and resultant APPL1 downregulation. Hypertension 2013, 61, 1028–1035. [Google Scholar] [CrossRef]
- Pravenec, M.; Kozich, V.; Krijt, J.; Sokolova, J.; Zidek, V.; Landa, V.; Simakova, M.; Mlejnek, P.; Silhavy, J.; Oliyarnyk, O.; et al. Folate deficiency is associated with oxidative stress, increased blood pressure, and insulin resistance in spontaneously hypertensive rats. Am. J. Hypertens. 2013, 26, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Leguisamo, N.M.; Lehnen, A.M.; Machado, U.F.; Okamoto, M.M.; Markoski, M.M.; Pinto, G.H.; Schaan, B.D. GLUT4 contents decreases along with insulin resistance and high levels of inflammatory markers in rats with metabolic syndrome. Cardiovasc. Diabetol. 2012, 11, 100. [Google Scholar] [CrossRef]
- Ou, H.Y.; Wu, H.T.; Yang, Y.C.; Wu, J.S.; Cheng, J.T.; Chang, C.J. Elevated retinol binding protein 4 contributes to insulin resistance in spontaneously hypertensive rats. Horm. Metab. Res. 2011, 43, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Bosse, J.D.; Lin, H.Y.; Sloan, C.; Zhang, Q.-J.; Abel, E.D.; Pereira, T.J.; Dolinsky, V.W.; Symons, J.D.; Jalili, T. A low-carbohydrate/high-fat diet reduces blood pressure in spontaneously hypertensive rats without deleterious changes in insulin resistance. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1733–H1742. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, H.; Pang, Z.; Shimosato, T.; Moritani, T.; Kurinami, H.; Koriyama, H.; Tenma, A.; Shimamura, M.; Morishita, R. The dipeptidyl peptidase-4 inhibitor teneligliptin improved endothelial dysfunction and insulin resistance in the SHR/NDmcr-cp rat model of metabolic syndrome. Hypertens. Res. 2014, 37, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, H.; Ushijima, K.; Arakawa, Y.; Aizawa, K.-I.; Fujimura, A. Effects of telmisartan and olmesartan on insulin sensitivity and renal function in spontaneously hypertensive rats fed a high fat diet. Pharmacol. Sci. 2016, 131, 190–197. [Google Scholar] [CrossRef]
- Sechi, L.A.; Griffin, C.A.; Sechi, G.; De Carli, S.; Bartoli, E. Expression of the insulin receptor gene in liver and kidney of rats with genetic hypertension. J. Hypertens. 1993, 11 (Suppl.5), S68–S69. [Google Scholar] [CrossRef]
- Sechi, L.A.; Griffin, C.A.; Giacchetti, G.; Zingaro, L.; Catena, C.; Bartoli, E.; Schambelan, M. Abnormalities of insulin receptors in spontaneously hypertensive rats. Hypertension 1996, 27, 955–961. [Google Scholar] [CrossRef]
- Kotchen, T.A.; Zhang, H.Y.; Covelli, M.; Blehschmidt, N. Insulin resistance and blood pressure in Dahl rats and in one-kidney, one-clip hypertensive rats. Am. J. Physiol. 1991, 261, E692–E697. [Google Scholar] [CrossRef]
- Shehata, M.F. Genetic and dietary salt contributors to insulin resistance in Dahl salt-sensitive (S) rats. Cardiovasc. Diabetol. 2008, 7, 7. [Google Scholar] [CrossRef]
- Tomiyama, H.; Kushiro, T.; Abeta, H.; Kurumatrani, H.; Taguchi, H.; Kuga, N.; Saito, F.; Kobayashi, F.; Otsuka, Y.; Kanmatsuse, K. Blood pressure response to hyperinsulinemia in salt-sensitive and salt-resistant rats. Hypertension 1992, 20, 596–600. [Google Scholar] [CrossRef]
- Somova, L.; Channa, M.L. Glucose metabolism and insulin sensitivity in Dahl hypertensive rats. Methods Find. Exp. Clin. Pharmacol. 1999, 21, 421–425. [Google Scholar] [CrossRef]
- Hattori, T.; Murase, T.; Takatsu, M.; Nagasawa, K.; Matsuura, N.; Watanabe, S.; Murohara, T.; Nagata, K. Dietary salt restriction improves cardiac and adipose tissue pathology independently of obesity in a rat model of metabolic syndrome. J. Am. Heart Ass. 2014, 3, e001312. [Google Scholar] [CrossRef] [PubMed]
- Sechi, L.A.; Griffin, C.A.; Zingaro, L.; Catena, C.; De Carli, S.; Schambelan, M.; Bartoli, E. Glucose metabolism and insulin receptor binding and mRNA levels in tissues of Dahl hypertensive rats. Am. J. Hypertens. 1997, 10, 1223–1230. [Google Scholar] [CrossRef]
- Ogihara, T.; Asano, T.; Ando, K.; Sakoda, H.; Anai, M.; Shojima, N.; Ono, H.; Onishi, Y.; Fujishiro, M.; Abe, M.; et al. High-salt diet enhances insulin signaling and induces insulin resistance in Dahl salt-sensitive rats. Hypertension 2002, 40, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.-S.; Ho, H.; Hoffman, B.B.; Reaven, G.M. Fructose-induced insulin resistance and hypertension in rats. Hypertension 1987, 10, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.T.; Yuen, V.G.; McNeill, J.H. The fructose-fed rat: A review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol. Cel. Biochem. 2009, 332, 145–159. [Google Scholar] [CrossRef]
- Tobey, T.A.; Mondon, C.E.; Zavaroni, I.; Reaven, G.M. Mechanisms of insulin resistance in fructose-fed rats. Metabolism 1982, 31, 608–612. [Google Scholar] [CrossRef]
- Catena, C.; Giacchetti, G.; Novello, M.; Colussi, G.; Cavarape, A.; Sechi, L.A. Cellular mechanisms of insulin resistance in rats with fructose induced hypertension. Am. J. Hypertens. 2003, 16, 973–978. [Google Scholar] [CrossRef]
- Deutsch, D.D.; Jen, K.L.; Grunberger, G. Regulation of hepatic insulin receptor tyrosine kinase in rat models of mild insulin resistance. J. Lab. Clin. Med. 1993, 122, 421–425. [Google Scholar]
- Bezerra, R.M.; Ueno, M.; Silva, M.S.; Tavares, D.Q.; Carvalho, C.R.; Saad, M.J. A high fructose diet affects the early steps of insulin action in muscle and liver of rats. J. Nutr. 2000, 130, 1531–1535. [Google Scholar] [CrossRef]
- Johnson, M.D.; Zhang, H.Y.; Kotchen, T.A. Sucrose does not raise blood pressure in rats maintained on a low-salt intake. Hypertension 1993, 21, 779–785. [Google Scholar] [CrossRef]
- Catena, C.; Cavarape, A.; Novello, M.; Giacchetti, G.; Sechi, L.A. Insulin receptors and renal sodium handling in hypertensive fructose-fed rats. Kidney Int. 2003, 64, 2163–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| WKY | SHR | ||
|---|---|---|---|
| Low-sodium | High-sodium | Low-sodium | High-sodium |
| 6.9 ± 1.0 | 4.5 ± 0.7 * | 6.5 ± 1.2 | 6.4 ± 1.1 |
| DSR | DSS | ||
| Low-sodium | High-sodium | Low-sodium | High-sodium |
| 7.5 ± 1.0 | 5.9 ± 1.6 | 6.3 ± 1.3 | 6.8 ± 1.6 |
| CTR | FFR | ||
| Low-sodium | High-sodium | Low-sodium | High-sodium |
| 6.5 ± 1.2 | 3.8 ± 1.7 * | 6.4 ± 1.1 | 6.5 ± 1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brosolo, G.; Da Porto, A.; Bulfone, L.; Vacca, A.; Bertin, N.; Scandolin, L.; Catena, C.; Sechi, L.A. Insulin Resistance and High Blood Pressure: Mechanistic Insight on the Role of the Kidney. Biomedicines 2022, 10, 2374. https://doi.org/10.3390/biomedicines10102374
Brosolo G, Da Porto A, Bulfone L, Vacca A, Bertin N, Scandolin L, Catena C, Sechi LA. Insulin Resistance and High Blood Pressure: Mechanistic Insight on the Role of the Kidney. Biomedicines. 2022; 10(10):2374. https://doi.org/10.3390/biomedicines10102374
Chicago/Turabian StyleBrosolo, Gabriele, Andrea Da Porto, Luca Bulfone, Antonio Vacca, Nicole Bertin, Laura Scandolin, Cristiana Catena, and Leonardo A. Sechi. 2022. "Insulin Resistance and High Blood Pressure: Mechanistic Insight on the Role of the Kidney" Biomedicines 10, no. 10: 2374. https://doi.org/10.3390/biomedicines10102374