
Re-Definition of the Epidemiology of Cardiac Amyloidosis

 ,
, 
 , , ,
, , ,
Abstract
:1. Introduction
2. Unveiling the Real Prevalence of CA: Reports from Scintigraphy with Bone Tracers Performed for Non Cardiac Reasons and Autopsies
3. Prevalence of CA in Different At-Risk Scenarios
- Carpal tunnel syndrome and CA
- b.
- Heart Failure and CA
- c.
- Aortic stenosis and CA
- d.
- Hypertrophic Cardiomyopathy misdiagnosis
4. Challenges in Diagnosis and Treatment in CA
- Do not forget al. Cardiac Amyloidosis
- b.
- When chronic inflammatory or infectious diseases causes Amyloidosis: Amyloid A (AA) Amyloidosis
- c.
- What is the role of endomyocardial biopsy?
- d.
- Impact of the advances in epidemiology on the recognition and diagnostic work-up of patients at suspicion of CA

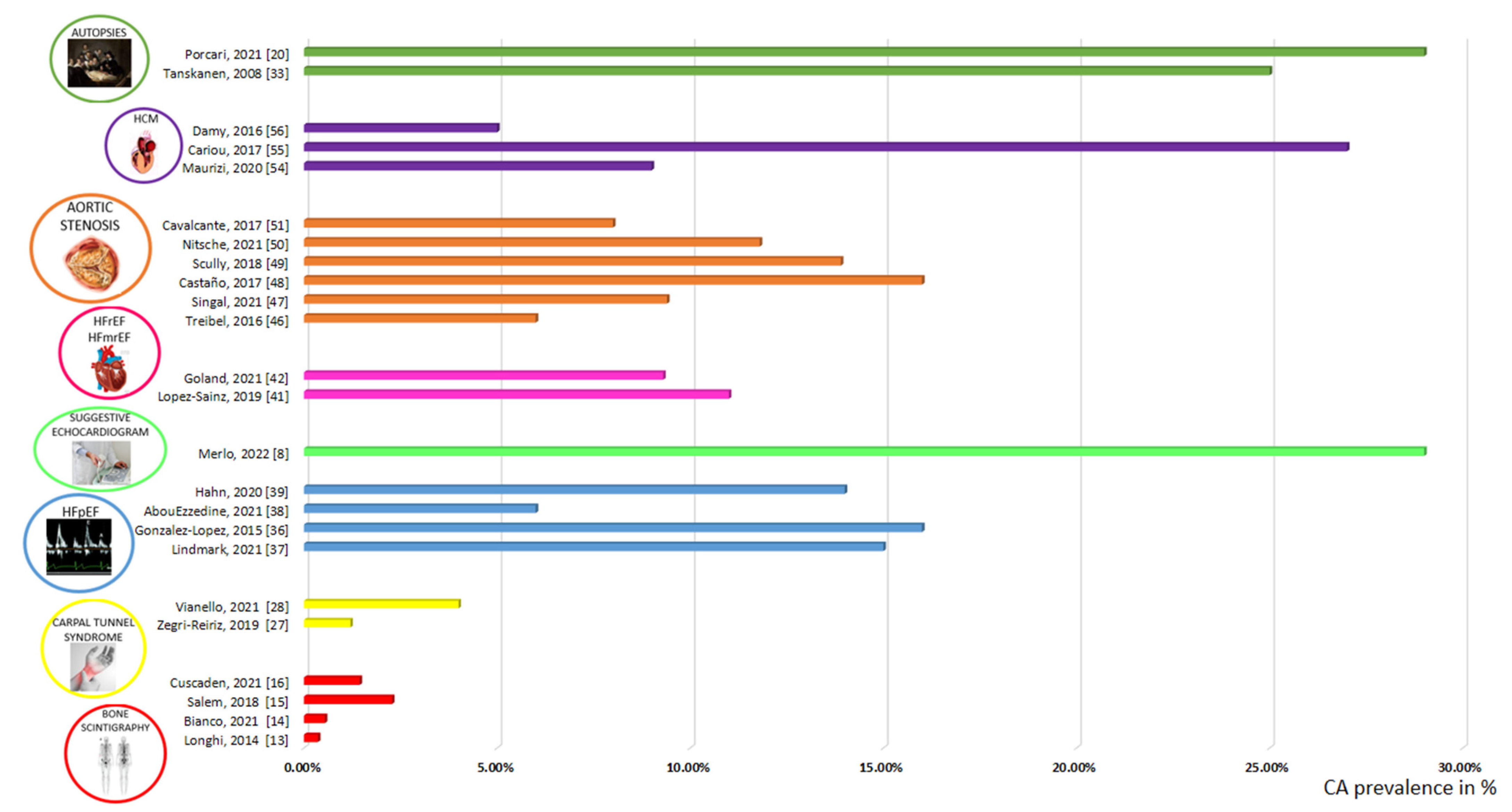{kind=link}
| Authors | Year | Indication to Screening | Setting | Population (n) | Mean/ Median Age | CA Prevalence (%) | Diagnostic Algorithm |
|---|---|---|---|---|---|---|---|
| Longhi S. et al. [13] | 2014 | Bone scan for non-cardiac reasons | Scintigraphy with bone tracers | 12,400 | 74 | 0,4 | Scintigraphy with bone tracers (EKG, echocardiography, EMB in selected patients with scintigraphy+) |
| Bianco M. et al. [14] | 2021 | Bone scan for any reasons | Scintigraphy with bone tracers | 4228 | N/A | 0,5 | Scintigraphy with bone tracers |
| Mohamed-Salem L. et al. [15] | 2018 | Bone scan for non-cardiac reasons, ≥75 years | Scintigraphy with bone tracers | 1114 | 81 | 2,8 | Scintigraphy with bone tracers |
| Cuscaden C. et al. [16] | 2021 | Bone scan for non-cardiac reasons | Scintigraphy with bone tracers | 6918 | N/A | 0,2 | Scintigraphy with bone tracers |
| Zegri-Reiriz I. et al. [27] | 2019 | CTS surgery, ≥60 years, LV wall thickness ≥ 12 mm | CTS | 101 | 69 | 3 | Scintigraphy with bone tracers, monoclonal protein, biopsy when needed |
| Vianello PF. et al. [28] | 2021 | Bilateral CTS surgery in male patients | CTS | 53 | 73 | 4 | Scintigraphy with bone tracers, monoclonal protein |
| Tanskanen M. et al. [33] | 2008 | Autopsy > 85 years | Autopsy | 256 | N/A | 25 | Histology |
| Mohammed S. et al. [34] | 2014 | Autopsy in HFpEF patients/control subjects | Autopsy/HFpEF | 109 (HFpEF)/131 (Control) | 76 (HFpEF)/69 (Control) | 17 (HFpEF)/5 (Control) | Histology |
| Porcari A. et al. [20] | 2021 | Autopsy ≥ 75 years | Autopsy | 56 | 86 | 43 (diffuse Amyloidosis in the LV: 29%) | Histology |
| Gonzalez-Lopez E. et al. [36] | 2015 | HF hospitalization, ≥60 years, LV wall thickness ≥ 12 mm | HFpEF | 120 | 86 | 13 | Scintigraphy, monoclonal protein, biopsy when needed |
| Lindmark K. et al. [37] | 2021 | HF clinic, LV wall thickness ≥14 mm | HFpEF | 86 | 77 | 15 | Scintigraphy, monoclonal protein, biopsy when needed |
| AbouEzze ddine OF. et al. [38] | 2021 | HF, LVEF ≥ 40%, LV wall thickness ≥ 12 mm, ≥60 years | HFpEF | 286 | 78 | 6 | Scintigraphy, monoclonal protein, biopsy when needed |
| Hahn VS. et al. [39] | 2020 | HFpEF | HFpEF | 108 | 66 | 14 | Histology (EMB) |
| Merlo M. et al. [8] | 2022 | LV wall thickness ≥ 12 mm, LVEDVi ≤ 85 mL/m2, LVEF ≥ 50%, ≥55 years, at least 1 echocardiographic red flag of CA | HFpEF/LVH | 217 | 75 | 29 (Apical sparing or a combination of ≥2 other echocardiographic red flags, excluding interatrial septum thickness, provided a diagnostic accuracy > 70%) | Scintigraphy, monoclonal protein, extra-cardiac histology, genetic test. CMR and cardiac histology (EMB) in selected patients, if needed. |
| Dungu JN [40] | 2016 | Afro-Caribbeans patients admitted with HF | HF | 211 | 71 | 11 (8,5% with Val122Ile mutation) | CMR, scintigraphy, monoclonal protein, biopsy when needed, genetic test |
| Lopez-Sainz A. et al. [41] | 2019 | HF hospitalization, LVEF < 50%, >60 years, LV wall thickness ≥ 12 mm | HFrEF/HF mrEF | 28 | 78 | 11 | Scintigraphy, monoclonal protein, biopsy when needed |
| Goland S. et al. [42] | 2021 | Unexplained LV systolic dysfunction | HFrEF/HF mrEF | 75 | 65 | 9 | Scintigraphy, monoclonal protein, biopsy when needed |
| Treibel TA. et al. [46] | 2016 | AS referred to SAVR, >65 years | AS | 146 | 71 | 4 | Histology (intraoperative biopsy) |
| Singal AK. et al. [47] | 2021 | AS referred to SAVR, >65 years | AS | 32 | 70 | 9 (no Amyloid in IVS biopsy, 72% in the aortic valve) | Scintigraphy, histology |
| Castano A. et al. [48] | 2017 | AS referred to TAVR | AS | 151 | 84 | 16 | Scintigraphy, monoclonal protein |
| Scully P. et al. [49] | 2018 | AS referred to TAVR, >75 years | AS | 101 | 86 | 14 | Scintigraphy, monoclonal protein |
| Nitsche C. et al. [50] | 2021 | AS referred to TAVR | AS | 407 | 83 | 12 | Scintigraphy, monoclonal protein |
| Cavalcante JL. et al. [51] | 2017 | Moderate/severe AS referred to CMR | AS | 113 | 70 | 8 | CMR (suspected CA) |
| Maurizi N. et al. [54] | 2019 | Initial diagnosis of HCM | HCM | 343 | 60 | 9 | Genetic test, if no TTR mutations but ≥1 CA red flag, monoclonal protein, abdominal fatbiopsy and/or scintigraphy and ApoAI sequencing |
| Cariou E. et al. [55] | 2017 | LV wall thickness ≥12 mm | HCM | 114 | 72 | 27 | Scintigraphy, monoclonal protein, CMR |
| Damy T. et al. [56] | 2016 | Initial diagnosis of HCM | HCM | 298 | 62 | 5 | TTR gene testing, then scintigraphy, CMR, biopsy |
| Helder MRK. et al. [59] | 2014 | HCM underwent septal myectomy | HCM | 1714 | N/A | 1 | Histology |

5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Merlini, G.; Bellotti, V. Molecular Mechanisms of Amyloidosis. N. Engl. J. Med. 2003, 349, 583–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcari, A.; Merlo, M.; Rapezzi, C.; Sinagra, G. Transthyretin amyloid cardiomyopathy: An uncharted territory awaiting discovery. Eur. J. Intern. Med. 2020, 82, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canepa, M.; Vianello, P.F.; Porcari, A.; Merlo, M.; Scarpa, M. Cardiac amyloidosis: A changing epidemiology with open challenges. Vessel Plus 2022, 6, 30. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef]
- Koike, H.; Okumura, T.; Murohara, T.; Katsuno, M. Multidisciplinary Approaches for Transthyretin Amyloidosis. Cardiol. Ther. 2021, 10, 289–311. [Google Scholar] [CrossRef]
- Merlo, M.; Porcari, A.; Pagura, L.; Cameli, M.; Vergaro, G.; Musumeci, B.; Biagini, E.; Canepa, M.; Crotti, L.; Imazio, M.; et al. A national survey on prevalence of possible echocardiographic red flags of amyloid cardiomyopathy in consecutive patients undergoing routine echocardiography: Study design and patients characterization-the first insight from the AC-TIVE Study. Eur. J. Prev. Cardiol. 2021, 29, e173–e177. [Google Scholar] [CrossRef]
- Merlo, M.; Pagura, L.; Porcari, A.; Cameli, M.; Vergaro, G.; Musumeci, B.; Biagini, E.; Canepa, M.; Crotti, L.; Imazio, M.; et al. Unmasking the Prevalence of Amyloid Cardiomyopathy in the Real World: Results from Phase 2 of AC-TIVE Study, an Italian Nationwide Survey. Eur. J. Heart Fail. 2022, 24. [Google Scholar] [CrossRef]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N. Engl. J. Med. 2018, 379, 1007–1016. [Google Scholar] [CrossRef]
- Coelho, T.; Maurer, M.S.; Suhr, O.B. THAOS–The Transthyretin Amyloidosis Outcomes Survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin. 2012, 29, 63–76. [Google Scholar] [CrossRef]
- Pozsonyi, Z.; Peskó, G.; Takács, H.; Csuka, D.; Nagy, V.; Szilágyi, Á.; Hategan, L.; Muk, B.; Csányi, B.; Nyolczas, N.; et al. Variant Transthyretin Amyloidosis (ATTRv) in Hungary: First Data on Epidemiology and Clinical Features. Genes 2021, 12, 1152. [Google Scholar] [CrossRef]
- Porcari, A.; Pagura, L.; Varrà, G.G.; Rossi, M.; Longo, F.; Saro, R.; Barbisan, D.; Cittar, M.; Rapezzi, C.; Merlo, M. Grey zones in the supportive treatments of cardiac amyloidosis. Vessel Plus 2022, 6, 33. [Google Scholar] [CrossRef]
- Longhi, S.; Guidalotti, P.L.; Quarta, C.C.; Gagliardi, C.; Milandri, A.; Lorenzini, M.; Potena, L.; Leone, O.; Bartolomei, I.; Pastorelli, F.; et al. Identification of TTR-related subclinical amyloidosis with 99mTc-DPD scintigraphy. JACC Cardiovasc. Imaging 2014, 7, 531–532. [Google Scholar] [CrossRef] [Green Version]
- Bianco, M.; Parente, A.; Biolè, C.; Righetti, C.; Spirito, A.; Luciano, A.; Destefanis, P.; Nangeroni, G.; Angusti, T.; Anselmino, M.; et al. The prevalence of TTR cardiac amyloidosis among patients undergoing bone scintigraphy. J. Nucl. Cardiol. 2021, 28, 825–830. [Google Scholar] [CrossRef]
- Mohamed-Salem, L.; Santos-Mateo, J.J.; Sanchez-Serna, J.; Hernández-Vicente, Á.; Reyes-Marle, R.; Sánchez, M.I.C.; Claver-Valderas, M.A.; Vioque, E.G.; Moral, F.J.H.-D.; García-Pavía, P.; et al. Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int. J. Cardiol. 2018, 270, 192–196. [Google Scholar] [CrossRef]
- Cuscaden, C.; Ramsay, S.C.; Prasad, S.; Goodwin, B.; Smith, J. Estimation of prevalence of transthyretin (ATTR) cardiac amyloidosis in an Australian subpopulation using bone scans with echocardiography and clinical correlation. J. Nucl. Cardiol. 2021, 28, 2845–2856. [Google Scholar] [CrossRef]
- Dorbala, S.; Ando, Y.; Bokhari, S.; Dispenzieri, A.; Falk, R.H.; Ferrari, V.A.; Fontana, M.; Gheysens, O.; Gillmore, J.D.; Glaudemans, A.W.J.M.; et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: Part 1 of 2-evidence base and standardized methods of imaging. J. Nucl. Cardiol. 2019, 26, 2065–2123. [Google Scholar] [CrossRef]
- Porcari, A.; Rossi, M.; Dore, F.; Imazio, M.; Fontana, M.; Merlo, M.; Sinagra, G. I dieci punti che il cardiologo deve conoscere su scintigrafia miocardica con traccianti ossei, amiloidosi e cuore. G Ital. Cardiol. 2022, 23, 424–432. [Google Scholar]
- Mattana, F.; Muraglia, L.; Girardi, F.; Cerio, I.; Porcari, A.; Dore, F.; Bonfiglioli, R.; Fanti, S. Clinical application of cardiac scintigraphy with bone tracers: Controversies and pitfalls in cardiac amyloidosis. Vessel Plus 2022, 6, 13. [Google Scholar] [CrossRef]
- Porcari, A.; Bussani, R.; Merlo, M.; Varrà, G.G.; Pagura, L.; Rozze, D.; Sinagra, G. Incidence and Characterization of Concealed Cardiac Amyloidosis Among Unselected Elderly Patients Undergoing Post-mortem Examination. Front. Cardiovasc. Med. 2021, 8, 749523. [Google Scholar] [CrossRef]
- Pourmemari, M.H.; Heliövaara, M.; Viikari-Juntura, E.; Shiri, R. Carpal tunnel release: Lifetime prevalence, annual incidence, and risk factors. Muscle Nerve 2018, 58, 497–502. [Google Scholar] [CrossRef]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Siepen, F.A.D.; Hein, S.; Prestel, S.; Baumgärtner, C.; Schönland, S.; Hegenbart, U.; Röcken, C.; Katus, H.A.; Kristen, A.V. Carpal tunnel syndrome and spinal canal stenosis: Harbingers of transthyretin amyloid cardiomyopathy? Clin. Res. Cardiol. 2019, 108, 1324–1330. [Google Scholar] [CrossRef]
- Maurer, M.S.; Hanna, M.; Grogan, M.; Dispenzieri, A.; Witteles, R.; Drachman, B.; Judge, D.P.; Lenihan, D.J.; Gottlieb, S.S.; Shah, S.J.; et al. Genotype and Phenotype of Transthyretin Cardiac Amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J. Am. Coll. Cardiol. 2016, 68, 161–172. [Google Scholar] [CrossRef]
- Fosbøl, E.; Rørth, R.; Leicht, B.P.; Schou, M.; Maurer, M.S.; Kristensen, S.L.; Kober, L.; Gustafsson, F. Association of Carpal Tunnel Syndrome With Amyloidosis, Heart Failure, and Adverse Cardiovascular Outcomes. J. Am. Coll. Cardiol. 2019, 74, 15–23. [Google Scholar] [CrossRef]
- Porcari, A.; Pagura, L.; Longo, F.; Sfriso, E.; Barbati, G.; Murena, L.; Longo, E.; Ramella, V.; Arnež, Z.M.; Rapezzi, C.; et al. Prognostic significance of unexplained left ventricular hypertrophy in patients undergoing carpal tunnel surgery. ESC Heart Fail. 2022, 9, 751–760. [Google Scholar] [CrossRef]
- Zegri-Reiriz, I.; Moral, F.J.D.H.-D.; Dominguez, F.; Salas, C.; de la Cuadra, P.; Plaza, A.; Krsnik, I.; Gonzalez-Lopez, E.; Garcia-Pavia, P. Prevalence of Cardiac Amyloidosis in Patients with Carpal Tunnel Syndrome. J. Cardiovasc. Transl. Res. 2019, 12, 507–513. [Google Scholar] [CrossRef]
- Vianello, P.F.; La Malfa, G.; Tini, G.; Mazzola, V.; Miceli, A.; Santolini, E.; Briano, S.; Porto, I.; Canepa, M. Prevalence of transthyretin amyloid cardiomyopathy in male patients who underwent bilateral carpal tunnel surgery: The ACTUAL study. Int. J. Cardiol. 2021, 329, 144–147. [Google Scholar] [CrossRef]
- Milandri, A.; Farioli, A.; Gagliardi, C.; Longhi, S.; Salvi, F.; Curti, S.; Foffi, S.; Caponetti, A.G.; Lorenzini, M.; Ferlini, A.; et al. Carpal tunnel syndrome in cardiac amyloidosis: Implications for early diagnosis and prognostic role across the spectrum of aetiologies. Eur. J. Heart Fail. 2020, 22, 507–515. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of heart failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Porcari, A.; Falco, L.; Lio, V.; Merlo, M.; Fabris, E.; Bussani, R.; Sinagra, G. Cardiac amyloidosis: Do not forget to look for it. Eur. Heart J. 2020, 22, H142–H147. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.-L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Mirzoyev, S.A.; Edwards, W.D.; Dogan, A.; Grogan, D.R.; Dunlay, S.M.; Roger, V.L.; Gertz, M.A.; Dispenzieri, A.; Zeldenrust, S.R.; et al. Left Ventricular Amyloid Deposition in Patients with Heart Failure and Preserved Ejection Fraction. JACC Heart Fail. 2014, 2, 113. [Google Scholar] [CrossRef]
- Gilstrap, L.G.; Dominici, F.; Wang, Y.; El-Sady, M.S.; Singh, A.; Di Carli, M.F.; Falk, R.H.; Dorbala, S. Epidemiology of Cardiac Amyloidosis Associated Heart Failure Hospitalizations Among Fee-for-Service Medicare Beneficiaries in the United States. Circ. Heart Fail. 2019, 12, e005407. [Google Scholar] [CrossRef]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Lindmark, K.; Pilebro, B.; Sundström, T.; Lindqvist, P. Prevalence of wild type transtyrethin cardiac amyloidosis in a heart failure clinic. ESC Heart Fail. 2021, 8, 745. [Google Scholar] [CrossRef]
- AbouEzzeddine, O.F.; Davies, D.R.; Scott, C.G.; Fayyaz, A.U.; Askew, J.W.; McKie, P.M.; Noseworthy, P.A.; Johnson, G.B.; Dunlay, S.M.; Borlaug, B.A.; et al. Prevalence of Transthyretin Amyloid Cardiomyopathy in Heart Failure With Preserved Ejection Fraction. JAMA Cardiol. 2021, 6, 1267. [Google Scholar] [CrossRef]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure with Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712. [Google Scholar] [CrossRef]
- Dungu, J.N.; Papadopoulou, S.A.; Wykes, K.; Mahmood, I.; Marshall, J.; Valencia, O.; Fontana, M.; Whelan, C.J.; Gillmore, J.D.; Hawkins, P.N.; et al. Afro-Caribbean Heart Failure in the United Kingdom: Cause, Outcomes, and ATTR V122I Cardiac Amyloidosis. Circ. Heart Fail. 2016, 9, e003352. [Google Scholar] [CrossRef]
- Sainz, A.L.; Moral, F.J.D.H.-D.; Dominguez, F.; Restrepo-Cordoba, A.; Amor-Salamanca, A.; Hernandez-Hernandez, A.; Ruiz-Guerrero, L.; Krsnik, I.; Cobo-Marcos, M.; Castro, V.; et al. Prevalence of cardiac amyloidosis among elderly patients with systolic heart failure or conduction disorders. Amyloid 2019, 26, 156–163. [Google Scholar] [CrossRef]
- Goland, S.; Volodarsky, I.; Fabricant, Y.; Livschitz, S.; Tshori, S.; Cuciuc, V.; Zilberman, L.; Fugenfirov, I.; Meledin, V.; Shimoni, S.; et al. Wild-type TTR amyloidosis among patients with unexplained heart failure and systolic LV dysfunction. PLoS ONE 2021, 16, e0254104. [Google Scholar] [CrossRef]
- Ternacle, J.; Krapf, L.; Mohty, D.; Magne, J.; Nguyen, A.; Galat, A.; Gallet, R.; Teiger, E.; Côté, N.; Clavel, M.-A.; et al. Aortic Stenosis and Cardiac Amyloidosis: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 2638–2651. [Google Scholar] [CrossRef]
- Kristen, A.V.; Schnabel, P.A.; Winter, B.; Helmke, B.M.; Longerich, T.; Hardt, S.; Koch, A.; Sack, F.-U.; Katus, H.A.; Linke, R.P.; et al. High prevalence of amyloid in 150 surgically removed heart valves--a comparison of histological and clinical data reveals a correlation to atheroinflammatory conditions. Cardiovasc. Pathol. 2010, 19, 228–235. [Google Scholar] [CrossRef]
- Audet, A.; Côté, N.; Couture, C.; Bossé, Y.; Després, J.-P.; Pibarot, P.; Mathieu, P. Amyloid substance within stenotic aortic valves promotes mineralization. Histopathology 2012, 61, 610–619. [Google Scholar] [CrossRef]
- Treibel, T.A.; Fontana, M.; Gilbertson, J.A.; Castelletti, S.; White, S.K.; Scully, P.R.; Roberts, N.; Hutt, D.F.; Rowczenio, D.M.; Whelan, C.J.; et al. Occult Transthyretin Cardiac Amyloid in Severe Calcific Aortic Stenosis: Prevalence and Prognosis in Patients Undergoing Surgical Aortic Valve Replacement. Circ. Cardiovasc. Imaging 2016, 9, e005066. [Google Scholar] [CrossRef] [Green Version]
- Singal, A.K.; Bansal, R.; Singh, A.; Dorbala, S.; Sharma, G.; Gupta, K.; Saxena, A.; Bhargava, B.; Karthikeyan, G.; Ramakrishnan, S.; et al. Concomitant Transthyretin Amyloidosis and Severe Aortic Stenosis in Elderly Indian Population: A Pilot Study. JACC CardioOncol. 2021, 3, 565–576. [Google Scholar] [CrossRef]
- Castaño, A.; Narotsky, D.L.; Hamid, N.; Khalique, O.K.; Morgenstern, R.; DeLuca, A.; Rubin, J.; Chiuzan, C.; Nazif, T.; Vahl, T.; et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 2017, 38, 2879–2887. [Google Scholar] [CrossRef] [Green Version]
- Scully, P.R.; Treibel, T.; Fontana, M.; Lloyd, G.; Mullen, M.; Pugliese, F.; Hartman, N.; Hawkins, P.N.; Menezes, L.; Moon, J.C. Prevalence of Cardiac Amyloidosis in Patients Referred for Transcatheter Aortic Valve Replacement. J. Am. Coll. Cardiol. 2018, 71, 463. [Google Scholar] [CrossRef]
- Nitsche, C.; Scully, P.R.; Patel, K.P.; Kammerlander, A.A.; Koschutnik, M.; Dona, C.; Wollenweber, T.; Ahmed, N.; Thornton, G.D.; Kelion, A.D.; et al. Prevalence and Outcomes of Concomitant Aortic Stenosis and Cardiac Amyloidosis. J. Am. Coll. Cardiol. 2021, 77, 128–139. [Google Scholar] [CrossRef]
- Cavalcante, J.L.; Rijal, S.; Abdelkarim, I.; Althouse, A.D.; Sharbaugh, M.S.; Fridman, Y.; Soman, P.; Forman, D.E.; Schindler, J.T.; Gleason, T.G.; et al. Cardiac amyloidosis is prevalent in older patients with aortic stenosis and carries worse prognosis. J. Cardiovasc. Magn. Reson. 2017, 19, 98. [Google Scholar] [CrossRef] [Green Version]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872. [Google Scholar] [CrossRef]
- Canepa, M.; Fumagalli, C.; Tini, G.; Vincent-Tompkins, J.; Day, S.M.; Ashley, E.A.; Mazzarotto, F.; Ware, J.S.; Michels, M.; Jacoby, D.; et al. Temporal Trend of Age at Diagnosis in Hypertrophic Cardiomyopathy: An Analysis of the International Sarcomeric Human Cardiomyopathy Registry. Circ. Heart Fail. 2020, 13, e007230. [Google Scholar] [CrossRef]
- Maurizi, N.; Rella, V.; Fumagalli, C.; Salerno, S.; Castelletti, S.; Dagradi, F.; Torchio, M.; Marceca, A.; Meda, M.; Gasparini, M.; et al. Prevalence of cardiac amyloidosis among adult patients referred to tertiary centres with an initial diagnosis of hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 300, 191–195. [Google Scholar] [CrossRef]
- Cariou, E.; Smires, Y.B.; Victor, G.; Robin, G.; Ribes, D.; Pascal, P.; Petermann, A.; Fournier, P.; Faguer, S.; Roncalli, J.; et al. Diagnostic score for the detection of cardiac amyloidosis in patients with left ventricular hypertrophy and impact on prognosis. Amyloid 2017, 24, 101–109. [Google Scholar] [CrossRef]
- Damy, T.; Costes, B.; Hagege, A.A.; Donal, E.; Eicher, J.-C.; Slama, M.; Guellich, A.; Rappeneau, S.; Gueffet, J.-P.; Logeart, D.; et al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur. Heart J. 2016, 37, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.K.; Tajik, A.J.; Edwards, W.D.; Bresnahan, J.F.; Kyle, R.A. Dynamic left ventricular outflow tract obstruction in cardiac amyloidosis detected by continuous-wave Doppler echocardiography. Am. J. Cardiol. 1987, 59, 1008–1010. [Google Scholar] [CrossRef]
- Mookadam, F.; Haley, J.H.; Olson, L.J.; Cikes, M.; Mookadam, M. Dynamic left ventricular outflow tract obstruction in senile cardiac amyloidosis. Eur. J. Echocardiogr. 2006, 7, 465–468. [Google Scholar] [CrossRef] [Green Version]
- Helder, M.R.; Schaff, H.V.; Nishimura, R.A.; Gersh, B.J.; Dearani, J.A.; Ommen, S.R.; Mereuta, O.M.; Theis, J.D.; Dogan, A.; Edwards, W.D. Impact of Incidental Amyloidosis on the Prognosis of Patients With Hypertrophic Cardiomyopathy Undergoing Septal Myectomy for Left Ventricular Outflow Tract Obstruction. Am. J. Cardiol. 2014, 114, 1396–1399. [Google Scholar] [CrossRef]
- Falk, R.H.; Alexander, K.M.; Liao, R.; Dorbala, S. AL (Light-Chain) Cardiac Amyloidosis: A Review of Diagnosis and Therapy. J. Am. Coll. Cardiol. 2016, 68, 1323–1341. [Google Scholar] [CrossRef]
- Comenzo, R.L. Out, Out—Making Amyloid’s Candle Briefer. N. Engl. J. Med. 2015, 373, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Witteles, R.M.; Bokhari, S.; Damy, T.; Elliott, P.; Falk, R.H.; Fine, N.M.; Gospodinova, M.; Obici, L.; Rapezzi, C.; Garcia-Pavia, P. Screening for Transthyretin Amyloid Cardiomyopathy in Everyday Practice. JACC Heart Fail. 2019, 7, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Larson, D.R.; Kurtin, P.J.; Kumar, S.; Cerhan, J.R.; Therneau, T.M.; Rajkumar, S.V.; Vachon, C.M.; Dispenzieri, A. Incidence of AL Amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin. Proc. 2019, 94, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Nardi, G.; Del Monaco, G.; Allinovi, M.; Gabriele, M.; Zocchi, C.; Casagrande, S.; Fumagalli, C.; Di Mario, C.; Olivotto, I.; et al. Changes in the perceived epidemiology of amyloidosis: 20 year-experience from a Tertiary Referral Centre in Tuscany. Int. J. Cardiol. 2021, 335, 123–127. [Google Scholar] [CrossRef]
- Tini, G.; Cappelli, F.; Biagini, E.; Musumeci, B.; Merlo, M.; Crotti, L.; Cameli, M.; Di Bella, G.; Cipriani, A.; Marzo, F.; et al. Current patterns of beta-blocker prescription in cardiac amyloidosis: An Italian nationwide survey. ESC Heart Fail. 2021, 8, 3369–3374. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and treatment of cardiac amyloidosis: A position statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef]
- Palladini, G.; Russo, P.; Bosoni, T.; Verga, L.; Sarais, G.; Lavatelli, F.; Nuvolone, M.; Obici, L.; Casarini, S.; Donadei, S.; et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin. Chem. 2009, 55, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Geller, H.I.; Singh, A.; Mirto, T.M.; Padera, R.; Mitchell, R.; Laubach, J.P.; Falk, R.H. Prevalence of Monoclonal Gammopathy in Wild-Type Transthyretin Amyloidosis. Mayo Clin. Proc. 2017, 92, 1800–1805. [Google Scholar] [CrossRef]
- Brunger, A.F.; Nienhuis, H.L.A.; Bijzet, J.; Hazenberg, B.P.C. Causes of AA amyloidosis: A systematic review. Amyloid 2020, 27, 1–12. [Google Scholar] [CrossRef]
- Dubrey, S.W.; Cha, K.; Simms, R.W.; Skinner, M.; Falk, R.H. Electrocardiography and Doppler echocardiography in secondary (AA) amyloidosis. Am. J. Cardiol. 1996, 77, 313–315. [Google Scholar] [CrossRef]
- Hassan, W.; Al-Sergani, H.; Mourad, W.; Tabbaa, R. Amyloid Heart Disease: New Frontiers and Insights in Pathophysiology, Diagnosis, and Management. Tex. Heart Inst. J. 2005, 32, 178. Available online: https://pubmed.ncbi.nlm.nih.gov/16107109/ (accessed on 29 April 2022).
- Sinagra, G.; Porcari, A.; Fabris, E.; Merlo, M. Standardizing the role of endomyocardial biopsy in current clinical practice worldwide. Eur. J. Heart Fail. 2021, 23, 1995–1998. [Google Scholar] [CrossRef]
- Porcari, A.; Allegro, V.; Pagura, L.; Longo, F.; Rossi, M.; Varra, G.G.; Dore, F.; Bussani, R.; Merlo, M.; Sinagra, G. 201 Clinical presentation profiles and natural history of cardiac amyloidosis: Long-term monocentric analysis (1990–2020). Eur. Heart J. 2021, 23, suab142.043. [Google Scholar] [CrossRef]
- Rapezzi, C.; Aimo, A.; Serenelli, M.; Barison, A.; Vergaro, G.; Passino, C.; Panichella, G.; Sinagra, G.; Merlo, M.; Fontana, M.; et al. Critical Comparison of Documents From Scientific Societies on Cardiac Amyloidosis: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 1288–1303. [Google Scholar] [CrossRef]
- Kittleson, M.M.; Maurer, M.S.; Ambardekar, A.V.; Bullock-Palmer, R.P.; Chang, P.P.; Eisen, H.J.; Nair, A.P.; Nativi-Nicolau, J.; Ruberg, F.L.; On behalf of the American Heart Association Heart Failure and Transplantation Committee of the Council on Clinical Cardiology. Cardiac Amyloidosis: Evolving Diagnosis and Management: A Scientific Statement from the American Heart Association. Circulation 2020, 142, E7–E22. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, M.; Varrà, G.G.; Porcari, A.; Saro, R.; Pagura, L.; Lalario, A.; Dore, F.; Bussani, R.; Sinagra, G.; Merlo, M. Re-Definition of the Epidemiology of Cardiac Amyloidosis. Biomedicines 2022, 10, 1566. https://doi.org/10.3390/biomedicines10071566
Rossi M, Varrà GG, Porcari A, Saro R, Pagura L, Lalario A, Dore F, Bussani R, Sinagra G, Merlo M. Re-Definition of the Epidemiology of Cardiac Amyloidosis. Biomedicines. 2022; 10(7):1566. https://doi.org/10.3390/biomedicines10071566
Chicago/Turabian StyleRossi, Maddalena, Guerino Giuseppe Varrà, Aldostefano Porcari, Riccardo Saro, Linda Pagura, Andrea Lalario, Franca Dore, Rossana Bussani, Gianfranco Sinagra, and Marco Merlo. 2022. "Re-Definition of the Epidemiology of Cardiac Amyloidosis" Biomedicines 10, no. 7: 1566. https://doi.org/10.3390/biomedicines10071566