Endocrine Risk
The human placenta integrates numerous sources of maternal stress signals, including cortisol, and responds with a dose-dependent release of placental CRH. The surge in placental CRH is produced by syncytial cells (which can be created in vitro by fusion of purified cytotrophoblast cells).[49] As described above, there is a positive or feed-forward loop between maternal cortisol and placental CRH, in contrast to the negative-feedback system controlling hypothalamic CRH synthesis and release. The positive relationship between cortisol and CRH release from the placenta is similar to the stimulating effect of cortisol on the amygdala release of CRH.[56] In placental tissue, glucocorticoids stimulate CRH gene expression by interacting with proteins that bind to the cAMP response site of the CRH promoter.[57]
Evidence suggests that the normal trajectory of placental CRH production over the course of gestation may be accelerated by an adverse intrauterine environment characterized by physiological stress. For example, elevated placental CRH has been observed in pregnancies complicated by preeclampsia, reduced utero–placental perfusion, intrauterine infection and in cases where fetal distress has led to elective preterm delivery.[58] A series of in vitro studies have shown that CRH is released from cultured human placental cells in a dose-response manner in response to all the major biological effectors of stress, including cortisol, catecholamines and proinflammatory cytokines.[49,59,60] The authors have in vivo evidence in humans that elevated levels of maternal cortisol early in gestation are associated with a more rapid rise in placental CRH concentrations.[15] Not only is placental CRH responsive to stress-related increases in maternal cortisol, but the administration of synthetic glucocorticoids for fetal lung maturation in pregnant women at risk for preterm delivery is similarly associated with significant increases in circulating placental CRH. Placental CRH concentrations increase by 1.5-fold within 12 h in response to the administration of synthetic glucocorticoids, such as betamethasone.[61,62]
The placental detection of stress or adversity results in a rapid rise in CRH and primes or advances the 'placental clock' and begins the cascade of events influencing myometria[54] and in extreme cases, precipitating preterm birth. Moreover, placental CRH has been shown to increase the placental production of estrogens and to inhibit the synthesis of progesterone.[63,64] Placental CRH is additionally released into the fetal compartment, where it stimulates the fetal adrenal to release dehydroepiandrosterone sulfate, an obligate precursor for placental estriol production.[65] Alterations in the production of progesterone and estriol may be one pathway by which placental CRH regulates the timing of delivery.[66]
There is evidence that it is the trajectory of placental CRH and other hormone production over gestation, rather than their absolute concentrations, that best predicts preterm delivery, suggesting that target cells are highly responsive to relative changes in circulating concentrations. The effects of HPA and placental axis hormones on gestational length are modulated further by the activities of binding proteins and enzymes. For example, concurrent with increases in circulating levels of placental CRH, a CRH-binding protein (CRH-BP) is produced in the liver and also in the trophoblast and intrauterine tissues during pregnancy, and binds to circulating CRH, reducing its biological action.[67–69] In contrast to the increasing levels of circulating placental CRH over the course of gestation, CRH-BP levels are constant in the first, second and early third trimester (and are not significantly different from nonpregnant levels), but fall by approximately 30% as birth approaches.[70] The consequence of these changes in the levels of CRH and CRH-BP is a large increase of free and bioactive CRH during the last part of gestation. Maternal plasma cortisol-binding globulin (CBG) levels also increase progressively with advancing gestation until the end of gestation, when there is a significant decline in CBG leading to an increase in bioactive cortisol.[71] Moreover, levels of placental 11β-HSD2 (which oxidizes cortisol into its inactive form, cortisone) also increase as gestation progresses before falling abruptly near term (Figure 3).[72] The decrease in CBG and the decrease in the activity of placental 11β-HSD2 toward the end of gestation increase fetal exposure to maternal cortisol, ensuring maturation of the fetal lungs, CNS and other organ systems in full-term births.[73,74] These exposures to elevated cortisol toward the end of gestation are normative and necessary for fetal maturation. Excessive exposures such as in the case of maternal stress or synthetic glucocorticoid treatment, however, are detrimental. Maternal stress may result in fetal exposure to excess cortisol both by increasing cortisol production and downregulating the activity of 11β-HSD2.[75]

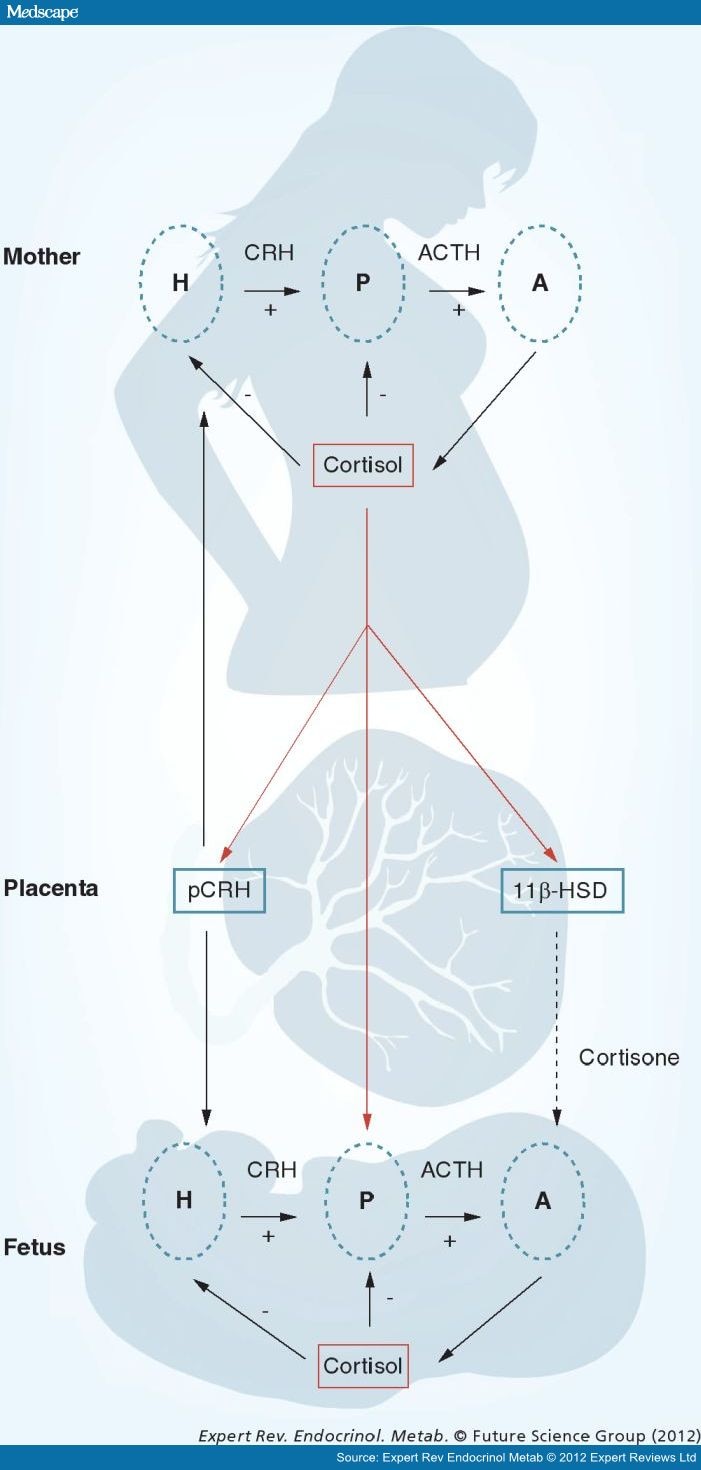
Figure 3.
The regulation of the hypothalamic–pituitary–adrenal axis changes dramatically over the course of gestation. Uniquely during primate pregnancy, cortisol stimulates the synthesis and release of placental corticotrophin-releasing hormone. These two hormones are important stress signals that can influence the timing of birth and alter the neurological development of the fetus.A: Adrenal gland; ACTH: Adrenocorticotropic hormone;CRH: Corticotrophin-releasing hormone; H: Hypothalamus;P: Pituitary gland; pCRH: Placental corticotrophin-releasing hormone.
Expert Rev Endocrinol Metab. 2012;7(4):445-459. © 2012 Expert Reviews Ltd.