
Abstract
Purpose
Aicardi syndrome is a rare disorder, affecting ~1 in 100 000 live births. Chorioretinal lacunae feature alongside agenesis of the corpus callosum and spasms in flexion to make up a diagnostic triad. Recently ophthalmic findings such as microphthalmia and optic disc anomalies have been recognised in association with Aicardi syndrome. This population study aims to determine the presence of ocular findings and identifies some novel associations in these patients.
Methods
A retrospective review of charts for seven patients with Aicardi syndrome was carried out.
Results
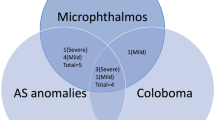
The incidence of Aicardi syndrome in Northern Ireland was found to be 1 in 110 000 live births. Four patients who had microphthalmus also had iris abnormalities; two patients with bilateral microphthalmus had partial aniridia and two patients with unilateral microphthalmus had iris coloboma in the same eye. Optic disc abnormalities were found in 11 eyes of six patients. Two patients were found to have areas of fibrovascular proliferation with a thickened white ridge and avascular zone beyond. Both of these patients developed retinal detachments.
Conclusions
Our review of patients with Aicardi syndrome in Northern Ireland has revealed some novel clinical findings, including aniridia in two cases. We also found a higher than previously reported rate of excavated disc anomalies of 50% in our cohort. We found two cases of peripheral retinal dysplasia, which has not been previously reported. This finding was associated with microphthalmus and severe optic disc abnormalities, and we feel this warrants early EUA to enable early treatment and hopefully result in better visual prognosis.
Similar content being viewed by others
Introduction
Jean Aicardi et al,1 first described the condition that is now known as Aicardi syndrome in 1965. It is a condition with a triad of features: agenesis of corpus callosum, ocular abnormalities, and spasms in flexion. Following a larger case series in 1969, Aicardi stated that the ocular findings of chorioretinal lacunae are an essential feature of the condition.2
The aetiology of Aicardi syndrome is not fully understood. However, it has always been thought to be a de novo mutation on the X chromosome as it is almost exclusively found in females and males with chromosome 47,XXY.3 However, findings have recently been published by Schrauwens et al who investigated 10 girls with Aicardi and their parents by exome or genome sequencing. In one case, a de novo mutation in autosomal gene TEAD1 was identified, which has previously been linked to chorioretinal atrophy, suggesting that Aicardi syndrome is a genetically heterogeneous disease and is not restricted to the X chromosome.4
It is a rare condition with a worldwide incidence of ~1 in 100 000 live births.5 Recently, prevalence in the United States was estimated at over 853 cases.5 Over the last 50 years the spectrum of this condition has been recognised as being more complex. Modified diagnostic criteria have been put forward to include other major and supporting features that are highly suggestive of Aicardi syndrome.6 Neurological findings now include cortical malformations, cysts, gross cerebral asymmetry, and ‘split-brain’ on electroencephalography. Ophthalmic features now recognised are optic nerve coloboma or hypoplasia and microphthalmia.6 Other features have been reported more sporadically, including nystagmus, detached retina, and hyperplastic primary vitreous.7 Although chorioretinal lacunae are virtually pathognomonic of Aicardi syndrome, they have been seen in other conditions including orofaciodigital syndrome type IX.6 To ensure correct diagnosis, it is important to be aware of the varied and asymmetrical ophthalmic signs that can occur in Aicardi patients.
Methods
A search for Aicardi patients was performed on the Paediatric Ophthalmology clinical database held in the Royal Victoria Hospital, Belfast, which is the tertiary referral centre for paediatric ophthalmology in Northern Ireland. This was cross-referenced with a search for Aicardi patients on a similar database held by the Regional Genetics department in the Belfast City Hospital.
Once all potential patients were identified, a retrospective review of paediatric and ophthalmic notes was performed. Full approval was obtained from the Belfast Health and Social Care Trust Standards, Quality and Audit department. Only patients with documented chorioretinal lacunae, agenesis of the corpus callosum, and seizure activity were included. Analysis of magnetic resonance imaging (MRI) findings of the brain and all ophthalmic findings was performed. Written consent for the use of fundal photographs was obtained.
Results
Nine patients identified from our database were assessed for Aicardi syndrome; seven of these patients, who all had classical chorioretinal lacunae and agenesis of the corpus callosum were included in our study (Table 1). All patients suffered from seizure activity, five of which were confirmed by EEG findings. One child had passed away before EEG examination and the other patient aged 31 had no official record of EEG as old notes had been destroyed. Two were excluded; one was found to have neuronal migration disorder without lacunae, the other had chorioretinal atrophy but normal brain structure.
Epidemiological data
All seven patients were female. Mean age at the time of data collection was 10.3 years (range 1–31 years). One patient had died at age 10 weeks. Five patients had the diagnosis made before 4 months of age. In one patient, diagnosis was not made until the age of 15. All patients were assessed by an Ophthalmologist, but only four went on to have an examination under anaesthetic (EUA). The majority of these seven patients presented with seizure activity with only one initially presenting to ophthalmology with ocular findings of microphthalmia and heterochromia noted by the parents.
Developmental delay and MRI findings
Four patients were officially assessed for developmental delay. Two were found to have severe delay and two were found to have moderate developmental delay. Of the three patients who were not officially assessed, one died at the age of 10 weeks but was felt to be significantly delayed. The other two patients were documented to have significant learning disabilities.
All seven patients had agenesis of the corpus callosum on their MRI brain scans. In addition, five patients also showed some degree of cortical dysgenesis. No apparent optic nerve or chiasm was evident in one patient.
Ophthalmic findings
All seven girls were examined by a paediatric ophthalmologist. Five were examined within the first 6 months of life. One was referred to ophthalmology at 4 years of age to confirm the diagnosis. One girl, with a delayed diagnosis, was assessed at age 15.
All patients had chorioretinal lacunae present in both eyes although the findings were asymmetrical. Four patients who had microphthalmus also had iris abnormalities; the two patients with bilateral microphthalmus had partial aniridia and the two patients with unilateral microphthalmus had iris coloboma in the same eye. Of the seven patients, two were also found to have a persistent tunica vasculosa; one bilateral and one unilateral. Only one patient developed raised intra-ocular pressure.
Optic disc abnormalities were found in 11 eyes of six patients. One patient had absence of the optic nerves and chiasm confirmed by MRI. Overall seven eyes had excavated discs and two had hypoplastic discs. All the patients with abnormal anterior segment findings were also found to have optic disc abnormalities. Two patients, neither of whom were born prematurely, had unilateral peripheral retinal changes. Both showed fibrovascular proliferation with a thickened white ridge and avascular zone beyond (Figures 1 and 2). One patient had already developed a detached retina at the time of initial presentation and, following discussions with the vitreoretinal team, the parents declined any surgical intervention. The other patient progressed to retinal detachment over a period of 2–3 weeks, despite peripheral retinal laser at the time of initial EUA and anti-VegF treatment 2 days later. Surgical repair was performed when retinal detachment was detected but this was unsuccessful and after consideration of the patient’s general condition no further surgery was pursued.

Patient 5. (a) Fluorescein angiogram: left eye at presentation showing morning glory disc, chorioretinal lacunae, fibrovascular ridge, and peripheral non-perfused retina. (b) Left eye 2 weeks later with inferotemporal retinal detachment. (c) Right eye at presentation with optic disc coloboma and chorioretinal lacunae.

Patient 6. (a) Right eye with normal sized-globe and pupil. (b) Left microphthalmic eye with irregular pupil and small inferior iris notch (obscured by light artefact). (c) Right eye with optic disc coloboma and chorioretinal lacunae. (d) Left eye with optic disc coloboma, chorioretinal lacunae, peripheral non-perfused retina, and retinal detachment at presentation.
Four patients underwent electrophysiology; only one of which was bilaterally normal. One showed absent ERG and VEP in one eye with the other having normal responses. The other two had unilateral gross attenuation over one hemisphere on VEP.
NPL was documented in five of the 14 eyes with two of these eyes being phthisical. In the remaining eyes, visual potential was limited by structural optic disc abnormalities, brain abnormalities or both.
Discussion
A study in 2008 of 408 cases of Aicardi syndrome found a prevalence of greater than 853 cases for the United States and several thousand worldwide.5 More recently an epidemiological study in Norway found their age-adjusted prevalence to be 0.63 per 100 000 females.8 Over the last 31 years, seven patients were diagnosed with Aicardi syndrome in Northern Ireland. All were female and aged 1–31 years at the time of our study. The average number of live births per year in Northern Ireland is 25 000.9 Extrapolating this over 31 years of our study, we can determine that the incidence for Aicardi is 1 per 110 000 live births in Northern Ireland. This appears to be comparable to the incidence rate of 1 per 105 000 live births in the United States and 1 per 93 000 live births in Holland.5, 10
Our age range of 0–31 years old appears to be in keeping with a recent study that analysed data from 28 countries.5 They found the age range from a sample of 408 Aicardi cases to be 0–42 years old. Interestingly, the life expectancy has improved from previous reports with survival rate of 62% at 27 years of age.5 This is likely due to improvements in healthcare in recent years.
Our review of patients with Aicardi syndrome in Northern Ireland has revealed some novel clinical findings. It has long been recognised that, along with the pathognomonic finding of chorioretinal lacunae, there are many other ophthalmic findings that are felt to be typical.7 Our study found some form of optic disc abnormality in 86% of patients, which is higher than that reported in the case series by Aicardi which described 50% of patients with optic disc anomalies.3 However Menezes et al also reported some form of optic disc anomaly in 86% of their study cohort but they included optic nerve drusen and pigmented discs as anomalies.7, 11 Excavated disc anomalies including coloboma and morning glory discs were found to be more prevalent in our cohort (50%) compared to findings by Fruhman and Menezes of 39 and 21%, respectively.7, 11 This may reflect our low threshold for EUA, which was performed in four of the seven patients. Microphthalmus can restrict full fundal examination and is felt to be associated with a high rate of posterior segment disease.12 Our study supports this finding with all six of the microphthalmic eyes in our cohort having optic disc abnormalities and two of the eyes having peripheral retinal changes. To our knowledge, patients described in recent study cohorts have not had EUA examinations to reliably determine associated posterior segment findings in microphthalmic eyes.7, 12
Four of our patients with microphthalmic eyes had associated iris abnormalities. Two had inferior iris colobomas, which have previously been described in the literature.7 However, two patients with bilateral microphthalmus had partial aniridia which, to the best of our knowledge, is a novel finding in patients with Aicardi syndrome.
Although a case of retinopathy of prematurity and a case of choroidal neovascularisation have both been reported in patients with Aicardi syndrome, we are not aware of any reports of eyes with peripheral retinal non-perfusion and anomalous retinal vasculature having been previously described in full-term infants with Aicardi syndrome (Figures 1 and 2).12 This is a novel finding with significant implications for visual prognosis in these patients. Both eyes with this finding had visual acuity of no perception of light (NPL), both developed chronic retinal detachment and one eye ultimately became phthisical. Both eyes had associated microphthalmus, iris colobomas or excavated discs, and the peripheral retinal changes were only discovered following EUA.
Shapiro et al13 have recently reported on the association of congenital optic nerve anomalies with peripheral retinal non-perfusion and the secondary complication of fibrovascular proliferation and tractional retinal detachment. Interestingly, both of our Aicardi patients who had peripheral retinal non-perfusion also had congenital excavated optic discs. Early examination under anaesthesia to allow adequate examination of the peripheral retina in Aicardi patients with optic disc anomalies would facilitate earlier intervention with laser therapy or possibly anti-VEGF therapy if peripheral non-perfusion was identified, which might ultimately prevent retinal detachment and blindness. Shapiro et al13 reported that the retina remained attached in all eyes in their cohort that had congenital disc anomalies and severe retinal non-perfusion when they had been treated with laser.
The developmental insult in Aicardi syndrome is thought to occur between 9–12 weeks due to the presence of persistant foetal vasculature and associated microphthalmus, persistant pupillary membrane, and epiretinal glial tissue.The developmental14 The findings in Aicardi syndrome patients also suggest developmental origins in the neuroectoderm, which is responsible for the pigmented epithelia of retina and iris, the sensory retina, optic nerve, iris sphincter and dilator muscles, and part of the vitreous. The developmental association of these ocular structures would suggest that patients with more severe anterior segment findings may be at higher risk of posterior findings and would therefore be most likely to benefit from examination under anaesthesia.
Strengths and limitations
Northern Ireland has only one Regional Genetic Centre, which allowed identification of all affected patients. However, given the rarity of Aicardi syndrome and the small population of Northern Ireland (1.8 million), our population-based cohort was consists of only seven patients. There were some limitations in data collection due to historical data loss when patients transferred from paediatric care to adult services. This study had a high rate of EUAs, which provided more detail on posterior segment findings then previous studies.
Conclusions
This study confirms that Aicardi syndrome is a rare disorder with our incidence calculated at 1 per 110 000 live births in Northern Ireland. It confirms previous suspicions that microphthalmus is associated with worse optic disc and posterior segment findings. We feel the association of microphthalmus and severe optic disc abnormalities with peripheral retinal dysplasia warrants early EUA to enable early treatment and hopefully result in better prognosis in these cases. And finally, we report two cases of aniridia in our Aicardi patients, which is a newly reported finding and may also be associated with more severe clinical findings and poor prognosis.

References
Aicardi J, Lefebvre J, Lerique-Koechlin A. Spasma in flextion, callosal agenesis, ocular abnormalities: a new syndrome. Electroencephalogr Clin Neurophysiol 1965; 19: 609–610.
Aicardi J, Chevrie JJ, Rousselie F. Spasma-in-flexion syndrome, callosal agenesis, chorioretinal abnormalities. Arch Fr Pediatr 1969; 26: 1103–1120.
Aicardi J. Aicardi syndrome. Brain Dev 2005; 27: 164–171.
Schrauwen I, Szelinger S, Siniard AL, Corneveaux JJ, Kurdoglu A, Richholt R et al. A de novo mutation in TEAD1 causes non–X-linked Aicardi syndrome. Invest Ophthalmol Vis Sci 2015; 56 (6): 3896–3904.
Kroner BL, Preiss LR, Ardini MA, Gaillard WD. New incidence, prevalence, and survival of Aicardi syndrome from 408 cases. J Child Neurol 2008; 23: 531–535.
Aicardi J. Aicardi syndrome: old and new findings. Int Pediatr 1999; 14: 5–8.
Fruhman G, Eble T, Gambhir N, Sutton VR, Van den Veyver IB, Lewis RA . Ophthalmologic findings in Aicardi syndrome. J AAPO 2012; 16: 238–241.
Lund C, Bjornvold M, Tuft M, Kostov H, Røsby O, Selmer KK . Aicardi syndrome: an epidemiologic and clinical study in Norway. Pediatr Neurol 2015; 52: 182–186.
Northern Ireland Statistics and Research agency. Population and household estimates for district electoral areas in Northern Ireland, 2015. Available at http://www.nisra.gov.uk/Census/2011_results_population.html (accessed September 2015).
United States Demographic Profile, 2014. Available at http://www.indexmundi.com/united_states/demographics_profile.html (accessed September 2015).
Menezes A, Lewis T, Buncic JR. Role of ocular involvement in the prediction of visual development and clinical prognosis in Aicardi syndrome. Br J Ophthalmol 1996; 80: 805–811.
Cabrera M, Winn B, Porco T, Strominger Z, Barkovich AJ, Hoyt CS et al. Laterality of brain and ocular lesions in Aicardi syndrome. Pediatr Neurol 2011; 45: 149–154.
Shapiro M, Chow C, Blair M, Kiernan DF, Kaufman LM . Peripheral nonperfusion and tractional retinal detachment associated with congenital optic nerve anomalies. Ophthalmology 2013; 120 (3): 607–615.
Brodsky MC. Pediatric Neuro-Ophthalmology. Chapter 2: Congenital Optic Disc Anomalies, XVIII Vol. Springer science and business Media LLC: New York, NY, USA, Philadelphia, PA, USA, 2010, pp 59–96.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Shirley, K., O'Keefe, M., McKee, S. et al. A clinical study of Aicardi syndrome in Northern Ireland: the spectrum of ophthalmic findings. Eye 30, 1011–1016 (2016). https://doi.org/10.1038/eye.2016.81
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2016.81
