
Abstract
Treatment for congenital adrenal hyperplasia (CAH) was introduced in the 1950s following the discovery of the structure and function of adrenocortical hormones. Although major advances in molecular biology have delineated steroidogenic mechanisms and the genetics of CAH, management and treatment of this condition continue to present challenges. Management is complicated by a combination of comorbidities that arise from disease-related hormonal derangements and treatment-related adverse effects. The clinical outcomes of CAH can include life-threatening adrenal crises, altered growth and early puberty, and adverse effects on metabolic, cardiovascular, bone and reproductive health. Standard-of-care glucocorticoid formulations fall short of replicating the circadian rhythm of cortisol and controlling efficient adrenocorticotrophic hormone-driven adrenal androgen production. Adrenal-derived 11-oxygenated androgens have emerged as potential new biomarkers for CAH, as traditional biomarkers are subject to variability and are not adrenal-specific, contributing to management challenges. Multiple alternative treatment approaches are being developed with the aim of tailoring therapy for improved patient outcomes. This Review focuses on challenges and advances in the management and treatment of CAH due to 21-hydroxylase deficiency, the most common type of CAH. Furthermore, we examine new therapeutic developments, including treatments designed to replace cortisol in a physiological manner and adjunct agents intended to control excess androgens and thereby enable reductions in glucocorticoid doses.
Key points
-
Challenges in the management of congenital adrenal hyperplasia (CAH) arise from multiple hormonal imbalances, the intrinsic tendency of the CAH-affected adrenal gland to overproduce androgens and limited treatment options, which often necessitate glucocorticoid excess.
-
The relationship between glucocorticoid and mineralocorticoid actions should be considered in the management of replacement therapies in CAH.
-
Patients are at risk of life-threatening adrenal crises with hypoglycaemia, most often triggered by infectious illnesses and exacerbated by adrenaline deficiency.
-
Traditional biomarkers vary with glucocorticoid dose or time of day and are not adrenal-specific, reflecting the need for new biomarkers; for example, the biologically active 11-oxygenated androgens, which are elevated in CAH.
-
Circadian glucocorticoid replacement and adjunct non-glucocorticoid therapies promise to enable glucocorticoid dose reduction; furthermore, the development of personalized gene and cellular therapies is under way.
Similar content being viewed by others
Introduction
Congenital adrenal hyperplasia (CAH) comprises a group of genetic disorders that affect the adrenal glands. Although CAH is rare, the most common form is caused by steroid 21-hydroxylase deficiency (21-OHD) and is an autosomal-recessive disorder of adrenal steroidogenesis that results from CYP21A2 mutations. There are two forms of 21-OHD CAH: classic CAH, which is severe, and non-classic CAH, which is mild. Classic 21-OHD CAH is a life-threatening condition owing to deficiencies of cortisol, aldosterone and adrenaline, which have essential roles in several homeostatic pathways, while compensatory mechanisms result in the overproduction of adrenal androgens1. Of note, classic CAH is included in neonatal screening programmes in the USA and over 50 other countries and regions2. Neonatal screening indicates that the classic form is a rare disease and occurs in 1 in 14,000 to 1 in 18,000 births worldwide1. Classic CAH is often subdivided into two forms on the basis of disease severity: salt-wasting CAH, associated with CYP21A2 mutations that ablate enzyme activity, and simple virilizing CAH, associated with CYP21A2 mutations that retain <5% of enzyme activity and some ability to make aldosterone. In the absence of early diagnosis and treatment (now possible with newborn baby screening programmes), infants with salt-wasting CAH experience a life-threatening adrenal crisis in the first 2 weeks of life and individuals with classic simple virilizing CAH present as toddlers with signs and symptoms of androgen excess such as pubic hair and growth acceleration3,4,5. The non-classic (mild) form is associated with CYP21A2 mutations that retain 20–50% of enzyme activity. This form can be asymptomatic and is quite common, with an estimated prevalence in the USA of 1 individual with non-classic CAH per 200 individuals6 and a carrier rate in several European countries of 4.0–7.5%7,8,9. Although a continuum of disease severity and phenotypic variations occurs with some CYP21A2 variants, this Review focuses on the rare classic form.
The earliest description of presumed classic 21-OHD CAH dates to 1865, when an Italian pathologist, Luigi De Crecchio, described the autopsy of a man with a 10-cm phallus, hypospadias, empty scrotum, vagina, uterus, fallopian tubes, ovaries and enlarged adrenals10. This individual died during a vomiting illness, potentially as the result of an adrenal crisis caused by a lack of cortisol. The first treatment of CAH did not occur until almost 100 years later, with the introduction of cortisone (a pregnane (21-carbon) steroid hormone that is converted to cortisol in the body) in the 1950s11,12. The introduction of glucocorticoid and mineralocorticoid replacement therapy allowed patients with classic CAH to survive and have a long lifespan; however, treatment has failed to normalize the growth and development of many children and adults often experience treatment-related iatrogenic Cushing syndrome or disease-related hyperandrogenism.
In the 1990s, studies began of alternative CAH therapies aimed at reducing daily glucocorticoid doses. For example, in children with CAH, adrenalectomy was investigated as well as peripheral blockade of androgen action and oestrogen production13,14. Alternative adrenal androgen pathways are now known that can produce excess androgens even when the classic androgen synthesis pathway seems well controlled15. Decades of advances in our understanding of the pathophysiology of CAH have led to the development and investigation of several alternative treatments, including circadian cortisol replacement and various approaches to lower androgen production, which promise to enable reductions in glucocorticoid dosing for patients.
This Review discusses the challenges of effectively managing patients with CAH and summarizes available and novel therapies in clinical trials or preclinical testing.
Pathophysiology
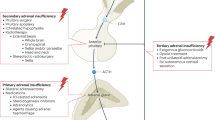
Complex hormonal imbalances result from 21-OHD in the adrenal cortex (Fig. 1). Decreased cortisol production alters the hypothalamic–pituitary–adrenal (HPA) feedback loop, with increased hypothalamic production of corticotropin-releasing factor (CRF) and pituitary production of adrenocorticotrophic hormone (ACTH). Low intra-adrenal levels of cortisol during development result in adrenomedullary dysplasia and adrenaline deficiency16. The degree of adrenaline deficiency is associated with the CYP21A2 genotype, with the largest impairment in enzyme function being associated with a salt-wasting phenotype17. Although the renin–angiotensin–aldosterone system is not directly under the influence of ACTH, volume depletion due to aldosterone insufficiency serves as an additional stimulus for ACTH production by indirectly stimulating vasopressin synthesis in the hypothalamus. Vasopressin, co-secreted with CRF, acts synergistically with CRF to augment ACTH release18.

In congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency, reduced circulating levels of cortisol increase the hypothalamic secretion of corticotropin-releasing factor (CRF) and pituitary production of adrenocorticotrophic hormone (ACTH), and decrease adrenomedullary adrenaline secretion. Elevated ACTH drives adrenocortical hyperplasia and uninhibited synthesis of adrenal androgens. The renin–angiotensin–aldosterone system regulates blood pressure as well as fluid and electrolyte balance and is not directly under the influence of ACTH. However, volume depletion and salt loss from aldosterone insufficiency in CAH leads to an increase in circulating levels of angiotensin II, which in turn stimulates vasopressin secretion. Vasopressin acts synergistically with CRF to augment ACTH release. The dashed lines indicate processes that are blunted in CAH. Plus symbols indicate processes that are enhanced in CAH; processes with three plus symbols are greatly enhanced and those with one plus symbol are mildly enhanced. ACE, angiotensin-converting enzyme.
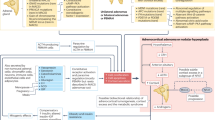
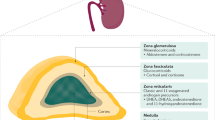
ACTH signalling through the melanocortin type 2 receptor (MC2R) drives adrenocortical steroidogenesis and simultaneously acts as an adrenal trophic factor. Excess ACTH leads to adrenocortical hyperplasia as well as the uninhibited synthesis of adrenal androgens and androgen precursors, including the traditional biomarkers of CAH 17-hydroxyprogesterone (17-OHP) and androstenedione19. This accumulation of androgen precursors and over-activity of the HPA axis leads to an intrinsic tendency of CAH-affected adrenal glands to overproduce androgens. Histological and biochemical profiles highlight zone-specific alterations in the adrenal cortex in CAH. For example, immunostaining of CAH-affected adrenal glands shows a strong presence of zona reticularis enzyme CYB5A compared with other zones20,21 (Fig. 2).

Nodular cortical adrenal hyperplasia is common in congenital adrenal hyperplasia (CAH). Intermingled zonation, adrenomedullary dysplasia and hyperplasia, predominantly involving the cells in the adrenocortical reticularis, are found in the untreated or undertreated CAH-affected adrenal. Immunostaining demonstrates strong expression of the zona reticularis enzyme CYB5A, which results in efficient androgen production. Increased CYB5A expression in the CAH-affected adrenal leads to increased activity of steroid 17α-hydroxylase-17,20-lyase (CYP17A1), a key enzyme involved in adrenocortical androgen production. The CAH-adrenal also efficiently makes biologically active 11-oxygenated (11-OH) androgens. Androgens and androgen precursors are highlighted in the deep blue boxes. Dashed lines indicate processes that are blunted in CAH. 17-OHP, 17-hydroxyprogesterone; DHEA, dehydroepiandrosterone; DHEA-S, DHEA sulfate.
This amalgam of hormonal imbalances, cortisol deficiency, androgen excess, and varying degree of aldosterone and adrenaline deficiency influences phenotype19. The predominant symptomatology is determined by the degree of enzyme impairment along with lifetime hormonal control.
Currently available treatments
Glucocorticoid therapy
Clinical management aims to re-set the multiple hormonal imbalances in classic CAH by replacing deficient hormones (that is, cortisol and aldosterone) and controlling adrenal androgen overproduction. The mainstay of treatment is glucocorticoid replacement. Currently available glucocorticoid preparations fail to replicate the physiological cortisol circadian rhythm (Fig. 3); therefore, adequate androgen suppression often requires supraphysiological doses of glucocorticoid therapy. Unsurprisingly, inadequate hormonal control is common, and approximately one-third of patients in two large cohort studies of patients with classic CAH had normal serum levels of androstenedione, reflecting good hormonal control22,23. Several oral glucocorticoid formulations are available for use in the clinical management of CAH, including short-acting, intermediate-acting and long-acting formulations24,25 (Table 1; Fig. 4). Currently, no well-designed, head-to-head trials that compare glucocorticoid regimens have been conducted and practices vary internationally26,27.

The physiological cortisol circadian rhythm has one peak in the morning at approximately 8.00 a.m. or upon awakening, with a gradual decline throughout the day112. By contrast, in patients with congenital adrenal hyperplasia (CAH) receiving hydrocortisone three times daily (the glucocorticoid of choice in all children and select adults) results in three cortisol peaks approximately 90 minutes following each dose, followed by rapid declines to undetectable levels108. Similarly, patients with CAH receiving once-daily dexamethasone or twice-daily prednisone have non-physiological glucocorticoid profiles48. Failure to mimic the circadian cortisol profile contributes to management challenges and supraphysiological doses of glucocorticoid are typically needed to adequately suppress the adrenocorticotrophic hormone (ACTH)-mediated androgen excess of CAH. Data presented are from 24-hour serial sampling studies and reflect varied treatment practices. Data remain inconclusive regarding recommended dosing practices, including morning versus evening dose weighting. Data are expressed as geometric means. Data sources: healthy volunteers (n = 33, aged 17–57 years; dark blue line)112; patients with classic 21-hydroxylase deficiency CAH (n = 14, aged 17–55 years) on thrice-daily oral hydrocortisone tablet regimen (10 mg at 8.00 a.m., 5 mg at 3.00 p.m., 15 mg at 10.00 p.m.; yellow line)108; patients with classic 21-hydroxylase deficiency CAH (n = 13) on an individualized dose thrice-daily oral hydrocortisone tablet regimen (8.00 a.m., 3.00 p.m. and 10.00 p.m.; orange line)25; patients with CAH receiving once-daily dexamethasone (n = 4, dose range: 0.25–0.5 mg, median dose time: 10.00 p.m.; light blue line) or twice-daily prednisone (n = 11, dose range: 2–7.5 mg (morning), 2–5 mg (night), median dose administration times 8.00 a.m. and 9.00 p.m.; green line)48.

Chemical structures for short-acting, intermediate-acting and long-acting glucocorticoids and mineralocorticoids used for the management of classic congenital adrenal hyperplasia.
Hydrocortisone is the preferred glucocorticoid in children with CAH owing to its short half-life and the fact that it has the lowest growth suppressing effect of available glucocorticoids24,28. The lowest available dose in oral hydrocortisone tablets is 5 mg (USA) or 10 mg (for example, Europe or Canada). The recommended body surface area-based hydrocortisone dose for infants and children is 10–15 mg/m2 per day, which is usually administered in three and sometimes four divided doses1,24. Paediatric dosing can be achieved by cutting whole tablets into sections but inconsistencies between doses are unavoidable. Of note, a hydrocortisone cypionate suspension (Pharmacia & UpJohn) was voluntarily recalled (July 2000)29 and inaccuracies of compounded hydrocortisone preparations made by pharmacies to enable lower doses can lead to unanticipated adverse effects30. In contrast with licensed formulations, compounded preparations are exempt from Good Manufacturing Practice and are not subject to regulatory health agency approval31,32. A survey in Germany evaluated the ‘real-world’ state of using compounding pharmacies and found that nearly 25% of 56 compounded hydrocortisone capsule batches did not meet the European Pharmacopoeia criteria owing to insufficiencies in net mass and drug content, highlighting the need for paediatric preparations33.
An immediate-release hydrocortisone sprinkle preparation, Infacort (brand name Alkindi, with four doses: 0.5 mg, 1 mg, 2 mg and 5 mg), enables direct oral administration as dry granules or mixed with a small amount of soft food and received EMA approval for use in children with adrenal insufficiency in 2018, followed by FDA approval in 2020 (ref.34). Results from the 2.5-year prospective extension study in children with adrenal insufficiency (n = 18, including 17 with CAH, aged 0–8 years) demonstrated good disease control with normal growth and no increased risk of adrenal crises35. However, the long-term use of Alkindi in the medical management of CAH has yet to be studied and challenges exist around its widespread use owing to cost.
The glucocorticoid regimens used in adults with CAH vary. A prospective UK cross-sectional CAH adult study (CaHASE) from 17 centres (n = 203)22 and a cross-sectional study from our USA centre (n = 244)23 highlight the wide variation in clinical practice. Approximately one-third of adults receive hydrocortisone and the remaining receive long-acting glucocorticoids (prednisolone, prednisone, dexamethasone or a combination).
Mineralocorticoid therapy
In patients with CAH, mineralocorticoid replacement is another important arm of treatment that corrects aldosterone deficiency. Patients with classic simple virilizing CAH have minimal aldosterone production but the levels are often insufficient to maintain normal intravascular volume36,37. A meta-analysis of adult height in classic CAH found that mineralocorticoid therapy during childhood was associated with a taller height outcome compared with those who did not receive mineralocorticoid therapy38. Achieving optimum sodium balance reduces ACTH and vasopressin, thus decreasing the dose requirements of glucocorticoid replacement therapy. Thus, mineralocorticoid therapy is recommended in all patients with classic CAH and the majority of non-hypertensive adults with classic CAH benefit from continued fludrocortisone treatment24 (Table 1; Fig. 4).
In CAH, desoxycorticosterone acetate was initially used in the 1950s as mineralocorticoid replacement with intramuscular injections or as desoxycorticosterone acetate subcutaneous pellets implanted every 6–12 months39. These parenteral approaches were eventually replaced with the only currently available synthetic mineralocorticoid, 9α-fludrocortisone1. Mineralocorticoid dose titration is based on plasma renin activity or direct measurement of plasma concentrations of renin (aiming for the age-specific normal range) but might also include electrolyte monitoring and blood pressure measurements, supplemented by a review of any symptoms that are suggestive of salt loss (for example, salt craving, postural hypotension and, in infants, poor weight gain and failure to thrive)40. A twice-daily regimen seems to be more effective than once-daily administration (D.P.M., unpublished work); thus, we propose that splitting the daily dose is the equivalent of a dose increase.
Given the physiological resistance to aldosterone in the kidneys of newborn babies, mineralocorticoid requirements are generally higher in early infancy than in older patients. Sodium supplementation of 1–2 g per day (4 mEq/kg per day) distributed in breast milk or formula throughout the day is commonly used24. In infants receiving high fludrocortisone doses, sodium supplementation might not be needed but high fludrocortisone risks hypertension41,42. In the first year of life, the maturing kidneys become increasingly sensitive to mineralocorticoid, increasing sodium retention and the risk of hypertension. In a large cohort of patients with CAH (n = 716, aged 3–18 years) in Germany, the prevalence of hypertension was 12.5% and it was more commonly observed in younger children (aged <18 months) than in adolescents (18.5% versus 4.9%). Fludrocortisone dose was a risk factor for hypertension in children aged <8 years43. Furthermore, this finding was confirmed in a 2021 report from our longitudinal CAH cohort that evaluated cardiovascular risk factors. Compared with the general population in the USA, higher rates of hypertension were found in both children (commonly aged <2 years) and adults with classic CAH, and high mineralocorticoid doses were associated with hypertension in children44. Close monitoring and judicious use of mineralocorticoids are recommended.
All glucocorticoids (except dexamethasone) have mineralocorticoid activity. Approximately 40 mg of hydrocortisone has the mineralocorticoid potency of 0.1 mg of fludrocortisone45. Furthermore, fludrocortisone also has some glucocorticoid activity and 0.1 mg of fludrocortisone has the glucocorticoid potency of 1 mg of hydrocortisone46. Elevated levels of 17-OHP and progesterone, as seen in poorly controlled CAH, exert an anti-mineralocorticoid effect47. Thus, the biological effects and interrelationship between glucocorticoid and mineralocorticoid actions and adrenal steroids must be accounted for in the management of CAH.
Management challenges
Biochemical monitoring
Standard biomarkers used in biochemical monitoring include adrenal androgen precursors (17-OHP and androstenedione), plasma renin activity, or plasma concentrations of renin and sometimes testosterone. All synthetic glucocorticoids have a narrow therapeutic index, and regimens or doses targeted to normalize or suppress 17-OHP in CAH can lead to over-replacement and associated metabolic comorbidities such as obesity and insulin resistance24. Hence, target levels of 17-OHP are above the normal range (for example, <1,200 ng/dl or <36 nmol/l)22,23.
Traditional biomarkers (17-OHP and androstenedione) are subject to considerable variability, acutely increase with stress and decrease following a dose of glucocorticoids, and are not synthesized exclusively by the adrenals. Additionally, the timing, dose and type of glucocorticoid drug influence biochemical results. Serial sampling data over 24 hours of 17-OHP and androstenedione in adults with classic CAH (n = 16) highlighted the intact circadian rhythms of these traditional adrenal biomarkers and demonstrated the influence of these rhythms by the timing and type of glucocorticoid regimen48.
In CAH, levels of dehydroepiandrosterone are typically low and the elevated levels of 17-OHP and androstenedione are diverted to an alternative 11-oxygenated pathway of androgen production (Fig. 2). These under-recognized 11-oxygenated adrenal metabolites are bioactive, dominant steroids in CAH15,20,49,50. Serum levels of 11-oxygenated androgens are higher in patients with 21-OHD CAH on replacement therapy when compared with age-matched and sex-matched control individuals20. In these patients, these increased levels derive primarily from the adrenals and high levels correlate well with poor long-term disease control and disease-specific comorbidities (for example, increased adrenal volume, testicular tumours of adrenal-like tissue termed testicular adrenal rest tumours (TART), menstrual irregularity or hirsutism)51. A retrospective analysis of >2,700 laboratory assessments in patients with CAH showed that 17% of samples had discrepant 17-OHP and androstenedione, and that elevated 11-oxygenated androgens could be used to identify those with poor disease control when traditional biomarkers are inconclusive52.
The optimal levels of 17-OHP and androstenedione are debatable; normalization of 11-ketotestosterone is desirable as it is a potent androgen with bioactivity similar to that of testosterone53,54. The use of 11-oxygenated androgens in the management of CAH has yet to be determined. Moreover, some adrenal steroid precursors that accumulate in CAH can bind to glucocorticoid and mineralocorticoid receptors as antagonists or agonists, complicating the biochemical management55,56.
Adrenal crisis
Low dose glucocorticoid therapy is associated with increased episodes of illness57 and adrenal crises58, and premature mortality in CAH might be primarily due to adrenal crisis. Adrenal crisis is estimated to be responsible for up to 42% of excess deaths in patients with CAH and patients with the salt-wasting form are especially at risk59. In a retrospective matched-cohort study in the UK, all-cause mortality was higher in patients with CAH compared with control individuals60. In a Swedish population-based study, adrenal crisis was reported as the leading cause of death in 588 patients with CAH59. The incidence of adrenal crisis in patients with adrenal insufficiency is estimated to be 5–10 adrenal crises per 100 patient-years, with mortality estimated to be 0.5 deaths per 100 patient-years61,62,63,64,65, mostly based on studies of adults. Although studies of children report similar findings62,66, a large international registry study (34 centres, n = 518 patients, 2,300 patient-years) reported 2.7 adrenal crises per 100 patient-years in children with CAH, with the majority of illness episodes managed at home57.
Life-threatening adrenal crisis occurs when there is insufficient cortisol to maintain homeostasis67. Aldosterone deficiency exacerbates adrenal crisis through sodium and water loss and potassium retention, and adrenomedullary dysfunction (adrenaline deficiency) contributes to the risk of cardiovascular instability and hypoglycaemia58,68. Life-threatening hypoglycaemia can be associated with seizures and can rarely result in permanent neurological sequelae in children with adrenal crises58,69,70. Having low blood levels of adrenaline in CAH is a risk factor for needing emergency care in children and is associated with increased illnesses in infants and adults58,68.
Infectious illnesses can trigger an adrenal crisis and gastrointestinal and upper respiratory tract infections are the most common precipitants at all ages57,58,62,71. There is no evidence that patients with CAH are more likely to develop a severe course of COVID-19 after SARS-CoV-2 infection, but the risk of adrenal crisis remains as with any notable infectious illness72. Prevention of adrenal crisis is best accomplished through repeated, structured patient education on stress dosing with glucocorticoids73,74.
Childhood growth and development
Adrenocortical androgen excess in CAH can advance epiphyseal bone maturation and lead to gonadotropin-releasing hormone (GnRH)-dependent precocious puberty. Prepubertal advanced bone maturation attenuates height gain during puberty. By altering growth plate chondrogenesis, glucocorticoids also exert direct adverse effects on linear growth as seen in states of clinical and subclinical hypercortisolism75. A meta-analysis of >1,000 patients with classic CAH found shorter than average adult height when adjusted for genetic potential (–1.03 standard deviation below expected height based on parental heights, ~7 cm)38.
CAH control during puberty can be particularly challenging owing to physiological and lifestyle changes. During puberty, an increase in circulating growth hormone (GH) and insulin-like growth factor 1 (IGF1) inhibits hepatic 11β-hydroxysteroid dehydrogenase 1 (11βHSD1), the enzyme responsible for converting cortisone to cortisol, which leads to alterations in cortisol kinetics that increase clearance. Additionally, a GH–IGF1-mediated increase in glomerular filtration rate increases cortisol clearance76. In general, physiological changes in the hormonal milieu during puberty necessitate glucocorticoid dose uptitrations76. However, adult height correlates negatively with glucocorticoid dose during puberty, especially doses of hydrocortisone exceeding 17 mg/m2 per day28. Adolescent lifestyle changes, including altered sleep–wake patterns and increasing independence with self-management, are commonly associated with suboptimal adherence to medications, which contributes to poor disease control. Retrospective studies highlight the importance of individualizing glucocorticoid therapy to avoid over-replacement or under-replacement77 as the undesirable effects of glucocorticoids seem to be dependent on dose and age78.
Children with classic CAH (n = 20) with predicted adult height two standard deviations below their MPH treated with GH alone or in combination with a GnRH analogue achieved taller adult height compared with their predicted height at baseline (mean age at study entry: 8.6 years)79,80. Overall, however, data on the benefits of GH with or without a GnRH analogue are limited to small, non-randomized studies79,80, which limits the recommendation of such growth-promoting therapies in regular clinical practice24.
Cardiometabolic morbidity
Patients with CAH have increased cardiometabolic morbidity22,81. A meta-analysis of 20 studies of CAH found an increased risk of insulin resistance, elevated blood pressure and carotid intima thickness, although the quality of evidence was low82. A Swedish population-based study of 588 patients with CAH found an increased prevalence of obesity, type 2 diabetes mellitus, obstructive sleep apnoea, hypertension, elevated lipids, atrial fibrillation and venous thromboembolism compared with control individuals83. Another population-based study (n = 272 patients with CAH, 200 control individuals) by the same group reported an increased prevalence of gestational diabetes mellitus in women with classic CAH84. A longitudinal study in the USA of 58 patients with classic CAH followed for a median of 18.6 years with 1,962 visits spanning childhood and adulthood found that metabolic morbidity started before puberty, which was associated with both mineralocorticoid and glucocorticoid treatments44.
The presence of obesity, insulin resistance and metabolic syndrome adds another tier of challenge as these chronic comorbidities influence glucocorticoid pharmacokinetics and hence hormonal management in CAH85. For example, glucocorticoid dose requirements increase with an increase in body weight86. Furthermore, the presence of hepatic steatosis might increase cortisol clearance87. Decreased insulin sensitivity, independent of body adipose mass and hepatic steatosis, also alters cortisol kinetics, resulting in increased cortisol clearance87,88. Alterations in cortisol–cortisone kinetics in the liver and adipose tissue contribute to increased cortisol clearance and increased glucocorticoid doses required for disease control, leading to a bidirectional maladapted loop87. Weight loss with improvements in both insulin sensitivity and liver lipid content can reverse these processes85.
Risk for low bone mineral density
Long-term use of supraphysiological doses of glucocorticoid increases the risk of impaired bone mineral density (BMD). The use of long-acting glucocorticoids seems to have more deleterious effects on BMD than the use of short-acting or intermediate-acting glucocorticoids. Glucocorticoids can directly alter the bone remodelling cycle (increased osteoclastogenesis and decreased osteoblastogenesis) and increase fracture risk, or indirectly affect BMD by altering calcium homeostasis in the kidney and the intestine89. A meta-analysis of nine case–control studies (n = 254 patients with CAH and 344 control individuals) showed a lower BMD in patients with CAH; however, a relationship with glucocorticoid dose was not found90. A population-based Swedish study (n = 714 patients with CAH and 71,400 control individuals) found an increased risk of fragility fractures in patients with CAH91. Overall, BMD studies in CAH are limited to either small cohorts or heterogeneous cohorts lacking hormonal data, thereby highlighting the need for rigorous prospective studies.
Tumour formation and infertility
The trophic effects of chronic ACTH elevation predispose to adrenal tumour formation92. Adrenal hyperplasia and nodularity are commonly seen in patients with CAH, with a high prevalence of benign adrenal tumours (29%) and myelolipomas (8.6%)93. CAH cohort studies demonstrate that increased adrenal volume is correlated with suboptimal disease control, based on elevated adrenal androgens, and other comorbidities, including adverse cardiovascular profile, hypogonadism and oligomenorrhoea51,94,95.
A common and important complication in individuals with male reproductive organs and CAH is the development of TART96,97. These benign, bilateral tumours are histologically similar to adrenocortical cells and are thought to originate from pluripotent cells or from cells of adrenal origin, which descend with the testis during embryogenesis and proliferate with ACTH stimulation98,99. TART are centrally located in the rete testis and easily identified by ultrasound. They can compress the seminiferous tubules, which leads to irreversible damage to the surrounding testicular tissue and results in gonadal dysfunction and infertility. Suppression of ACTH with an increased glucocorticoid dose can decrease the size of TART and might reverse infertility100. Ovarian adrenal rest tumours are also seen in individuals with female reproductive organs and CAH; however, they are less commonly reported as they are not regularly seen on conventional imaging101.
In CAH, elevated adrenal androgens and the resulting alterations of the hypothalamic–pituitary–gonadal axis can lead to hypogonadotropic hypogonadism and sub-fertility in both sexes102. Although TART is thought to be the main cause of male infertility in CAH, improvements have been observed in male fertility in those born after the introduction of neonatal screening, which suggests that early diagnosis and treatment might benefit gonadal development and function103. In individuals with female reproductive organs and CAH, adverse effects of elevated progesterone on the uterine lining, combined with secondary development of polycystic ovaries with menstrual irregularity, increases the risk of sub-fertility19,102. Reduced fertility has been reported mostly in women with the salt-wasting subtype84. In both men and women, sub-fertility can improve with an increased glucocorticoid dose but at the expense of the adverse effects associated with glucocorticoid excess. The anatomical and psychological factors that contribute to female sub-fertility in CAH are beyond the scope of this Review.
Novel therapies
New ways to replace glucocorticoids
In efforts to improve patient outcomes and minimize glucocorticoid exposure, novel therapies are being developed, including new ways of replacing glucocorticoids (Fig. 5; Box 1). For example, Plenadren (Shire Services BVBA, Belgium) is a dual-release oral hydrocortisone preparation designed as an immediate-release coat over an extended-release core that is administered once daily. This therapy induces a physiological cortisol profile in the early part of the day and received EMA approval (2011) for the treatment of adrenal insufficiency104,105. However, the overnight cortisol-free interval falls short of addressing the overnight ACTH-driven androgen excess that occurs in CAH and data in patients with CAH are lacking.

Novel treatment approaches include new ways to deliver circadian cortisol replacement (novel glucocorticoids) as well as various adjunct therapies to decrease adrenocortical androgen production, thereby enabling glucocorticoid dose reduction. These include direct adrenocortical steroidogenesis inhibitors and agents to suppress the hypothalamic–pituitary–adrenal axis (such as corticotropin-releasing factor (CRF) receptor 1 (CRF1) antagonists, an adrenocorticotropic hormone (ACTH)-specific monoclonal antibody and melanocortin type 2 receptor (MC2R) antagonists). Preclinical studies are exploring the role of restorative cell-based therapies. A first-in-human recombinant adeno-associated virus-based gene therapy in classic 21-hydroxylase deficiency congenital adrenal hyperplasia (CAH) is also in development. Dashed arrows indicate processes blunted in CAH or targeted by abiraterone, a CYP17A1 inhibitor. Androgens and androgen precursors are highlighted in the deep blue boxes. Therapies are shown in yellow boxes. 17-OHP, 17-hydroxyprogesterone; DHEA, dehydroepiandrosterone; DHEA-S, DHEA sulfate.
To address the ACTH-driven rise in adrenal androgens that is characteristic of CAH, a modified-release hydrocortisone formulation was first developed as an oral, delayed-release and sustained-release tablet (Chronocort, Phoqus Pharmaceuticals plc, Kent, UK)106,107. This modified-release hydrocortisone tablet (30 mg administered once at night) demonstrated a nocturnal cortisol profile that mimics physiology, with peak cortisol occurring around 6.00 a.m. in 14 patients with classic CAH (aged 17–55 years)108; however, falling cortisol levels and subsequent afternoon elevations in adrenal androgens occurred. Owing to issues of scaling manufacturing, further development was halted107,108.
To tackle the challenge of mimicking the overnight cortisol dynamic profile, a modified-release hydrocortisone capsule formulation, also named Chronocort (Diurnal Limited, UK, referred to here as Chronocort capsules) was developed using a scalable, multi-layered, micro-particulate technology107. A delayed-release, pH-triggered enteric coat demonstrated good bioavailability and a twice-daily regimen (20 mg at 11.00 p.m. and 10 mg at 7.00 a.m.) approximated the physiological cortisol rhythm in dexamethasone-treated, adrenal-suppressed healthy volunteers107. A phase II, open-label, 6-month, non-randomized study of Chronocort capsules in 16 adults (aged 18–60 years) with classic CAH enabled dose adjustment based on androstenedione, 17-hydroxyprogesterone and clinical symptomatology. Chronocort capsules demonstrated control of adrenal androgens throughout the day despite down-titration, with no serious adverse events109. The 24-hour urine steroid metabolome profile obtained by mass spectrometry highlighted the differential effect of Chronocort capsules in comparison to standard glucocorticoid therapies, with improved 24-hour control of the traditional adrenal biomarker 17-OHP and alternative adrenal androgen pathway metabolites110.
With promising results from the phase II study, Chronocort capsules were further investigated in a multicentre (10 sites across the UK, Europe and USA (NIH)), phase III, randomized, parallel arm study in 122 adults with classic CAH conducted over 24 weeks, with dose titration performed by two blinded investigators111. The trial missed its primary end point as Chronocort was not superior to standard glucocorticoid therapy at study completion based on the 24-hour profile of serum levels of 17-OHP. However, compared with standard glucocorticoid therapy, patients receiving Chronocort had improved 17-OHP and androstenedione levels in the morning and early afternoon, with a decrease in hormonal fluctuations throughout the day at 24 weeks. At 18 months extension, patients receiving Chronocort reported events of therapeutic benefit, including improved menstruation (n = 4), patient or partner pregnancy (n = 5) and being more attentive (n = 11), despite receiving a reduced daily dose of Chronocort following the phase III clinical trial111. Chronocort (brand name Efmody) received marketing approval in 2021 in the UK and Europe for patients with CAH aged 12 years and older. The long-term, safety-extension phase of Chronocort is ongoing and additional studies are planned in the USA.
To provide circadian glucocorticoid replacement, continuous subcutaneous hydrocortisone infusion (CSHI) therapy has been developed with the use of a programmable pump112. A phase II, open-label clinical trial (6 months followed by a 12-month extension) in eight adults (aged 19–42 years) with classic CAH113,114 with poor disease control at study entry showed improved 24-hour serum androgen biomarker and plasma ACTH levels, health-related quality of life and fatigue compared with baseline oral glucocorticoid therapy of a similar daily dose113. Improvements in serum 11-oxygenated androgens were also observed115. Improved androgen control and health-related quality of life were maintained at 18 months114. However, no notable improvements were observed in BMI and cardiometabolic comorbidities, which reflects the challenges of reversing longstanding comorbidities. Although CSHI therapy is attractive as an improved method for glucocorticoid delivery, it is labour intensive, with practical concerns (such as equipment failure, discomfort, local skin irritation and infections, and cost) limiting its use.
Following 18 months of physiological cortisol replacement, adrenal androgen control was maintained on lower doses of glucocorticoid than those used with standard glucocorticoid replacement therapy for both oral (Chronocort capsules) and CSHI therapies. This finding suggests that adrenal volume reduction and/or resetting of the HPA axis might have contributed to improved disease control. Circadian cortisol replacement has also called for re-assessment of the biochemical evaluation of CAH. When 17-OHP was well controlled in patients receiving Chronocort capsules, serum levels of androstenedione were low, reflecting that high 17-OHP levels are needed to drive androstenedione and adrenal androgen production. As such, androstenedione, commonly used in the management of CAH, might not be an optimal biomarker in the setting of improved disease control. In patients receiving circadian glucocorticoid therapy with CSHI, 11-oxygenated androgen metabolites provided the largest difference compared with oral glucocorticoids115. Serum levels of 11-oxygenated androgens were also decreased with Chronocort capsule therapy110.
Corticosterone, which is the primary glucocorticoid in rodents, is a minor glucocorticoid in humans and circulates at low concentrations (5–10%) compared with the major glucocorticoid cortisol116. Differential tissue expression of the glucocorticoid transmembrane transporters has led to the consideration of corticosterone as an alternative therapeutic option in treating CAH, with potentially decreased effects on peripheral tissues and enhanced influence on the HPA axis117. A proof-of-concept, double-blind, randomized crossover trial compared the acute effects of cortisol and deuterated corticosterone (D8-corticosterone) in 14 adults with CAH. In this study, intravenous D8-corticosterone was acutely effective in suppressing circulating levels of ACTH and adrenal androgens without increasing circulating levels of insulin, unlike hydrocortisone (that is, cortisol)118. Given the favourable metabolic effects and matched efficacy of adrenal androgen suppression, further studies are warranted to evaluate corticosterone as a potential glucocorticoid-based approach in the treatment of CAH.
New ways to suppress adrenal androgens
Inhibiting key enzymes involved in androgen biosynthesis is an attractive option to address the androgen excess component of CAH. Improved control of androgens could enable glucocorticoid dose reduction, thereby resulting in minimization of adverse outcomes. However, the concomitant inhibition of sex steroid synthesis in the gonads restricts this adjunct, glucocorticoid-sparing approach to prepubertal children, adult women on oral contraceptives or men on testosterone replacement therapy. The potential implications of elevated circulating levels of ACTH and progesterone, especially in the context of fertility outcomes and tumour formation, need to be considered with this approach.
Historically, the first attempt to block excessive adrenal hormones in CAH was in the study of an androgen receptor antagonist in combination with an aromatase inhibitor in children. In a 2-year randomized, parallel study, children with classic CAH (n = 28) receiving low dose hydrocortisone and fludrocortisone combined with the experimental regimen of flutamide (an anti-androgen) and testolactone (an aromatase inhibitor) maintained normal growth velocity and skeletal maturation despite elevations in adrenal androgens14. A long-term study following children with classic CAH until adult height will soon be completed119.
Elevations in 17-OHP increase substrate availability for CYP17A1, the critical enzyme necessary for androgen and oestrogen biosynthesis116. Abiraterone is a potent CYP17A1 inhibitor that efficiently inhibits testosterone production120 and is used to treat prostate cancer. In men with prostate cancer, CYP17A1 inhibition leads to accumulation of the mineralocorticoid 11-deoxycorticosterone; however, in patients with classic 21-OHD CAH treated with abiraterone, 11-deoxycorticosterone does not accumulate owing to the block in the pathway leading to 11-deoxycorticosterone biosynthesis. Abiraterone was studied as a phase I, non-randomized, open-label, multiple-dose (6 days), sequential dose-escalation (100–250 mg once-daily) trial in six women (aged 19–46 years) with classic CAH121. At study entry, all had >1.5-times elevated serum levels of androstenedione (greater than 345 ng/dl or 12 nmol/l) and continued on hydrocortisone 8 mg/m2 per day (maximum dose of 20 mg per day), a combined oral contraceptive pill and fludrocortisone as well as the study dose of abiraterone. On the higher dose of abiraterone, five of six patients demonstrated normalization of androstenedione, which decreased from a mean serum concentration of 664 ng/dl to a mean concentration of 126 ng/dl (23.2 nmol/l to 4.4 nmol/l). Abiraterone also reduced serum levels of testosterone and 11-oxygenated androgens and urinary androgen metabolites121,122. This adjunct therapy was safe and well tolerated over the short-term study duration121. A phase I–II study in prepubertal children is under way123.
Another oral, steroidogenic inhibitor studied in CAH is Nevanimibe124, a selective acyl-coenzyme A:cholesterol O-acyltransferase 1 (ACAT1) inhibitor. ACAT1 is an enzyme that converts free cholesterol to cholesterol esters for storage, and ACAT1 inhibition impairs all adrenocortical steroidogenesis125. In a multicentre, phase II single-blind, dose-escalation study, ten adults (18–61 years) with uncontrolled CAH (defined as serum levels of 17-OHP more than four times the upper limit of normal) were enrolled124. Patients continued on their usual glucocorticoid and fludrocortisone medication regimen. Nevanimibe was administered twice-daily for 2 weeks, followed by a 2-week placebo washout, with sequential dose-escalation (125 mg, 250 mg, 500 mg, 750 mg and 1,000 mg). Although a drug effect was evident in 80% of patients during the treatment phase, only two patients (20%) met the primary end point of 17-OHP more than two times the upper limit of normal. Gastrointestinal-related adverse effects were common (30%). A longer-duration study was terminated following an interim data review.
HPA axis suppression
Drug discovery over the past few decades in small molecules and monoclonal antibody-based targeted therapies have gained momentum in the field of endocrinology. Potential strategies to address the drivers of excess androgen synthesis in CAH include small molecules that antagonize the action of CRF1, a monoclonal antibody to ACTH and a selective MC2R antagonist.
The first proof-of-concept study of an oral small-molecule (Neurocrine Biosciences, USA) CRF1 antagonist was a phase Ib, single-blind, placebo-controlled, fixed-sequence (placebo, NBI-77860 300 mg and 600 mg), single-dose trial in eight women (aged 18–58 years) with classic CAH, which showed a dose-dependent reduction of serum levels of ACTH and/or 17-OHP126. A similar oral CRF1 antagonist (crinecerfont, NBI-74788, Neurocrine Biosciences, USA) was evaluated in an open-label, sequential, dose-finding, phase II study in 18 adults (aged 18–50 years) with classic CAH127. Two weeks of crinecerfont therapy induced dose-dependent reductions in ACTH (54–69%), 17-OHP (55–75%) and androstenedione (21–64%). At the highest dose of crinecerfont (100 mg twice-daily), at least a 50% reduction in ACTH, 17-OHP and androstenedione was observed in 75% of patients compared with baseline, with a favourable safety profile127. Subsequently, a crinecerfont 100 mg twice-daily regimen was chosen for a globally registered phase III, double-blind, placebo-controlled study to evaluate the efficacy, safety and tolerability of crinecerfont in adults with CAH for a 24-week duration, followed by a 1-year extension128. A 28-week paediatric study of children with CAH (aged 2–17 years) with a 24-week extension is also under way129.
A second newly developed oral CRF1 antagonist, tildacerfont (SPR001; LY2371712, Spruce Biosciences, USA), was investigated in two multicentre, open-label phase II studies in adults with CAH130. A phase IIa, multiple-dose, dose-escalation study consisted of three cohorts receiving different doses either once or twice daily. All patients (n = 24, aged 19–67 years) had a serum concentration of 17-OHP ≥800 ng/dl at study entry. In addition, a phase IIb study investigated tildacerfont 400 mg once-daily (n = 11) over 12 weeks. Efficacy analysis for both trials was performed based on baseline androstenedione criteria (poor disease control was defined as androstenedione levels more than two times the upper limit of normal and good disease control as less than the upper limit of normal) and excluded 11 patients on dexamethasone owing to drug–drug interactions. In comparison with baseline in patients with poorly controlled disease, tildacerfont treatment induced average reductions of 74% for circulating levels of ACTH, 82% for levels of 17-OHP and 55% for levels of androstenedione. By contrast, ACTH and androgen biomarkers remained stable for patients with good disease control; overall, tildacerfont was well tolerated130. Two multicentre, randomized, double-blind, placebo-controlled, long-term (52 weeks) clinical trials in adults with CAH are under way to study the effect of tildacerfont on outcomes such as improved adrenal androgen levels in patients with poor disease control who are on stable glucocorticoid doses131 as well as the potential of reducing glucocorticoid dose with this add-on therapy in patients with good disease control132. Furthermore, a paediatric study is in development133.
ALD1613 (Alder BioPharmaceuticals, USA) is a specific, high-affinity, long-acting humanized, neutralizing, monoclonal antibody to ACTH that has been tested in preclinical studies134,135. Pharmacokinetic–pharmacodynamic studies in rodents and cynomolgus monkeys demonstrated substantial and durable reductions in plasma levels of glucocorticoid134.
A selective, non-peptide MC2R antagonist (Crinetics Pharmaceuticals, USA) demonstrated in vitro MC2R antagonistic activity136,137. In vivo studies have been performed in dogs and rodents that demonstrated good oral bioavailability. Efficacy studies in rats demonstrated dose-dependent acute suppression of plasma levels of corticosterone and reversal of the sustained ACTH exposure-induced phenotype and adrenal hypertrophy137,138. These promising preclinical results have led to the investigation of CRN04894, an oral MC2R antagonist, in a double-blind, randomized, placebo-controlled phase I study139. Other MC2R peptide antagonists are in preclinical development140,141.
Surgical adrenalectomy
Case reports and smaller case series suggest that adrenalectomy might be a therapeutic option to tackle the excess androgen feature of the disease, thereby reducing the need for supraphysiological glucocorticoid replacement therapy142,143,144. A meta-analysis evaluated the short-term and long-term outcomes of bilateral adrenalectomy as a treatment option in CAH and identified 48 patients (age at surgery 0.25–56 years) from 32 articles145. The two most common indications for surgery were virilization and/or iatrogenic Cushing syndrome (n = 30, 62%), followed by less common indications including benign adrenal tumour (n = 8). Over the median follow-up of 27 months post-adrenalectomy, the mean daily hydrocortisone dose equivalency was reduced by >30% with most patients (n = 34, 71%) reporting improvements in features of hyperandrogenism, including onset or restoration of menses, and improvements in features typical of Cushing disease with subjective improvements in body image. All three patients undergoing adrenalectomy for the indication of primary infertility had a positive fertility outcome. Immediate postoperative complications were seen in 10% (n = 5) and long-term complications included adrenal crises in 17% (n = 8) and ectopic adrenal rest tissue in 10% (n = 5). Patients undergoing adrenalectomy require strict adherence to CAH replacement therapy, stress dosing at times of acute illness143,145, and ongoing clinical and biochemical monitoring for hyperandrogenism owing to the risk of activation of ectopic adrenal rest tissues143,146,147,148. Given the associated risks, this approach, which was proposed in the 1990s for young children with double null mutations, is not recommended13,24. However, adrenalectomy can be considered the last therapeutic resort in select individuals who are not responding to medical management, especially when there is a desire for fertility.
Chemical adrenolytic therapy
Mitotane is a teratogenic agent derived from the well-known endocrine disruptor dichlorodiphenyltrichloroethane149, which downregulates steroidogenesis and is a potent adrenolytic with time-dependent and dose-dependent effects. Mitotane is the only agent approved by the FDA and EMA as the first-line agent in managing metastatic adrenocortical carcinoma for over six decades150. Mitotane is also rarely used (off-label) in the management of Cushing disease151 and in CAH in the context of infertility due to TART152,153.
Successful restoration of fertility was first achieved with mitotane therapy in a 29-year-old man with classic CAH and bilateral TARTs associated with hypogonadism and azoospermia152. Importantly, mitotane is a potent inducer of CYP3A4, which increases glucocorticoid clearance. Patients with CAH who receive mitotane might require a 50–100% increase in their glucocorticoid dose153. A retrospective case series highlights the long-term effects (60-months) of mitotane in five men with classic CAH and TART-associated infertility153. Given the narrow therapeutic index and toxicity, serum levels of mitotane were closely monitored (aimed at <14 mg/l) with dose titration and the glucocorticoid dose was increased to prevent adrenal insufficiency. Adverse effects included weight loss (n = 3), adrenal crises (n = 2), gastrointestinal disturbance with nausea and diarrhoea (causing one patient to discontinue treatment prematurely), and minor hepatic impairment (n = 4, which resolved with treatment discontinuation). In addition, TART showed resolution by ultrasonography in two patients, with improvements in sperm counts that enabled cryopreservation for future use. Mitotane is a highly lipophilic agent that accumulates in adipose tissue and, given its long half-life (median 53 days)154, close monitoring of glucocorticoid therapy is essential as the effects of mitotane can last for several months after discontinuation153. Given the teratogenic effects of mitotane, women should be counselled to avoid pregnancy for a minimum of 5 years following therapy155. Mitotane therapy in women with ovarian adrenal rest has not been studied. Similar to surgical adrenalectomy, chemical adrenolytic approaches can be considered as a last therapeutic resort in select individuals who are not responding to medical management.
Cell-based therapies
The discovery of cellular reprogramming to induce pluripotency has expanded the field of adrenal research, with the generation of induced pluripotent stem cells from mature cells in skin, urine or blood. Lineage conversion (direct reprogramming) attempts to generate steroidogenic adrenocortical-like cells were successful in the generation of human induced steroidogenic cells (hiSCs). These cells were generated by forced expression of progenitor transcription factors (steroidogenic factor 1) and activation of key signalling pathways (protein kinase A and luteinizing hormone-releasing hormone) in somatic cells derived from skin fibroblasts, urine and blood156. The reprogrammed hiSCs demonstrated de novo expression of steroidogenic enzymes and had the functional ability to synthesize adrenal steroids following physiological (that is, ACTH) or pharmacological stimuli. By introducing wild-type CYP21A2 into hiSCs derived from patients with CAH, in vitro dose-dependent increases in cortisol secretion and decreases in androgen production occurred156. However, the explants demonstrated limited in vivo cell viability when transplanted into mice.
Biomaterial engineering strategies have enabled the development of 3D in vitro cell models that mimic the in vivo environment. This research is a step towards creating a bioartificial adrenal cortex, which could mitigate the limitations of organ transplantation and offers a potential path forward for cell replacement therapies157. Xenogeneic adrenocortical cells from cows encapsulated in a microenvironment that supports cell viability and immune isolation were developed and showed encouraging results, including maintenance of steroidogenesis, when implanted into adrenalectomized rats158. The combination of reprogrammed hiSCs with novel biomaterials represents the early development of tailored cell-based therapies for CAH.
Gene therapy
Advances in adeno-associated virus (AAV) vector-based therapies have expanded the gene therapy landscape to include CAH. The first feasibility study used a replication-deficient adenovirus containing human CYP21A2 in 21-OHD CAH knockout mice. An intra-adrenal injection of the vector demonstrated successful morphological and functional restoration of adrenocortical function up to 40 days159. A subsequent study of an intravenous injection of an AAV-CYP21A2 vector in CAH mice demonstrated phenotypic changes (for example, weight gain) and restoration of steroidogenesis for more than 15 weeks160 and, in a separate but similar study in CAH mice, for 8 weeks161. Intramuscular injection of an AAV vector with mouse Cyp21a1 in CAH mice resulted in enzyme expression for more than 6 months162.
BBP-631 (Adrenas Therapeutics, USA) is an AAV5 vector encoding human CYP21A2. Preclinical durability studies of a single intravenous injection in CAH mice and cynomolgus monkeys demonstrated adrenal tropism of the vector, with robust, dose-dependent, human CYP21A2 RNA expression through to 12 weeks and durable expression of vector genome copies up to 24 weeks163,164. A phase I–II, open-label, dose-escalation trial investigating the safety and efficacy of gene therapy for adults with classic CAH is under way165.
Conclusions
Multiple hormonal imbalances complicate the management of CAH. Treatment involves balancing glucocorticoid and mineralocorticoid doses to effectively address both adrenal insufficiency and androgen excess. Enlarged hyperplastic adrenals efficiently produce androgens, which complicates the management of CAH and often necessitates supraphysiological glucocorticoid therapy to adequately suppress the ACTH-driven androgen excess. The overarching treatment goals are to prevent life-threatening adrenal crises across all ages, prevent virilization, optimize linear growth and development in affected children, and prevent long-term adverse clinical and patient-reported outcomes from glucocorticoid under-treatment or over-treatment. Adequacy of therapy is monitored clinically and biochemically; however, existing therapies fail to address disease-specific hormonal imbalances and traditional disease biomarkers are wrought with variabilities. 11-Oxygenated androgens require further study in patients with CAH as the profound ability of the CAH-affected adrenal to produce androgens includes these under-recognized, bioactive androgens. Future research in CAH aims to tailor therapy to maximize clinical benefits and minimize long-term adverse outcomes. The multitude of new advances in glucocorticoid replacement therapy, glucocorticoid-sparing adjuvant therapies, and cell-based and gene-based therapies promises improvement of patient outcomes.
References
Claahsen-van der Grinten, H. L. et al. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr. Rev. 43, 91–159 (2022).
White, P. C. Neonatal screening for congenital adrenal hyperplasia. Nat. Rev. Endocrinol. 5, 490–498 (2009).
Chiou, S. H., Hu, M. C. & Chung, B. C. A missense mutation at Ile172–Asn or Arg356–Trp causes steroid 21-hydroxylase deficiency. J. Biol. Chem. 265, 3549–3552 (1990).
Hu, M. C. & Chung, B. C. Expression of human 21-hydroxylase (P450c21) in bacterial and mammalian cells: a system to characterize normal and mutant enzymes. Mol. Endocrinol. 4, 893–898 (1990).
Tusie-Luna, M. T., Traktman, P. & White, P. C. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J. Biol. Chem. 265, 20916–20922 (1990).
Hannah-Shmouni, F. et al. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet. Med. 19, 1276–1279 (2017).
Ezquieta, B., Ruano, M. L., Dulín, E., Arnao, D. R. & Rodríguez, A. Prevalence of frequent recessive diseases in the Spanish population through DNA analyses on samples from the neonatal screening. Med. Clin. 125, 493–495 (2005).
Phedonos, A. A. et al. High carrier frequency of 21-hydroxylase deficiency in Cyprus. Clin. Genet. 84, 585–588 (2013).
Baumgartner-Parzer, S. M., Nowotny, P., Heinze, G., Waldhäusl, W. & Vierhapper, H. Carrier frequency of congenital adrenal hyperplasia (21-hydroxylase deficiency) in a middle European population. J. Clin. Endocrinol. Metab. 90, 775–778 (2005).
Delle Piane, L., Rinaudo, P. F. & Miller, W. L. 150 Years of congenital adrenal hyperplasia: translation and commentary of De Crecchio’s classic paper from 1865. Endocrinology 156, 1210–1217 (2015).
Wilkins, L., Lewis, R. A., Klein, R. & Rosemberg, E. The suppression of androgen secretion by cortisone in a case of congenital adrenal hyperplasia. Bull. Johns. Hopkins Hosp. 86, 249–252 (1950).
Miller, W. L. The hypothalamic-pituitary-adrenal axis: a brief history. Horm. Res. Paediatr. 89, 212–223 (2018).
Van Wyk, J. J. et al. The use of adrenalectomy as a treatment for congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 81, 3180–3190 (1996).
Merke, D. P. et al. Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 85, 1114–1120 (2000).
Kamrath, C., Wettstaedt, L., Boettcher, C., Hartmann, M. F. & Wudy, S. A. Androgen excess is due to elevated 11-oxygenated androgens in treated children with congenital adrenal hyperplasia. J. Steroid Biochem. Mol. Biol. 178, 221–228 (2018).
Merke, D. P. et al. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N. Engl. J. Med. 343, 1362–1368 (2000).
Charmandari, E. et al. Adrenomedullary function may predict phenotype and genotype in classic 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 87, 3031–3037 (2002).
Arvat, E. et al. Interaction between glucagon and human corticotropin-releasing hormone or vasopressin on ACTH and cortisol secretion in humans. Eur. J. Endocrinol. 143, 99–104 (2000).
Merke, D. P. & Auchus, R. J. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N. Engl. J. Med. 383, 1248–1261 (2020).
Turcu, A. F. et al. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur. J. Endocrinol. 174, 601–609 (2016).
Kolli, V. et al. Morphologic and molecular characterization of adrenals and adrenal rest affected by congenital adrenal hyperplasia. Front. Endocrinol. 12, 1150 (2021).
Arlt, W. et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J. Clin. Endocrinol. Metab. 95, 5110–5121 (2010).
Finkielstain, G. P. et al. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 97, 4429–4438 (2012).
Speiser, P. W. et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 103, 4043–4088 (2018).
Melin, J. et al. Pharmacokinetic/pharmacodynamic evaluation of hydrocortisone therapy in pediatric patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 105, e1729–e1740 (2020).
Bacila, I. et al. International practice of corticosteroid replacement therapy in congenital adrenal hyperplasia: data from the I-CAH registry. Eur. J. Endocrinol. 184, 553–563 (2021).
Ng, S. M., Stepien, K. M. & Krishan, A. Glucocorticoid replacement regimens for treating congenital adrenal hyperplasia. Cochrane Database Syst. Rev. 3, CD012517 (2020).
Bonfig, W., Bechtold, S., Schmidt, H., Knorr, D. & Schwarz, H. P. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J. Clin. Endocrinol. Metab. 92, 1635–1639 (2007).
Merke, D. P., Cho, D., Anton Calis, K., Keil, M. F. & Chrousos, G. P. Hydrocortisone suspension and hydrocortisone tablets are not bioequivalent in the treatment of children with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 86, 441–445 (2001).
Barillas, J. E., Eichner, D., Van Wagoner, R. & Speiser, P. W. Iatrogenic cushing syndrome in a child with congenital adrenal hyperplasia: erroneous compounding of hydrocortisone. J. Clin. Endocrinol. Metab. 103, 7–11 (2018).
Gudeman, J., Jozwiakowski, M., Chollet, J. & Randell, M. Potential risks of pharmacy compounding. Drugs R D 13, 1–8 (2013).
Guharoy, R., Noviasky, J., Haydar, Z., Fakih, M. G. & Hartman, C. Compounding pharmacy conundrum. Chest 143, 896–900 (2013).
Neumann, U. et al. Quality of compounded hydrocortisone capsules used in the treatment of children. Eur. J. Endocrinol. 177, 239–242 (2017).
Neumann, U. et al. Absorption and tolerability of taste-masked hydrocortisone granules in neonates, infants and children under 6 years of age with adrenal insufficiency. Clin. Endocrinol. 88, 21–29 (2018).
Neumann, U. et al. A prospective study of children aged 0–8 years with CAH and adrenal insufficiency treated with hydrocortisone granules. J. Clin. Endocrinol. Metab. 106, e1433–e1440 (2021).
Frisch, H. et al. Salt wasting in simple virilizing congenital adrenal hyperplasia. J. Pediatr. Endocrinol. Metab. 14, 1649–1655 (2001).
Nimkarn, S., Lin-Su, K., Berglind, N., Wilson, R. C. & New, M. I. Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 92, 137–142 (2007).
Muthusamy, K. et al. Clinical review: adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J. Clin. Endocrinol. Metab. 95, 4161–4172 (2010).
Wilkins, L. in The Diagnosis and Treatment of Endocrine Disorders in Childhood and Adolescence Ch. 15, 368-381 (Charles C. Thomas, 1965).
Pofi, R. et al. Plasma renin measurements are unrelated to mineralocorticoid replacement dose in patients with primary adrenal insufficiency. J. Clin. Endocrinol. Metab. 105, 314–326 (2020).
Bonfig, W. et al. Sodium chloride supplementation is not routinely performed in the majority of german and austrian infants with classic salt-wasting congenital adrenal hyperplasia and has no effect on linear growth and hydrocortisone or fludrocortisone dose. Horm. Res. Paediatr. 89, 7–12 (2018).
Bonfig, W. & Schwarz, H. P. Blood pressure, fludrocortisone dose and plasma renin activity in children with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency followed from birth to 4 years of age. Clin. Endocrinol. 81, 871–875 (2014).
Bonfig, W. et al. Blood pressure in a large cohort of children and adolescents with classic adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. Am. J. Hypertens. 29, 266–272 (2016).
Torky, A. et al. Cardiovascular disease risk factors and metabolic morbidity in a longitudinal study of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 106, e5247–e5257 (2021).
Thorn, G. W., Renold, A. E., Morse, W. I., Goldfien, A. & Reddy, W. J. Highly potent adrenal cortical steroids: structure and biologic activity. Ann. Intern. Med. 43, 979–1000 (1955).
Liu, D. et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 9, 30 (2013).
Mooij, C. F. et al. Influence of 17-hydroxyprogesterone, progesterone and sex steroids on mineralocorticoid receptor transactivation in congenital adrenal hyperplasia. Horm. Res. Paediatr. 83, 414–421 (2015).
Debono, M. et al. Hormonal circadian rhythms in patients with congenital adrenal hyperplasia: identifying optimal monitoring times and novel disease biomarkers. Eur. J. Endocrinol. 173, 727–737 (2015).
Storbeck, K. H. et al. Steroid metabolome analysis in disorders of adrenal steroid biosynthesis and metabolism. Endocr. Rev. 40, 1605–1625 (2019).
Turcu, A. F., Rege, J., Auchus, R. J. & Rainey, W. E. 11-Oxygenated androgens in health and disease. Nat. Rev. Endocrinol. 16, 284–296 (2020).
Turcu, A. F. et al. 11-Oxygenated androgens are biomarkers of adrenal volume and testicular adrenal rest tumors in 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 102, 2701–2710 (2017).
Jha, S. et al. 11-Oxygenated androgens useful in the setting of discrepant conventional biomarkers in 21-hydroxylase deficiency. J. Endocr. Soc. 5, bvaa192 (2021).
Pretorius, E. et al. 11-ketotestosterone and 11-ketodihydrotestosterone in castration resistant prostate cancer: potent androgens which can no longer be ignored. PLoS One 11, e0159867 (2016).
Rege, J. et al. 11-Ketotestosterone is the dominant circulating bioactive androgen during normal and premature adrenarche. J. Clin. Endocrinol. Metab. 103, 4589–4598 (2018).
Engels, M. et al. Glucocorticoid activity of adrenal steroid precursors in untreated patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 104, 5065–5072 (2019).
Pijnenburg-Kleizen, K. J. et al. Adrenal steroid metabolites accumulating in congenital adrenal hyperplasia lead to transactivation of the glucocorticoid receptor. Endocrinology 156, 3504–3510 (2015).
Ali, S. R. et al. Real-world estimates of adrenal insufficiency-related adverse events in children with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 106, e192–e203 (2021).
El-Maouche, D. et al. Longitudinal assessment of illnesses, stress dosing, and illness sequelae in patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 103, 2336–2345 (2018).
Falhammar, H. et al. Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 99, E2715–E2721 (2014).
Jenkins-Jones, S. et al. Poor compliance and increased mortality, depression and healthcare costs in patients with congenital adrenal hyperplasia. Eur. J. Endocrinol. 178, 309–320 (2018).
Rushworth, R. L., Torpy, D. J., Stratakis, C. A. & Falhammar, H. Adrenal crises in children: perspectives and research directions. Horm. Res. Paediatr. 89, 341–351 (2018).
Reisch, N. et al. Frequency and causes of adrenal crises over lifetime in patients with 21-hydroxylase deficiency. Eur. J. Endocrinol. 167, 35–42 (2012).
Hahner, S. et al. High incidence of adrenal crisis in educated patients with chronic adrenal insufficiency: a prospective study. J. Clin. Endocrinol. Metab. 100, 407–416 (2015).
Hahner, S. et al. Epidemiology of adrenal crisis in chronic adrenal insufficiency: the need for new prevention strategies. Eur. J. Endocrinol. 162, 597–602 (2010).
Smans, L. C., Van der Valk, E. S., Hermus, A. R. & Zelissen, P. M. Incidence of adrenal crisis in patients with adrenal insufficiency. Clin. Endocrinol. 84, 17–22 (2016).
Odenwald, B. et al. Children with classic congenital adrenal hyperplasia experience salt loss and hypoglycemia: evaluation of adrenal crises during the first 6 years of life. Eur. J. Endocrinol. 174, 177–186 (2016).
Rushworth, R. L., Torpy, D. J. & Falhammar, H. Adrenal crisis. N. Engl. J. Med. 381, 852–861 (2019).
Weber, J. et al. Low adrenomedullary function predicts acute illness in infants with classical congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 107, e264–e271 (2022).
Donaldson, M. D. et al. Presentation, acute illness, and learning difficulties in salt wasting 21-hydroxylase deficiency. Arch. Dis. Child. 70, 214–218 (1994).
Abe, Y. et al. Manifestations and characteristics of congenital adrenal hyperplasia-associated encephalopathy. Brain Dev. 38, 638–647 (2016).
Tresoldi, A. S. et al. Increased infection risk in addison’s disease and congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 105, 418–429 (2020).
Arlt, W., Baldeweg, S. E., Pearce, S. H. S. & Simpson, H. L. Endocrinology in the time of COVID-19: management of adrenal insufficiency. Eur. J. Endocrinol. https://doi.org/10.1530/eje-20-0361 (2020).
Hahner, S. & Allolio, B. Therapeutic management of adrenal insufficiency. Best. Pract. Res. Clin. Endocrinol. Metab. 23, 167–179 (2009).
Repping-Wuts, H. J., Stikkelbroeck, N. M., Noordzij, A., Kerstens, M. & Hermus, A. R. A glucocorticoid education group meeting: an effective strategy for improving self-management to prevent adrenal crisis. Eur. J. Endocrinol. 169, 17–22 (2013).
Nilsson, O., Marino, R., De Luca, F., Phillip, M. & Baron, J. Endocrine regulation of the growth plate. Horm. Res. 64, 157–165 (2005).
Charmandari, E., Hindmarsh, P. C., Johnston, A. & Brook, C. G. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: alterations in cortisol pharmacokinetics at puberty. J. Clin. Endocrinol. Metab. 86, 2701–2708 (2001).
Merke, D. P. & Poppas, D. P. Management of adolescents with congenital adrenal hyperplasia. Lancet Diabetes Endocrinol. 1, 341–352 (2013).
Stikkelbroeck, N. M. M. L., Van’T Hof-Grootenboer, B. A. E., Hermus, A. R. M. M., Otten, B. J. & Van’T Hof, M. A. Growth inhibition by glucocorticoid treatment in salt wasting 21-hydroxylase deficiency: in early infancy and (pre)puberty. J. Clin. Endocrinol. Metab. 88, 3525–3530 (2003).
Quintos, J. B., Vogiatzi, M. G., Harbison, M. D. & New, M. I. Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 86, 1511–1517 (2001).
Lin-Su, K., Harbison, M. D., Lekarev, O., Vogiatzi, M. G. & New, M. I. Final adult height in children with congenital adrenal hyperplasia treated with growth hormone. J. Clin. Endocrinol. Metab. 96, 1710–1717 (2011).
Subbarayan, A., Dattani, M. T., Peters, C. J. & Hindmarsh, P. C. Cardiovascular risk factors in children and adolescents with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin. Endocrinol. 80, 471–477 (2014).
Tamhane, S. et al. Cardiovascular and metabolic outcomes in congenital adrenal hyperplasia: a systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 103, 4097–4103 (2018).
Falhammar, H. et al. Increased cardiovascular and metabolic morbidity in patients with 21-hydroxylase deficiency: a Swedish population-based national cohort study. J. Clin. Endocrinol. Metab. 100, 3520–3528 (2015).
Hirschberg, A. L. et al. Reproductive and perinatal outcomes in women with congenital adrenal hyperplasia: a population-based cohort study. J. Clin. Endocrinol. Metab. 106, e957–e965 (2021).
Mallappa, A. et al. Alterations in hydrocortisone pharmacokinetics in a patient with congenital adrenal hyperplasia following bariatric surgery. J. Endocr. Soc. 1, 994–1001 (2017).
Mah, P. M. et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin. Endocrinol. 61, 367–375 (2004).
Ahmed, A. et al. A switch in hepatic cortisol metabolism across the spectrum of non alcoholic fatty liver disease. PLoS One 7, e29531 (2012).
Holt, H. B. et al. Cortisol clearance and associations with insulin sensitivity, body fat and fatty liver in middle-aged men. Diabetologia 50, 1024–1032 (2007).
Buckley, L. & Humphrey, M. B. Glucocorticoid-induced osteoporosis. N. Engl. J. Med. 379, 2547–2556 (2018).
Rangaswamaiah, S., Gangathimmaiah, V., Nordenstrom, A. & Falhammar, H. Bone mineral density in adults with congenital adrenal hyperplasia: a systematic review and meta-analysis. Front. Endocrinol. 11, 493 (2020).
Falhammar, H. et al. Increased prevalence of fractures in congenital adrenal hyperplasia: a Swedish population-based national cohort study. J. Clin. Endocrinol. Metab. 107, e475–e486 (2022).
Mallappa, A. & Merke, D. P. in Management of Adrenal Masses in Children and Adults 207–224 (Springer, 2017).
Nermoen, I. & Falhammar, H. Prevalence and characteristics of adrenal tumors and myelolipomas in congenital adrenal hyperplasia: a systematic review and meta-analysis. Endocr. Pract. 26, 1351–1365 (2020).
El-Maouche, D. et al. Adrenal morphology and associated comorbidities in congenital adrenal hyperplasia. Clin. Endocrinol. 91, 247–255 (2019).
Reisch, N. et al. Total adrenal volume but not testicular adrenal rest tumor volume is associated with hormonal control in patients with 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 95, 2065–2072 (2010).
Bouvattier, C. et al. Clinical outcome, hormonal status, gonadotrope axis, and testicular function in 219 adult men born with classic 21-hydroxylase deficiency. a French national survey. J. Clin. Endocrinol. Metab. 100, 2303–2313 (2015).
Mendes-Dos-Santos, C. T. et al. Prevalence of testicular adrenal rest tumor and factors associated with its development in congenital adrenal hyperplasia. Horm. Res. Paediatr. 90, 161–168 (2018).
Engels, M. et al. Testicular adrenal rest tumors: current insights on prevalence, characteristics, origin, and treatment. Endocr. Rev. 40, 973–987 (2019).
Smeets, E. E. et al. Molecular characterization of testicular adrenal rest tumors in congenital adrenal hyperplasia: lesions with both adrenocortical and Leydig cell features. J. Clin. Endocrinol. Metab. 100, E524–E530 (2015).
Tanaka, M., Enatsu, N., Chiba, K. & Fujisawa, M. Two cases of reversible male infertility due to congenital adrenal hyperplasia combined with testicular adrenal rest tumor. Reprod. Med. Biol. 17, 93–97 (2018).
Stikkelbroeck, N. M. et al. Prevalence of ovarian adrenal rest tumours and polycystic ovaries in females with congenital adrenal hyperplasia: results of ultrasonography and MR imaging. Eur. Radiol. 14, 1802–1806 (2004).
Claahsen-van der Grinten, H. L., Stikkelbroeck, N., Falhammar, H. & Reisch, N. Management of endocrine disease: gonadal dysfunction in congenital adrenal hyperplasia. Eur. J. Endocrinol. 184, R85–R97 (2021).
Falhammar, H. et al. Reduced frequency of biological and increased frequency of adopted children in males with 21-hydroxylase deficiency: a Swedish population-based national cohort study. J. Clin. Endocrinol. Metab. 102, 4191–4199 (2017).
Johannsson, G. et al. Improving glucocorticoid replacement therapy using a novel modified-release hydrocortisone tablet: a pharmacokinetic study. Eur. J. Endocrinol. 161, 119–130 (2009).
Johannsson, G. et al. Improved cortisol exposure-time profile and outcome in patients with adrenal insufficiency: a prospective randomized trial of a novel hydrocortisone dual-release formulation. J. Clin. Endocrinol. Metab. 97, 473–481 (2012).
Newell-Price, J. et al. Modified-release hydrocortisone for circadian therapy: a proof-of-principle study in dexamethasone-suppressed normal volunteers. Clin. Endocrinol. 68, 130–135 (2008).
Whitaker, M. et al. An oral multiparticulate, modified-release, hydrocortisone replacement therapy that provides physiological cortisol exposure. Clin. Endocrinol. 80, 554–561 (2014).
Verma, S. et al. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin. Endocrinol. 72, 441–447 (2010).
Mallappa, A. et al. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 100, 1137–1145 (2015).
Jones, C. M. et al. Modified-release and conventional glucocorticoids and diurnal androgen excretion in congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 102, 1797–1806 (2017).
Merke, D. P. et al. Modified-release hydrocortisone in congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 106, e2063–e2077 (2021).
Debono, M. et al. Modified-release hydrocortisone to provide circadian cortisol profiles. J. Clin. Endocrinol. Metab. 94, 1548–1554 (2009).
Nella, A. A. et al. A phase 2 study of continuous subcutaneous hydrocortisone infusion in adults with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 101, 4690–4698 (2016).
Mallappa, A. et al. Long-term use of continuous subcutaneous hydrocortisone infusion therapy in patients with congenital adrenal hyperplasia. Clin. Endocrinol. 89, 399–407 (2018).
Turcu, A. F. et al. 24-Hour profiles of 11-oxygenated C(19) steroids and Δ(5)-steroid sulfates during oral and continuous subcutaneous glucocorticoids in 21-hydroxylase deficiency. Front. Endocrinol. 12, 751191 (2021).
Miller, W. L. & Auchus, R. J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 32, 81–151 (2011).
Nixon, M. et al. ABCC1 confers tissue-specific sensitivity to cortisol versus corticosterone: a rationale for safer glucocorticoid replacement therapy. Sci. Transl. Med. 8, 352ra109 (2016).
Kyle, C. et al. Comparison of acute effects of corticosterone versus cortisol (hydrocortisone) infusion in adults with congenital adrenal hyperplasia. Endocr. Abstr. https://doi.org/10.1530/endoabs.59.OC2.2 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00001521 (1995).
De Bono, J. S. et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 364, 1995–2005 (2011).
Auchus, R. J. et al. Abiraterone acetate to lower androgens in women with classic 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 99, 2763–2770 (2014).
Wright, C., O’Day, P., Alyamani, M., Sharifi, N. & Auchus, R. J. Abiraterone acetate treatment lowers 11-oxygenated androgens. Eur. J. Endocrinol. 182, 413–421 (2020).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02574910 (2021).
El-Maouche, D. et al. A phase 2, multicenter study of nevanimibe for the treatment of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 105, 2771–2778 (2020).
Lapensee, C. R. et al. ATR-101, a selective and potent inhibitor of acyl-CoA acyltransferase 1, induces apoptosis in H295R adrenocortical cells and in the adrenal cortex of dogs. Endocrinology 157, 1775–1788 (2016).
Turcu, A. F. et al. Single-dose study of a corticotropin-releasing factor receptor-1 antagonist in women with 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 101, 1174–1180 (2016).
Auchus, R. J. et al. Crinecerfont lowers elevated hormone markers in adults with 21-hydroxylase deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 107, 801–812 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04490915 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04806451 (2021).
Sarafoglou, K. et al. Tildacerfont in adults with classic congenital adrenal hyperplasia: results from two phase 2 studies. J. Clin. Endocrinol. Metab. 106, e4666–e4679 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04457336 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04544410 (2021).
Spruce Biosciences. Pediatric Classic CAH, Pipeline https://sprucebiosciences.com/pipeline/indications/pediatric-classic-cah/ (2021).
Feldhaus, A. L. et al. ALD1613, a novel long-acting monoclonal antibody to control ACTH-driven pharmacology. Endocrinology https://doi.org/10.1210/en.2016-1455 (2016).
Gehrand, A. L., Phillips, J., Malott, K. & Raff, H. A long-acting neutralizing monoclonal ACTH antibody blocks corticosterone and adrenal gene responses in neonatal rats. Endocrinology 160, 1719–1730 (2019).
Kusnetzow, A. et al. SAT-364 nonpeptide orally-bioavailable ACTH antagonists: suppression of ACTH-induced corticosterone secretion and adrenal hypertrophy in rats. J. Endocr. Soc. https://doi.org/10.1210/js.2019-SAT-364 (2019).
Kusnetzow, A. K. et al. MON-176 Discovery and Identification of Late Stage Selective Nonpeptide ACTH Antagonists for the Treatment of Cushing’s Disease, Ectopic ACTH secreting tumors, and congenital adrenal hyperplasia. J. Endocr. Soc. https://doi.org/10.1210/jendso/bvaa046.690 (2020).
Markison, S. et al. OR19-03 effects of nonpeptide orally bioavailable ACTH antagonists on adrenal gland size and function in rats. J. Endocr. Soc. https://doi.org/10.1210/jendso/bvaa046.699 (2020).
Crinetics. Crinetics Pharmaceuticals Lead ACTH Antagonist for Congenital Adrenal Hyperplasia and Cushing’s Disease (Crn04894) Advances into Phase 1 Study https://crinetics.com/lead-acth-antagonist-crn04894-enters-phase1-study/ (2021).
Goldenberg, A. J. et al. Effect of a melanocortin type 2 receptor (MC2R) antagonist on the corticosterone response to hypoxia and ACTH stimulation in the neonatal rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 315, R128–R133 (2018).
Sanders, K., Mol, J. A., Kooistra, H. S. & Galac, S. Melanocortin 2 receptor antagonists in canine pituitary-dependent hypercortisolism: in vitro studies. Vet. Res. Commun. 42, 283–288 (2018).
Dagalakis, U., Mallappa, A., Elman, M., Quezado, M. & Merke, D. P. Positive fertility outcomes in a female with classic congenital adrenal hyperplasia following bilateral adrenalectomy. Int. J. Pediatr. Endocrinol. 2016, 10 (2016).
Van Wyk, J. J. & Ritzen, E. M. The role of bilateral adrenalectomy in the treatment of congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 88, 2993–2998 (2003).
Ogilvie, C. M., Rumsby, G., Kurzawinski, T. & Conway, G. S. Outcome of bilateral adrenalectomy in congenital adrenal hyperplasia: one unit’s experience. Eur. J. Endocrinol. 154, 405–408 (2006).
MacKay, D., Nordenstrom, A. & Falhammar, H. Bilateral adrenalectomy in congenital adrenal hyperplasia: a systematic review and meta-analysis. J. Clin. Endocrinol. Metab. 103, 1767–1778 (2018).
Crocker, M. K. et al. Use of PET/CT with cosyntropin stimulation to identify and localize adrenal rest tissue following adrenalectomy in a woman with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 97, E2084–E2089 (2012).
Burman, P., Falhammar, H., Waldenström, E., Sundin, A. & Bitzén, U. 11C-Metomidate PET/CT detected multiple ectopic adrenal rest tumors in a woman with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 106, e675–e679 (2021).
Tiosano, D. et al. Ovarian adrenal rest tumor in a congenital adrenal hyperplasia patient with adrenocorticotropin hypersecretion following adrenalectomy. Horm. Res. Paediatr. 74, 223–228 (2010).
Waszut, U., Szyszka, P. & Dworakowska, D. Understanding mitotane mode of action. J. Physiol. Pharmacol. 68, 13–26 (2017).
Kiseljak-Vassiliades, K. et al. American Association of Clinical Endocrinology disease state clinical review on the evaluation and management of adrenocortical carcinoma in an adult: a practical approach. Endocr. Pract. 26, 1366–1383 (2020).
Tritos, N. A. & Biller, B. M. K. Medical therapy for Cushing’s syndrome in the twenty-first century. Endocrinol. Metab. Clin. North. Am. 47, 427–440 (2018).
Bry-Gauillard, H., Cartes, A. & Young, J. Mitotane for 21-hydroxylase deficiency in an infertile man. N. Engl. J. Med. 371, 2042–2044 (2014).
Bachelot, A. et al. Effects of mitotane on testicular adrenal rest tumors in congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a retrospective series of five patients. Eur. J. Endocrinol. 184, 365–371 (2021).
U.S. Food and Drug Administration LYSODREN® (mitotane) Tablets https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/016885s023lbl.pdf (2017).
Tritos, N. A. & Biller, B. M. K. Medical management of Cushing’s disease. J. Neurooncol. 117, 407–414 (2014).
Ruiz-Babot, G. et al. Modeling congenital adrenal hyperplasia and testing interventions for adrenal insufficiency using donor-specific reprogrammed cells. Cell Rep. 22, 1236–1249 (2018).
Bornstein, S. R. et al. New horizons: novel adrenal regenerative therapies. J. Clin. Endocrinol. Metabol. https://doi.org/10.1210/clinem/dgaa438 (2020).
Balyura, M. et al. Transplantation of bovine adrenocortical cells encapsulated in alginate. Proc. Natl Acad. Sci. USA 112, 2527–2532 (2015).
Tajima, T. et al. Restoration of adrenal steroidogenesis by adenovirus-mediated transfer of human cytochromeP450 21-hydroxylase into the adrenal gland of21-hydroxylase-deficient mice. Gene Ther. 6, 1898–1903 (1999).
Perdomini, M., Dos Santos, C., Goumeaux, C., Blouin, V. & Bougneres, P. An AAVrh10-CAG-CYP21-HA vector allows persistent correction of 21-hydroxylase deficiency in a Cyp21(-/-) mouse model. Gene Ther. 24, 275–281 (2017).
Markmann, S. et al. Biology of the adrenal gland cortex obviates effective use of adeno-associated virus vectors to treat hereditary adrenal disorders. Hum. Gene Ther. 29, 403–412 (2018).
Naiki, Y. et al. Extra-adrenal induction of Cyp21a1 ameliorates systemic steroid metabolism in a mouse model of congenital adrenal hyperplasia. Endocr. J. 63, 897–904 (2016).
Eclov, R. J. et al. OR25-01 Durable CYP21A2 gene therapy in non-human primates for treatment of congenital adrenal hyperplasia. J. Endocr. Soc. https://doi.org/10.1210/jendso/bvaa046.899 (2020).
Merke, D. P. et al. Design of a phase 1/2 open-label, dose-escalation study of the safety and efficacy of gene therapy in adults with classic congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency through administration of an adeno-associated virus (AAV) serotype 5-based recombinant vector encoding the human CYP21A2 Gene. J. Endocr. Soc. 5, A82–A82 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04783181 (2020).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
D.P.M. received research funds from Diurnal Limited through the National Institutes of Health Cooperative Research and Development Agreement. During the writing of this manuscript, A.M. took up employment at AstraZeneca and is currently employed there.
Peer review
Peer review information
Nature Reviews Endocrinology thanks Antonio Balsamo, who co-reviewed the manuscript with Rita Ortolano, Anna Nordenström and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mallappa, A., Merke, D.P. Management challenges and therapeutic advances in congenital adrenal hyperplasia. Nat Rev Endocrinol 18, 337–352 (2022). https://doi.org/10.1038/s41574-022-00655-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41574-022-00655-w
This article is cited by
-
Increased risk of nephrolithiasis: an emerging issue in children with congenital adrenal hyperplasia due to 21-hydroxylase deficiency
Endocrine (2024)
-
EndoBridge 2023: highlights and pearls
Hormones (2024)
-
Genetic control of typical and atypical sex development
Nature Reviews Urology (2023)
-
The Spectrum of Endocrine Pathology
Endocrine Pathology (2023)
