
Abstract
Immune-mediated inflammatory diseases (IMIDs) are characterized by excessive and uncontrolled inflammation and thrombosis, both of which are responsible for organ damage, morbidity and death. Platelets have long been known for their role in primary haemostasis, but they are now also considered to be components of the immune system and to have a central role in the pathogenesis of IMIDs. In patients with IMIDs, platelets are activated by disease-specific factors, and their activation often reflects disease activity. Here we summarize the evidence showing that activated platelets have an active role in the pathogenesis and the progression of IMIDs. Activated platelets produce soluble factors and directly interact with immune cells, thereby promoting an inflammatory phenotype. Furthermore, platelets participate in tissue injury and promote abnormal tissue healing, leading to fibrosis. Targeting platelet activation and targeting the interaction of platelets with the immune system are novel and promising therapeutic strategies in IMIDs.
Similar content being viewed by others

Introduction
Immune-mediated inflammatory diseases (IMIDs) such as systemic lupus erythematosus (SLE), rheumatoid arthritis and psoriasis are highly prevalent, for example affecting 3–8% of the population in high-income countries, and they remain a major cause of morbidity and death worldwide despite therapeutic advances1. IMIDs include autoimmune diseases characterized by autoantibodies and autoreactive T cells, such as SLE or systemic sclerosis, and inflammatory diseases such as psorias or inflammatory bowel disease. For most of these diseases, corticosteroids and nonspecific (antiproliferative) immunosuppressive drugs such as methotrexate or mycophenolate mofetil are the standard of care. These drugs reduce disease activity but have significant side effects, including myelosuppression and increased risk of infection2. Furthermore, the risk of cardiovascular complications is increased significantly in individuals with IMIDs, as a result of both the disease itself and some of the drugs used to treat it (such as corticosteroids). For example, the risk of myocardial infarction is 50 times higher in young women with SLE than in age-matched and sex-matched controls3. The increased risk of death from cardiovascular complications is shared across different IMIDs4, which positions IMIDs as an independent risk factor for cardiovascular disease. A disease feature that has been identified as having a major contribution to the increased risk of thrombosis and cardiovascular disease is platelet activation. In the past 20 years, interest in understanding the immune roles of platelets has grown exponentially; in addition to their well-characterized role in primary haemostasis, platelets have been found to have important roles in immunity and inflammation.
Platelets are small (2–5 µm) anucleate cells that originate from megakaryocytes (Box 1). They are the most prevalent blood component after red blood cells, and they patrol the circulatory system to seal areas of damage to the vessel wall through platelet aggregation. The lack of a nucleus, which is suggested to provide platelets with the ability to spread optimally and to better resist shear flow in the case of blood vessel damage, is partially compensated for by the presence of coding and non-coding RNAs in the cytoplasm, as well as cellular organelles such as ribosomes for protein translation and mitochondria for energy production. Platelets contain two types of granules. Dense granules contain serotonin, nucleotides (adenosine triphosphate (ATP) and adenosine diphosphate (ADP)) and calcium, which increase platelet activation in autocrine and paracrine manners5. α-Granules contain molecules that promote haemostasis such as von Willebrand factor and CXCL4 (also known as platelet factor 4), as well as molecules associated with the immunological functions of platelets, such as CD40 ligand (CD40L), P-selectin (also known as CD62P) and complement5.
During wounding, platelets rapidly recognize the damaged vasculature, leading to activation and aggregation of platelets and their expression of receptors that promote the recruitment of immune cells. This process, which contributes to preventing the invasion of the bloodstream by microbial agents, exemplifies the role of platelets as a front line of immune defence. Some clinical examples reinforce this notion. For example, during sepsis (resulting from severe infection), platelets may become overactivated and form clots in small vessels — a phenomenon known as disseminated intravascular coagulation — to limit the haematogenous spread of the infectious agent. Although disseminated intravascular coagulation and, more generally, sepsis-associated thrombocytopenia are associated with a severe prognosis, the inhibition of disseminated intravascular coagulation in mice is linked to greater lethality from infection with Gram-negative bacteria6, such that this function of platelets is now known as immunothrombosis. Furthermore, platelets migrate to the site of bacterial infection and can trap microorganisms on their surface by mechanically scavenging fibrin and bacteria, which ultimately facilitates neutrophil activation and phagocytosis to clear the infection7. More recent findings that we discuss here indicate that in addition to these ‘mechanical’ roles in immune defence, platelets also have roles in linking the innate and adaptive immune responses and may promote inflammation and organ damage.
This Review summarizes current evidence as to the mechanisms by which platelets become activated in IMIDs and change their phenotype to secrete soluble factors and express surface glycoproteins through which they alter the function of immune cells. Activated platelets interact with and influence the phenotype of both innate and adaptive immune cells, making them a driver of immune dysregulation in IMIDs. Furthermore, owing to their increased aggregation, activated platelets contribute to the increased cardiovascular disease risk that characterizes IMIDs and to the promotion of end organ damage by magnifying local inflammation and fibrosis. The evidence that links platelets to the pathogenesis of IMIDs is rapidly evolving (reviewed in refs. 8,9), and several strategies targeting platelet activation or platelet-derived factors are being developed and tested in patients.
Mechanisms of platelet activation
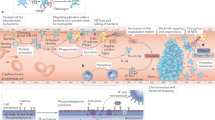
Circulating platelets patrol the bloodstream in a resting state (Fig. 1a), waiting to encounter stimuli such as vessel wall injury or bacteria to become activated. Platelet activation is a multistep and complex event leading to several outcomes (Fig. 1b). Following activation, platelets lose their discoid shape and acquire a spherical conformation with lamellipodia that promote interaction with other cells5. This reshaping is secondary to intraplatelet calcium release, leading to the activation of the actin–myosin cytoskeleton5. In clinical practice, this reshaping may be observed as an increased mean platelet volume, which has been reported in patients with active SLE, inflammatory bowel disease (IBD) and rheumatoid arthritis10,11,12. Platelet activation also results in the exposure on the platelet surface of negatively charged phospholipids, which support the binding and activation of coagulation factors together with tissue factor and promote thrombosis5. Activated platelets release dense granules and α-granules, leading to further amplification of platelet activation through ATP or ADP and complement release13. The relocalization of glycoproteins such as CD40L and P-selectin from cytoplasmic granules to the surface of activated platelets contributes to interaction with immune cells14,15. The production of platelet-derived extracellular vesicles containing numerous molecules from activated platelets can disseminate platelet components to fluids and tissues that are normally inaccessible to platelets16, such as the lymphatic system, as has been shown in a mouse model of inflammatory arthritis17. In summary, activation reshapes the phenotype and morphology of platelets, which may promote their interaction with immune cells. In the setting of IMIDs, several factors can induce such platelet activation (Table 1).

a, Resting platelets express platelet-specific markers such as the glycoproteins GPIIb/IIIa, GPIb and GPVI. Platelets also express Fcγ receptor (FcγR), which enables them to scavenge circulating immune complexes. Resting platelets may inhibit CD8+ T cells by the expression of MHC class I molecules in the absence of co-stimulatory molecules, as well as by expressing the ectonucleosidase CD73, which catalyses the production of anti-inflammatory adenosine from adenosine monophosphate (AMP). b, Activated platelets release cytokines such as IL-1, and molecules such as soluble P-selectin (sP-selectin), soluble CD40 ligand (sCD40L) and serotonin. They also release damage-associated molecular patterns (DAMPs), such as calprotectin and high mobility group box 1 (HMGB1), and extrude mitochondria and mitochondrial DNA (mtDNA) into the extracellular milieu. Activated platelets relocalize to their surface glycoproteins such as P-selectin and CD40L that are initially present in cytoplasmic granules, which promotes interaction with immune cells. Platelet antigens and scavenged immune complexes are captured by antigen-presenting cells (APCs) for processing and immune presentation. Furthermore, platelets express MHC class I molecules together with the co-stimulatory molecule CD86, and may directly present antigen to CD8+ T cells and promote their activation. Finally, activated platelets produce platelet-derived extracellular vesicles that contain platelet-derived molecules such as P-selectin, CD40L, HMGB1 and IL-1. TCR, T cell receptor.
Immune complexes through Fc receptors
Fcγ receptor IIA (FcγRIIA; also known as CD32) recognizes the Fc (crystallizable fragment) region of circulating IgG and is constitutively expressed by human platelets (although absent in mice). Immune complexes purified from patients with SLE promote platelet activation and the production of platelet-derived extracellular vesicles, through an FcγRIIA-dependent mechanism14,18. Transgenic NZB × NZW(F1) lupus-prone mice expressing the human FCGR2A transgene develop more severe lupus-like nephritis and have a worse prognosis than wild-type NZB × NZW(F1) mice18. Upon activation, FcγRIIA-expressing platelets produce platelet-derived extracellular vesicles, interact with circulating neutrophils and precipitate thrombosis in several organs18. Furthermore, FcγRIIA-stimulated platelets release mitochondria, either free or embedded in platelet-derived extracellular vesicles, which function as autoantigens and damage-associated molecular patterns (DAMPs), thereby promoting an (auto)immune response (as discussed later)19. This mechanism of platelet activation is likely to apply to many IMIDs, as IgG-containing immune complexes are found in a wide range of these diseases, including rheumatoid arthritis, systemic sclerosis, vasculitis and Sjögren syndrome20,21. Furthermore, human platelets also express FcαRI (also known as CD89) and FcεR (also known as CD23), which recognize the Fc regions of IgA and IgE, respectively22,23. As IgA-containing or IgE-containing immune complexes are also prevalent in patients with IMIDs such as rheumatoid arthritis, Sjögren syndrome and SLE (reviewed in ref. 24), it is plausible that FcαRI and FcεR constitute an additional pathway of platelet activation in these diseases. Collectively, this body of evidence underscores the importance of immune complex–Fc receptor interactions in platelet activation in human IMIDs.
Platelet-targeting autoantibodies
A subset of IMIDs are characterized by the presence of a large panel of autoantibodies targeting a wide range of autoantigens. Among them, antiphospholipid antibodies, which are a hallmark of antiphospholipid syndrome (APS), are present in one-third of patients with SLE25, as well as in patients with systemic sclerosis26, Sjögren syndrome27 and other IMIDs. The presence of antiphospholipid antibodies is linked to thrombosis and a worse prognosis in connective tissue disorders25,26,27. These antibodies can bind platelets directly through phospholipids present in the cell membrane or through the platelet glycoprotein Ibα (GPIbα) component of GPIb (also known as CD42), which forms part of the receptor for von Willebrand factor, and they promote platelet activation and aggregation28. Importantly, antiphospholipid antibodies also bind lysobisphosphatidic acid presented by endothelial protein C receptor (EPCR) on monocytes and dendritic cells (DCs), which promotes interferon signalling downstream of Toll-like receptor 7 (TLR7) activation by immune complexes, thereby linking the prothrombotic pathway with autoimmune responses29. Consequently, patients with antiphospholipid antibodies have increased circulating levels of markers of platelet activation, such as soluble CD40L and platelet-derived chemokines30,31. In addition to antiphospholipid antibodies, antiplatelet antibodies can bind platelets directly, resulting in their activation and/or immune-mediated destruction through complement-dependent cytotoxicity or antibody-dependent phagocytosis32. Antiplatelet antibodies target mainly the platelet glycoprotein receptors GPIb–IX (consisting of GPIbα, GPIbβ and GPIX) and GPIa in conjunction with the GPIIb/IIIa complex (also known as CD41/CD61)33, and are found in up to 88% of patients with SLE who have immune thrombocytopenia (ITP)34. However, these antibodies are also present in up to 17% of patients with SLE and 22% of patients with systemic sclerosis who have normal platelet counts34,35, which suggests that antiplatelet antibodies might have a role in a wider group of patients with IMIDs.
Toll-like receptors
A large body of literature indicates that platelets express all TLRs at the protein level8,36,37, making platelets the most prevalent innate immune sensors in the bloodstream. In IMIDs, circulating levels of various pathogen-associated molecular patterns and DAMPs can be increased, for example in response to viral reactivation or tissue damage, and can activate platelet TLRs. For example, platelets express functional TLR4 (ref. 38), leading to platelet activation and accumulation in the lungs of mice exposed to lipopolysaccharide37,38, although conflicting data exist regarding the ability of TLR4 agonists to activate platelets39. Alternatively, high mobility group box 1 (HMGB1) may also cause platelet activation through TLR4 (ref. 40). TLR7 is an endosomal TLR constitutively expressed in human and mouse platelets that recognizes single-stranded RNA, and its importance in antiviral immunity has been shown in mice41. Indeed, treatment of mice with a TLR7 agonist led to platelet activation, thrombocytopenia and increased circulating levels of platelet–neutrophil aggregates in a manner dependent on P-selectin binding to P-selectin glycoprotein ligand 1 (PSGL1; encoded by the SELPLG gene)41. Similar platelet activation through TLRs was reported when mice were infected with encephalomyocarditis virus or when human platelets were exposed to encephalomyocarditis virus in vitro. Importantly, the depletion of platelets precipitated the death of mice infected with encephalomyocarditis virus, which was prevented following the injection of wild-type platelets but not Tlr7−/− platelets41. Furthermore, a gain-of-function mutation in the TLR7 gene was discovered in one family with severe and early-onset SLE, and a mouse model carrying this mutation developed severe thrombocytopenia, which is suggestive of platelet activation42. As antibodies directed against mitochondrial RNA are reported in patients with SLE and APS43,44, they may be an additional source of immune complexes capable of promoting antigen internalization and activation of endosomal TLR7 in these diseases. Platelets also express TLR9, which promotes platelet activation in response to CpG dinucleotides, such as those found in the DNA of pathogens37,45, and mitochondrial DNA, which has been reported in the sera of patients with SLE or rheumatoid arthritis19,46. Chronic infection with DNA viruses such as Epstein–Barr virus (EBV) has long been associated with autoimmunity, with recent emphasis on the role of EBV in multiple sclerosis47. Transient EBV reactivation (with measurable viraemia) is relatively common in autoimmune diseases48 and in patients with Long COVID, who may present with autoimmune manifestations49. Indeed, patients with SLE are defective in controlling EBV48, and EBV reactivation correlates with disease flares50. EBV may activate platelets through TLR9 after endocytosis of viral particles51, as well as through the direct binding of EBV to complement receptor type 2 (CR2)52. Therefore, reactivation of viruses such as EBV may promote platelet activation and ultimately disease flares in IMIDs.
Other platelet-activating stimuli
Gain-of-function mutations in components of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome are well known in the context of autoinflammatory diseases characterized by sterile inflammation owing to increased IL-1β production53. Human and mouse platelets express NLRP3, which participates in their activation and some of their immune functions54. Indeed, the NLRP3 inflammasome may be activated through several stimuli, such as RNA virus infection (through mitochondrial antiviral signalling protein (MAVS)), oxidated mitochondrial DNA, mitochondrial reactive oxygen species (ROS)53 and platelet-derived HMGB1 signalling through TLR4 (ref. 55), which increases platelet activation and drives IL-1β production.
In pathological conditions, collagen is abnormally exposed on the damaged blood vessel endothelium or joints and may interact with GPVI or the GPIa/IIa complex on platelets. In mice, a GPVI agonist leads to the production of platelet-derived extracellular vesicles in vitro, and knockdown of Gp6 leads to reduced disease in the K/B×N serum transfer model of arthritis56. In systemic sclerosis, which is characterized by tissue fibrosis (in other words, collagen production), platelets have upregulated GPVI expression57. In SLE, autoantibodies targeting GPVI have been identified and shown to promote platelet activation58. Upon activation, platelets release ATP and ADP, which function as paracrine factors promoting further platelet activation through the ligation of purine receptors (P2X for ATP and P2Y for ADP)59. In the setting of autoimmunity, ATP also functions as a DAMP, which promotes neutrophil chemotaxis through the activation of P2X receptors59. ATP is normally degraded to anti-inflammatory adenosine by ectonucleosidases (CD39 and CD73) expressed by various immune cells60 and platelets themselves61 (Fig. 1a); however, in IMIDs such as SLE or psoriasis, it has been reported that there is defective activity62 or expression63 of CD73, leading to a dysregulated ATP–adenosine balance and immune activation. It is not known whether this mechanism is also responsible for increased platelet activation through ATP–P2X.
Physical factors may also activate platelets. Raynaud phenomenon is characterized by peripheral vasoconstriction of the extremities, which results in a local painful ischaemia of digits and toes. Raynaud phenomenon is found in all patients with systemic sclerosis and also commonly in patients with SLE and other connective tissue diseases. Localized ischaemia–reperfusion injury damages endothelial cells, leading to the exposure of collagen and the release of ROS, which synergize with collagen binding to GPVI to induce platelet activation64,65. Indeed, individuals with primary Raynaud phenomenon are characterized by increased serum levels of markers of platelet activation (such as P-selectin), as well as increased circulating levels of platelet–leukocyte aggregates66,67.
Overall, several inflammatory, haemostatic and physical stimuli can activate platelets in IMIDs. These stimuli often use different signalling pathways to activate platelets68, which might account for the heterogeneity of platelet phenotypes observed in these diseases69. The various stimuli may also have cumulative effects on platelet activation.
Effects of platelet activation
Many studies have confirmed that platelet activation occurs in multiple IMIDs70,71,72,73,74, through the mechanisms described in the previous section. For example, platelet-derived extracellular vesicles were identified in the synovial fluid of patients with rheumatoid arthritis, likely having reached the joint and spread through the lymphatic system, thereby pointing to platelet activation as occurring in the inflamed joints56,75. Here we describe the effects of platelet activation in terms of the release of immune-activating stimuli, antigen presentation and interaction with immune cells (Fig. 1b).
Release of cytokines and signalling molecules
Platelet-derived extracellular vesicles found in the joints and lymphatic system of patients with rheumatoid arthritis17 were found to express surface IL-1α and IL-1β75 and to stimulate fibroblast-like synoviocytes to produce pro-inflammatory CXCL8 (also known as IL-8) in an IL-1α/β-dependent manner, allowing the recruitment of neutrophils into the inflammatory joint and thereby driving disease56. Furthermore, platelet-derived extracellular vesicles in the synovial fluid form aggregates with leukocytes, including neutrophils, which may promote inflammation76 (see later). The IL-1β released by human platelets upon stimulation with serum from patients with SLE stimulates endothelial cell activation through increased expression of adhesion molecules and pro-inflammatory cytokines77.
Serotonin (also known as 5-hydroxytryptamine) is a neurotransmitter derived from tryptophan that is scavenged in the circulation by platelets and concentrated in dense granules, making platelets the largest reservoir of the peripheral serotonin pool78. Serotonin derived from activated platelets in a mouse model of autoimmune arthritis promoted leakage of the joint vasculature, which may promote egress of platelet-derived extracellular vesicles and other molecules from blood into the inflamed joint and the lymphatic system17,79. As most immune cells express at least one type of serotonin receptor, serotonin released from activated platelets may accordingly affect immune cell function. Indeed, mice lacking serotonin synthesis (Tph1−/− mice) have decreased neutrophil migration to inflammatory sites upon infectious challenge as a result of impaired diapedesis80. Furthermore, serotonin promotes the activation of naive T cells (in mice) and proliferation of CD4+ T helper cells (in humans) through 5-HT7 and 5-HT1B receptors, respectively81,82.
Platelets contain a wide range of chemokines stored in α-granules, which are released upon activation to promote the local migration of immune cells. These chemokines include CXCL4, CCL5, CXCL12 and CXCL16 (ref. 83). The importance of platelet-derived chemokines such as CXCL4 and CCL5 has been widely demonstrated in atherosclerosis (a major complication of IMIDs), where these chemokines drive immune cell recruitment to the plaques and promote a pro-inflammatory and atherogenic milieu83. Importantly, the role of these chemokines now extends beyond chemotaxis as they can also function as superinducers of type I interferon production by plasmacytoid DCs (pDCs) through a TLR9-dependent mechanism and therefore participate in the development of autoimmunity84. Similarly, thrombocidins, which are antibacterial peptides that originate from carboxy-terminal deletion of platelet chemokines, are released from α-granules upon platelet activation85. Platelets may also secrete other effector molecules, such as β-defensins86, as well as complement factors that can accumulate on the platelet membrane and participate in the development of the immune response (reviewed in ref. 87).
Release of DAMPs
Considering the likely bacterial origin of mitochondria and their similarity to pathogen-associated molecular patterns, when present in the extracellular environment mitochondria are recognized as DAMPs by immune cells through pattern recognition receptors such as TLRs88. Upon activation, mouse and human platelets extrude mitochondria into the extracellular environment, either free or embedded in the membrane of extracellular vesicles19. Mitochondrial DNA is recognized by endosomal TLR9 expressed by immune cells, especially when mitochondrial DNA is oxidized88,89, leading to the activation of pDCs and neutrophils89. In patients with SLE, platelets are an important source of circulating mitochondrial DNA in the blood19, which can result in immune activation through TLR9 as well as the production of autoantibodies.
S100A8–S100A9 (also known as calprotectin), which is a TLR4 agonist, is another DAMP that has increased serum levels in several IMIDs, including SLE, rheumatoid arthritis and IBD90. In SLE and vasculitis, platelets upregulate S100A8–S100A9 expression and release it from their granules upon activation91,92. In patients with acute myocardial infarction, levels of platelet–monocyte aggregates (which are a marker of platelet activation) correlate with S100A8–S100A9 levels93, which supports the claim that platelets are a major source of circulating S100A8–S100A9. S100A8–S100A9 activates neutrophils through TLR4 (ref. 94) and promotes their migration into the tissues by increasing endothelial cell permeability94. Of note, S100A8–S100A9 can also activate platelets through GPIbα, thereby promoting the formation of procoagulant platelets95.
HMGB1 is a highly conserved DNA-binding protein that functions as a DAMP when released by dying cells or by activated platelets in sepsis96 or in IMIDs such as systemic sclerosis and IBD97,98. Platelet-derived HMGB1 is released as a soluble form or at the surface of platelet-derived extracellular vesicles, and links inflammation with the haemostasis system. Mice with a platelet-specific deletion of HMGB1 (Pf4–Cre Hmgb1fl/fl mice) have shown that HMGB1 increases platelet aggregation and thrombus formation by promoting platelet activation in a TLR4-dependent manner40. Furthermore, platelet-derived HMGB1 promotes surface expression of P-selectin on platelets, leading to increased interaction between platelets and neutrophils and increased ROS production and antibacterial activity by neutrophils in a mouse model of sepsis96. Importantly, post-translational modifications of HMGB1, such as its oxidation (to disulfide HMGB1), enhance its pro-inflammatory and prothrombotic properties99,100. In patients with rheumatoid arthritis101, SLE102 or Sjögren syndrome103, circulating and tissue levels of HMGB1 are increased, suggesting a role in the pathogenesis of these IMIDs.
Antigen presentation
Although MHC class I molecules are found on the surface of resting platelets, most of this is adsorbed from circulating MHC class I molecules in plasma104. On activation, platelets redistribute intracellular MHC class I molecules to the surface and, together with other molecules necessary for the formation of a synapse with T cells, such as the co-stimulatory molecule CD86, these MHC molecules can present antigens and promote T cell activation and proliferation105. The machinery necessary for antigen processing and presentation can also be transferred to platelet-derived extracellular vesicles, which can promote T cell activation in a model of antigen presentation using ovalbumin106. In contrast to platelets, which do not express MHC class II, megakaryocytes can both present antigens using MHC class I (ref. 107) and, a subpopulation of megakaryocytes found in the lung, can promote CD4+ T cell activation and proliferation through expression of MHC class II (ref. 108).
Platelet-derived extracellular vesicles in the synovial fluid of patients with rheumatoid arthritis are a source of citrullinated autoantigens, such as citrullinated fibrinogen and vimentin75, and thus are decorated with autoantibodies. In addition, activated platelets may be phagocytosed by antigen-presenting cells such as DCs and subsequently processed as foreign antigens109, thereby promoting the development of antiplatelet antibodies such as those that are present in the sera of most patients with ITP32, in the sera of some patients with SLE and in some other IMIDs34,35.
Interaction with endothelial cells
Platelets activated in the circulation engage in a crosstalk with endothelial cells to promote the local immune response and limit pathogen invasion through immunothrombosis110. For example, platelet-derived IL-1β activates endothelial cells and increases the permeability of the endothelium, which promotes the local immune response and tissue healing77,111,112. Activated platelets physically interact with endothelial cells through P-selectin binding to endothelial PSGL1 as well as through other glycoproteins (such as GPIb binding to endothelial von Willebrand factor), which promotes immune cell migration and participates in the growth of the thrombus110. Furthermore, the injection of human platelet-derived extracellular vesicles into immunodeficient mice leads to diffuse endothelial injury, at least in part through HMGB1 present on the vesicle surface leading to activation of neutrophils113.
Interaction with immune cells
In their basal (resting) state, platelets have minimal ability to stimulate the immune system and may even have inhibitory effects through the expression of MHC class I molecules in the absence of co-stimulatory molecules such as CD80 and CD86 (ref. 114). By contrast, activated platelets express adhesion molecules that promote their interaction with immune cells. Of these, the best described is P-selectin, a lectin that relocalizes to the platelet surface upon activation and that binds its ligand PSGL1 only when the latter is post-translationally modified to include the sialyl Lewis X (sLeX; also known as CD15s) motif115. The presence of the sLeX motif depends on cellular expression of fucosyltransferase 7 (FUT7)115, which allows the preferential binding of platelets to select immune cells that express FUT7 in humans (neutrophils and regulatory T cells (Treg cells))15,116. Other relevant adhesion molecules involved in platelet–immune cell interactions in humans include the leukocyte integrin Mac-1 (also known as αMβ2 integrin or CD11b–CD18), which interacts directly with platelet GPIbα117 or indirectly with GPIIb/IIIa through leukocyte–fibrinogen aggregates118; platelet CD40L, which mediates physical interaction and signalling with immune cells expressing CD40 (ref. 14); and CD84, a member of the signalling lymphocyte activation molecule (SLAM) family that is expressed by both leukocytes and platelets and is involved in homophilic binding119.
Patients with SLE, rheumatoid arthritis or APS have higher circulating levels of platelet–monocyte, platelet–neutrophil and platelet–lymphocyte aggregates than healthy controls15,120. Similar observations have been described in other IMIDs, such as IBD and type 1 diabetes121,122. Patients with these diseases are also characterized by increased plasma levels of platelet-derived extracellular vesicles and soluble P-selectin, which are indicative of platelet activation and suggest that this activation could be the driving factor for increased levels of platelet–leukocyte aggregates in IMIDs15,123. Indeed, neutrophils actively scan the circulation for activated (P-selectin-positive) platelets, which preferentially interact with the neutrophil uropod by binding PSGL1, and promote tissue damage in mouse models of organ injury124.
Leukocyte–platelet aggregates are also characterized by enhanced rolling on the endothelium compared with non-bound leukocytes125. Intravital microscopy in a mouse model of arthritis showed that the impaired vascular adhesion of leukocytes in P-selectin-deficient mice (owing to the lack of endothelial P-selectin expression and of platelet–leukocyte aggregates) was partially rescued by the infusion of wild-type platelets, particularly at sites of inflammation126. These results indicate that platelets provide help for the migration of immune cells towards inflammatory sites to promote the clearance of pathogens. In addition to this role in promoting immune cell migration, recent data indicate that platelet aggregation with immune cells affects their activation, phenotype and function.
Effects of platelet–immune cell interaction
A seminal study from 2003 identified that mouse platelets modulate adaptive immune responses in vitro and in vivo in the setting of viral infection127. Platelet-depleted mice had a dampened immune response to adenovirus, and platelet infusion rescued humoral and cellular responses127. Mechanistically, platelet CD40L was found to be instrumental in B cell isotype switching and in CD8+ T cell activation (through the modulation of DC phenotype) in mice during viral challenge127. Twenty years later, our understanding of the effects of platelets on immune cells has expanded and encompasses their interactions with both innate and adaptive immune cells (Fig. 2).

Activated platelets and platelet-derived extracellular vesicles physically interact with immune cells and stimulate an inflammatory response. a, Innate immunity. Platelets aggregate with plasmacytoid dendritic cells (pDCs) through CD40 ligand (CD40L)–CD40 interactions and stimulate interferon-α (IFNα) production in response to circulating immune complexes. Platelets also interact with monocytes and neutrophils through the P-selectin–P-selectin glycoprotein ligand 1 (PSGL1) axis, leading to the maturation of monocytes to antigen-presenting cells (APCs) and the priming of neutrophils. Platelets release mitochondria and mitochondrial DNA (mtDNA), which induce neutrophil activation and the production of neutrophil extracellular traps (NETosis). This leads to the release of autoantigens that are processed by APCs and presented to lymphocytes. b, Adaptive immunity. Activated platelets express membrane CD40L and soluble CD40L (sCD40L), which stimulate B cell responses and the production of autoantibodies. P-selectin-positive platelets and platelet-derived extracellular vesicles interact with regulatory T cells (Treg cells), leading to FOXP3 downregulation and Treg cell dysfunction. BCR, B cell receptor; FcγR, Fcγ receptor; sP-selectin, soluble P-selectin; TCR, T cell receptor.
Innate immune cells
Neutrophils are the most prevalent circulating immune cell subset and are involved in the pathogenesis of a wide variety of IMIDs, including SLE, antineutrophil cytoplasmic antibody-associated vasculitis, IBD and type 1 diabetes128. The role of neutrophils in the pathogenesis of IMIDs (reviewed in ref. 129) involves direct tissue damage and the release of autoantigens through immunogenic cell death, such as NETosis89 or ferroptosis130. Patients with sickle cell disease are characterized by increased levels of platelet–neutrophil aggregates, which are reduced upon treatment with a platelet inhibitor (clopidogrel) or an antibody to P-selectin116. Furthermore, ischaemia–reperfusion injury, which is commonly observed in Raynaud phenomenon, induces platelet activation and increased platelet–neutrophil aggregation in vivo116. Importantly, ex vivo mouse neutrophils aggregated with platelets are characterized by an activated phenotype (higher levels of CD11b expression) and an increased oxidative burst in response to hypoxia–reoxygenation116. These findings suggest that platelet binding to neutrophils induces a form of outside-in signalling that affects their phenotype.
Evidence in mice indicates that the P-selectin–PSGL1 axis precipitates NETosis, which may provide another link between platelet–neutrophil aggregates and autoimmunity and thrombosis131,132 (Fig. 2a). A recent study of APS identified that human or mouse neutrophils exposed in vivo to antiphospholipid antibodies became activated, upregulated expression of tissue factor and had increased sensitivity to NETosis, leading to increased arterial or venous thrombus in vivo133. Interestingly, this prothrombotic phenotype was reversed in vivo upon blockade of PSGL1 (ref. 133). Furthermore, platelet-derived HMGB1 becomes oxidized in vivo and subsequently facilitates NETosis in mice and participates in venous thrombosis99. This indicates a potential sequence of events involving platelet activation by antiphospholipid antibodies, followed by platelet–neutrophil aggregation, then NETosis and finally thrombosis.
In the context of immune complex-mediated inflammation in mice, platelet activation through GPVI, which is targeted by antiplatelet antibodies, is responsible for increased recruitment of neutrophils to the site of inflammation and their increased degranulation and release of elastase and matrix metalloproteases, leading to tissue damage134. Further research investigating the mechanisms of platelet-induced neutrophil activation may open the way to therapies designed to disrupt the platelet–neutrophil interaction.
pDCs have been implicated in the pathogenesis of several IMIDs, such as SLE, psoriasis and rheumatoid arthritis135. Levels of platelet–pDC aggregates are increased in patients with active SLE, and the interaction leads to increased interferon-α (IFNα) production by pDCs in response to immune complexes through the CD40L–CD40 axis14 (Fig. 2a). Furthermore, global depletion of platelets or treatment with a platelet P2Y12 inhibitor reduced the severity of lupus-like disease in the NZB × NZW(F1) mouse model, emphasizing the role of platelets and platelet-derived CD40L in autoimmunity.
Mouse monocytes also express PSGL1 and interact with activated platelets in vitro and in vivo136. Furthermore, P-selectin drives the maturation of monocytes to activated DCs with increased expression of CD80 and CD86, which results in increased T cell activation, including greater antitumour activity of CD8+ T cells in vitro136. In the context of viral infection, activated platelets interact with monocytes through P-selectin–PSGL1 and CD40L–CD40, and promote the differentiation of monocytes towards a pro-inflammatory DC phenotype with increased production of the inflammatory cytokine IL-1β137 (Fig. 2a). Importantly, platelets also contain large quantities of immunomodulatory molecules such as transforming growth factor-β (TGFβ), and several reports indicate that the incubation of monocytes with platelet lysates or their secretome induces the differentiation of DCs with deficient immunostimulatory activities138,139. Overall, the interactions between platelets and innate immune cells, including DCs, lead to multiple and complex effects on adaptive immune cells.
Adaptive immune cells
In the case of ITP, patients with active disease (low count of activated platelets) have impaired Treg cell function, and treatment that leads to the normalization of the platelet compartment also restores Treg cell functions140, through incompletely understood mechanisms. It was recently shown that although all subsets of human CD4+ T cells express PSGL1, only Treg cells and T follicular regulatory cells have PSGL1 containing the sLeX motif15. Upon P-selectin engagement with sLeX-containing PSGL1, PSGL1 recruits and phosphorylates spleen tyrosine kinase (SYK), leading to SYK-dependent calcium signalling. In Treg cells, P-selectin engagement rewires the human Treg cell transcriptional programme, involving downregulation of the TGFβ axis, which ultimately leads to impaired immunosuppressive function of Treg cells and T follicular regulatory cells15 (Fig. 2b). Furthermore, inhibition of the P-selectin–PSGL1 axis with a monoclonal antibody to P-selectin resulted in milder lupus-like disease in the Dnase1l3−/− mouse model of SLE, emphasizing the importance of P-selectin–PSGL1 interaction in SLE pathogenesis15. Ptenfl/flPf4–Cre mice, which have platelet-specific PTEN deficiency leading to constitutive platelet activation, develop spontaneously lethal autoimmune and lymphoproliferative disease141. Their phenotype is characterized by increased levels of platelet–CD4+ T cell aggregates with an expanded compartment of T follicular helper cells, expanded germinal centres in lymph nodes and increased titres of autoantibodies15. Although the mechanisms underlying this observation remain unclear, these studies together indicate that dysregulated platelet activation affects T cell phenotype and promotes autoimmunity.
Platelet–B cell interactions are less common than interactions of platelets with other immune cells, mainly because B cells do not express PSGL1 (ref. 15). However, a descriptive study in humans found that levels of platelet–B cell aggregates were increased in patients with SLE and correlated with circulating levels of preswitched memory B cells and immunoglobulins142. Mechanistically, in vitro data suggest that co-culture of human B cells with platelets leads to increased immunoglobulin production and antibody class-switching, most likely through the CD40L–CD40 axis143. Indeed, platelets are the main source of circulating CD40L144, either as a soluble form or on their membrane, and the CD40L–CD40 axis is instrumental for the development of a T cell-dependent B cell response. In IMIDs such as ITP, platelet expression of CD40L is increased and drives the development of pathogenic antibodies to GPIIb/IIIa145.
Role of platelets in end organ damage
By interacting with both innate and adaptive immune cells, platelets affect their phenotype and promote inflammatory and autoimmune responses (Fig. 3). As discussed earlier in this article, platelet-mediated modulation of immune cell function occurs through both paracrine–endocrine soluble factors released by activated platelets and physical interaction by ligand–receptor coupling. In addition to these effects on immune cells, platelets also impact the pathogenesis of IMIDs by promoting organ damage.

Inflammatory disease activity promotes platelet activation through increased levels of immune complexes, autoantibodies and damage-associated molecular patterns. Upon activation, platelets express P-selectin on their surface, release soluble P-selectin and produce P-selectin-positive platelet-derived extracellular vesicles. P-selectin subsequently interacts with immune cells through binding to its ligand P-selectin glycoprotein ligand 1 (PSGL1). In regulatory T (Treg) cells and T follicular regulatory (TFR) cells, P-selectin–PSGL1 interaction induces FOXP3 downregulation and cell dysfunction, leading to immune dysregulation, production of autoantibodies and further platelet activation. In neutrophils, P-selectin–PSGL1 interaction promotes the production of neutrophil extracellular traps (NETosis), leading to the release of autoantigens that are processed by antigen-presenting cells (APCs). P-selectin promotes the differentiation of monocytes to pro-inflammatory APCs expressing high levels of MHC class II, CD80 and CD86 to efficiently prime and co-stimulate T cells. BCR, B cell receptor; CD40L, CD40 ligand; TCR, T cell receptor; TFH, T follicular helper.
Tissue injury
Multiple sclerosis is an autoimmune disease affecting the central nervous system (CNS) that results in white matter and grey matter injury, leading to neurological symptoms and disability. In the experimental autoimmune encephalomyelitis mouse model of multiple sclerosis, platelet depletion prevents the onset of clinical disease146. Interestingly, platelets infiltrate the CNS several days before T cells during disease onset, and platelet depletion prevents the invasion of T cells and subsequent CNS damage. These results suggest that platelets have a role in the homing of inflammatory cells to the CNS and therefore in promoting immune-mediated CNS damage. Furthermore, platelets may also promote CNS damage by inducing apoptosis through FASL (also known as CD95L)147 and the local release of inflammatory cytokines148.
The kidneys are the target of several IMIDs, and their involvement in disease is linked to worse prognosis. Aggregation of activated platelets may cause thrombi in the kidney microcirculation, leading to kidney damage. Such microthrombi are found in a wide range of systemic autoimmune diseases, such as SLE with or without APS, systemic sclerosis and vasculitis149. In haemolytic–uraemic syndrome, a condition characterized by severe kidney injury caused by diffuse microthrombi, platelet activation mediated by the lipopolysaccharide of enterohaemorrhagic Escherichia coli drives tissue damage150. Of note, expression of the human FCGR2A transgene in lupus-prone mice accelerated the accumulation of platelets in the kidneys and led to worse pathology, which was consistent with the increased ability of platelets to firmly adhere to kidney-deposited immune complexes in an FcγRIIA-dependent manner18,19. In a model of anti-glomerular basement membrane (GBM) antibody-mediated kidney injury, platelet depletion resulted in decreased glomerular P-selectin expression, decreased neutrophil migration and ultimately decreased kidney injury151. P-selectin-deficient (Selp−/−) mice were also characterized by a milder phenotype after anti-GBM injection, and the disease was accelerated (increased neutrophil migration and local ROS production) by infusion of wild-type platelets but not Selp−/− platelets151,152. Furthermore, CD40L+ platelets stimulate human tubular renal cells to secrete IL-6 and stimulate podocytes to produce matrix metalloproteinase 9, which promote an inflammatory environment and tissue scarring, respectively153,154. Platelet-derived CD40L also stimulates the local production of CCL2 (also known as MCP1) by human mesangial cells in vitro, which may promote tissue injury by increased immune cell infiltration155. Finally, platelet-derived growth factor and TGFβ, both of which are released by activated platelets, promote mesangial cell proliferation and scarring, which impair the integrity of glomeruli156.
Tissue fibrosis
The activity of platelets has been implicated in fibrosis progression in many organs, such as heart, kidney, lung and skin157. Platelets and platelet-derived extracellular vesicles are the main sources of circulating TGFβ, which is contained in their α-granules and is released upon activation. It is widely accepted that TGFβ promotes fibrosis in the skin and the lung by inducing fibroblast proliferation, extracellular matrix degradation and the upregulation of profibrotic genes such as those encoding collagen A1, collagen A2 and fibrillin, which have demonstrated roles in the pathogenesis of SLE and systemic sclerosis158,159. Other platelet-derived molecules, such as thymic stromal lymphopoietin and serotonin, are associated with the progression of skin fibrosis in systemic sclerosis112,160. Platelet-derived β-thromboglobulin and CXCL4 have been associated with fibrosis in experimental models, and they are found at increased levels in the lungs of patients with systemic sclerosis who have lung fibrosis161. However, conflicting data suggest that platelets may be necessary to reprogramme macrophages in the lungs to an anti-inflammatory phenotype by stimulating Treg cell production of IL-10, which leads to the resolution of inflammation and the prevention of fibrosis162.
Therapeutic targeting of platelets
Considering the role of platelets in promoting both inflammation and tissue damage, targeting platelet activation and targeting platelet–immune cell interaction might be valuable strategies for the development of therapies for IMIDs (Fig. 4).

Two strategies may be used to target platelets in immune-mediated inflammatory disease: inhibiting platelet activation or inhibiting the interaction between platelets and immune cells. a, Platelet activation can be inhibited by blocking agonist engagement (for example, blocking adenosine diphosphate (ADP) binding to its receptor P2Y12 with clopidogrel), inhibiting platelet cyclooxygenase (COX) activity using aspirin, inhibiting Toll-like receptor 7 (TLR7) activation using hydroxychloroquine, inhibiting Fcγ receptor (FcγR) engagement or activation using high-dose intravenous immunoglobulin (IVIg), inhibiting the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome with colchicine and inhibiting intracellular signalling (for example, targeting AMP-activated protein kinase (AMPK) with metformin, spleen tyrosine kinase (SYK) with fostamatinib or Bruton’s tyrosine kinase (BTK) with a specific inhibitor). b, Platelet–immune cell interactions can be inhibited by targeting ligand–receptor pairs that mediate these interactions. For example, antibody to P-selectin prevents P-selectin binding to P-selectin glycoprotein ligand 1 (PSGL1) on neutrophils and other immune cells, and antibodies targeting CD40 or CD40 ligand (CD40L) prevent interaction with B cells. Fab, antigen-binding fragment.
Targeting platelet activation
Inhibition of platelet activation in cardiovascular diseases using anti-aggregation therapy is an effective strategy to prevent arterial thrombosis (coronary or cerebrovascular events). However, in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, which is associated with thrombotic events and a dysregulated inflammatory response163, platelet inhibition with a P2Y12 inhibitor led to worse COVID-19 outcomes164. As previously discussed, activated platelets drive a type I interferon response14,41, the lack of which has been linked to severe COVID-19 and death165. Nevertheless, these results confirm that platelet inhibition can alter the immune response in a clinically meaningful manner in humans.
In the setting of autoimmunity, inhibiting platelet activation is also currently being investigated as a therapeutic strategy (Fig. 4a). A proof-of-concept study in 18 patients with SLE who were treated with the P2Y12 inhibitor clopidogrel for 12 weeks showed that the percentage of activated CD40L+, P-selectin-positive platelets and the mean platelet volume decreased significantly after initiation of clopidogrel therapy but that these differences did not persist over time166. A placebo-controlled randomized study of clopidogrel is currently being conducted in patients at very high risk of developing systemic sclerosis to prevent disease onset (ClinicalTrials.gov identifier NCT05098704). Indeed, platelets are likely to have a crucial role in systemic sclerosis considering that its pathogenesis involves vasculopathy, endothelial dysfunction and autoimmunity112. Other P2Y12 inhibitors such as ticagrelor or prasugrel or the GPIIb/IIIa inhibitor abciximab are also highly inhibitory to platelets and may be useful in IMIDs. However, the increased bleeding risk associated with their use could counterbalance their potential benefit. Moreover, these drugs were designed to inhibit platelet activation in conditions promoting thrombosis, so they may not similarly affect the functions of platelets as stimulated by immune receptors or that involve different intracellular signalling pathways.
Aspirin, an irreversible cyclooxygenase inhibitor, is also being used in the clinic to prevent platelet activation and cardiovascular disease. However, considering that aspirin has lesser inhibitory effects than P2Y12 inhibitors on the surface expression of P-selectin and CD40L by platelets and on platelet–leukocyte aggregation167, and given that the response to aspirin is attenuated in patients with SLE168, it is unlikely that aspirin monotherapy would have a meaningful immunological effect in these patients.
The inhibition of FcγRIIA-mediated platelet activation is an appealing strategy. This goal may be reached by developing specific FcγRIIA inhibitors or by the use of high-dose intravenous immunoglobulin169, as is currently prescribed in some patients with severe manifestations of IMID. FcγRIIA signals partly through SYK170, and the use of a SYK inhibitor such as fostamatinib, which is approved for use in patients with ITP and functions by decreasing platelet phagocytosis by splenic macrophages32, may also dampen platelet activation.
Hydroxychloroquine (HCQ) is the mainstay treatment for SLE and other connective tissue diseases171. HCQ functions through several mechanisms, including by limiting endosome acidification, leading to impaired endosomal TLR activation and IFNα production. In addition, HCQ has been shown to reduce platelet activation after ex vivo stimulation with ADP or antiphospholipid antibodies. Furthermore, patients treated with HCQ have lower levels of platelet activation and P-selectin expression on platelets172. These results corroborate the abundant literature linking HCQ treatment with decreased risk of thrombosis in patients with SLE and APS173. Furthermore, HCQ ‘strips’ MHC class I molecules from the platelet membrane174, which limits the potential for interactions with CD8+ T cells.
Metformin, which is widely used for the treatment of type 2 diabetes, functions by activating the AMP-activated protein kinase (AMPK) metabolic rheostat. Its ability to affect immune cell metabolism has been shown in vitro, in animal models175,176 and in promising proof-of-concept studies in patients with SLE177 or multiple sclerosis178. Interestingly, metformin inhibits the activation of platelets in vitro and prevents their release of mitochondrial DNA179, a mechanism that is involved in the pathogenesis of SLE and other IMIDs. Furthermore, use of metformin led to decreased arterial and venous thrombosis risk in in vivo models15 and in patients with diabetes mellitus180, which reinforces its potential clinical benefit in IMIDs characterized by an increased cardiovascular disease risk.
Colchicine is a drug classically prescribed to patients with gout and in some IMIDs such as SLE pericarditis. It inhibits microtubule assembly and inflammasome activation. Interestingly, colchicine use is linked to reduced cardiovascular disease risk in a meta-analysis of randomized clinical trials181. Multiple mechanisms are responsible for this effect, including inhibition of inflammasome assembly and IL-1β production, and a direct effect on platelets. Indeed, colchicine prevents platelet activation in vitro and reduces the number of platelet–neutrophil and platelet–monocyte aggregates in healthy humans182. Furthermore, the pharmacological inhibition of Bruton’s tyrosine kinase (BTK) has been identified as an efficient approach to inhibit the NLRP3 inflammasome183, and may therefore limit platelet activation.
Targeting platelet–immune cell interaction
P-selectin blockade prevents the formation of platelet–leukocyte aggregates in humans and therefore is a promising approach to treat IMIDs118 (Fig. 4b). Crizanlizumab, a human monoclonal antibody targeting P-selectin, is approved for use in patients with sickle cell disease184. Crizanlizumab decreases the frequency of vaso-occlusive crisis in these patients and has a good safety profile across several studies and 1,545 patient-years of treatment185. Importantly, no increased risk of infection was reported, which might have been expected because platelets, as discussed earlier in this article, facilitate the migration of leukocytes to fight pathogens.
In the case of SLE, P-selectin blockade with a monoclonal antibody reduces hallmark disease features of the lupus-prone Dnase1l3−/− mouse model (such as antibodies to double-stranded DNA, proteinuria and kidney damage), at least in part by restoring Treg cell functions15. Furthermore, another study showed that P-selectin inhibition led to the reduction of proteinuria and kidney injury in lupus-prone MRL/faslpr mice, together with a reduction of hypoxia in the renal tissue186. Of note, other inhibitors of P-selectin could be considered in addition to crizanlizumab, such as a PSGL1 glycomimetic that was shown to reduce venous thrombosis in an in vivo model187. P-selectin inhibition is a promising therapeutic strategy in IMIDs owing to its multimodal immunological effects, the absence of immunosuppression and the potential protective effect on thrombosis.
Monoclonal antibodies to CD40L have also been tested in SLE but resulted in severe thrombotic events, likely as a result of antibody-mediated crosslinking of CD40L on the surface of activated platelets, causing further platelet activation and aggregation through FcγRIIA ligation. More recently, new anti-CD40L compounds that cannot ligate FcγRIIA, such as an anti-CD40L Fab with a pegylated tail (dapirolizumab)188 or a monoclonal antibody devoid of Fc receptor-binding capacity, are being tested in autoimmune diseases. Although the phase II trial of dapirolizumab in SLE missed its primary end point by a small margin, treated patients showed improvement in terms of several clinical and biological end points. As a result, a phase III study of dapirolizumab is currently recruiting and is expected to end in early 2024 (ClinicalTrials.gov identifier NCT04294667). Finally, monoclonal antibodies targeting CD40 (expressed by B cells and T cells) have also been developed, such as BI655064, but with disappointing results in patients with lupus nephritis189.
Conclusions and future perspectives
In this Review, we have summarized the current evidence for platelet activation in IMIDs and the multiple effects of platelets in promoting disease activity and organ damage. Understanding the pathophysiological mechanisms that underlie platelet–immune cell interactions may lead to the identification of new therapeutic targets in IMIDs. However, several open questions remain that should be addressed to realize this therapeutic potential.
First, we are only beginning to uncover the variety of immune cells with which platelets interact and how these interactions alter their function. Furthermore, the mechanisms that mediate such crosstalk are still only partially understood, and novel molecular players need to be identified. Second, recent data indicate that some immune cells, such as neutrophils, may be activated in the periphery (for example, the skin) and then remigrate to internal organs to promote injury. Platelet–neutrophil interaction promotes the migration of neutrophils to inflammatory sites, and the potential role of platelets in promoting neutrophil remigration should also be investigated. Finally, the best timing and best strategy to inhibit platelet-mediated immune dysregulation need to be investigated. Although the global inhibition of platelet activation is plausible, the benefit–risk ratio of this strategy is conditioned by the increased risk of bleeding. Therefore, molecules that more specifically target the molecular mechanisms identified as drivers of platelet-mediated immune dysfunction need to be identified. These molecules may limit platelet–immune cell interaction by the blockade of adhesion molecules (such as antibody to P-selectin), impair platelet co-stimulatory molecules (such as CD40L) or limit the release of pro-inflammatory molecules (DAMPs). As platelet activation can precede organ damage in some IMIDs, it will be important to test the ability of platelet-targeted treatments to prevent the development and spread of inflammation-mediated organ damage.
Change history
21 March 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41577-023-00869-7
References
Scherlinger, M. et al. Worldwide trends in all-cause mortality of auto-immune systemic diseases between 2001 and 2014. Autoimmun. Rev. 19, 102531 (2020).
Broen, J. C. A. & van Laar, J. M. Mycophenolate mofetil, azathioprine and tacrolimus: mechanisms in rheumatology. Nat. Rev. Rheumatol. 16, 167–178 (2020).
Manzi, S. M. et al. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am. J. Epidemiol. 145, 408–415 (1997).
Conrad, N. et al. Autoimmune diseases and cardiovascular risk: a population-based study on 19 autoimmune diseases and 12 cardiovascular diseases in 22 million individuals in the UK. Lancet 400, 733–743 (2022). This large population-based study reports that multiple IMIDs are associated with increased risk of a wide range of cardiovascular diseases.
Gremmel, T., Frelinger, A. & Michelson, A. Platelet physiology. Semin. Thromb. Hemost. 42, 191–204 (2016).
Luo, D. et al. Protective roles for fibrin, tissue factor, plasminogen activator inhibitor-1, and thrombin activatable fibrinolysis inhibitor, but not factor XI, during defense against the gram-negative bacterium Yersinia enterocolitica. J. Immunol. 187, 1866–1876 (2011).
Gaertner, F. et al. Migrating platelets are mechano-scavengers that collect and bundle bacteria. Cell 171, 1368–1382.e23 (2017). This study shows that platelets actively patrol blood vessels to bind and bundle circulating bacteria, and promote neutrophil activation in infection.
Maouia, A., Rebetz, J., Kapur, R. & Semple, J. W. The immune nature of platelets revisited. Transfus. Med. Rev. 34, 209–220 (2020).
Marcoux, G., Laroche, A., Espinoza Romero, J. & Boilard, E. Role of platelets and megakaryocytes in adaptive immunity. Platelets 32, 340–351 (2021).
Bai, M. et al. Mean platelet volume could reflect disease activity of adult patients with systemic lupus erythematosus. Clin. Lab. 62, 1317–1322 (2016).
Khodashahi, M., Saadati, N., Rezaieyazdi, Z., Sahebari, M. & Saremi, Z. Evaluation of mean platelet volume in patients with rheumatoid arthritis and its relation with severity of disease. Rheumatol. Res. 4, 121–126 (2019).
Masoumi, M. et al. Correlation of clinical signs and symptoms of Behçet’s disease with mean platelet volume (MPV) and red cell distribution width (RDW). Orphanet J. Rare Dis. 15, 297 (2020).
Stenberg, P. E., Shuman, M. A., Levine, S. P. & Bainton, D. F. Redistribution of alpha-granules and their contents in thrombin- stimulated platelets. J. Cell Biol. 98, 748–760 (1984).
Duffau, P. et al. Platelet CD154 potentiates interferon- secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci. Transl. Med. 2, 47ra63 (2010). This study shows that platelets interact with pDCs in SLE and promote their production of type I interferon through the CD40L–CD40 axis.
Scherlinger, M. et al. Selectins impair regulatory T cell function and contribute to systemic lupus erythematosus pathogenesis. Sci. Transl. Med. 13, eabi4994 (2021). This study shows that platelets from patients with SLE interact with Treg cells through the P-selectin–PSGL1 axis, which results in FOXP3 downregulation and Treg cell dysfunction.
Puhm, F., Boilard, E. & Machlus, K. R. Platelet extracellular vesicles: beyond the blood. Arterioscler. Thromb. Vasc. Biol. 41, 87–96 (2021).
Tessandier, N. et al. Platelets disseminate extracellular vesicles in lymph in rheumatoid arthritis. Arterioscler. Thromb. Vasc. Biol. 40, 929–942 (2020).
Melki, I. et al. FcγRIIA expression aggravates nephritis and increases platelet activation in systemic lupus erythematosus in mice. Blood https://doi.org/10.1182/blood.2020004974 (2020).
Melki, I. et al. Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci. Transl. Med. 13, eaav5928 (2021). This study reports that activated platelets release mitochondria, either free or embedded in extracellular vesicles, that activate the immune system and participate in immune dysregulation in SLE.
Ohyama, K. et al. Proteomic profiling of antigens in circulating immune complexes associated with each of seven autoimmune diseases. Clin. Biochem. 48, 181–185 (2015).
Hubbard, J. J. et al. FcRn is a CD32a coreceptor that determines susceptibility to IgG immune complex-driven autoimmunity. J. Exp. Med. 217, e20200359 (2020).
Qian, K. et al. Functional expression of IgA receptor FcαRI on human platelets. J. Leukoc. Biol. 84, 1492–1500 (2008).
Joseph, M. et al. Expression and functions of the high-affinity IgE receptor on human platelets and megakaryocyte precursors. Eur. J. Immunol. 27, 2212–2218 (1997).
Chalayer, E. et al. Fc receptors gone wrong: a comprehensive review of their roles in autoimmune and inflammatory diseases. Autoimmun. Rev. 21, 103016 (2022).
Love, P. E. Antiphospholipid antibodies: anticardiolipin and the lupus anticoagulant in systemic lupus erythematosus (SLE) and in non-SLE disorders: prevalence and clinical significance. Ann. Intern. Med. 112, 682 (1990).
Assous, N. et al. Prevalence of antiphospholipid antibodies in systemic sclerosis and association with primitive pulmonary arterial hypertension and endothelial injury. Clin. Exp. Rheumatol. 23, 199–204 (2005).
Fauchais, A. L. et al. Antiphospholipid antibodies in primary Sjögren’s syndrome: prevalence and clinical significance in a series of 74 patients. Lupus 13, 245–248 (2004).
Zhang, W. et al. Anti-β2 glycoprotein I antibodies in complex with β2 glycoprotein I induce platelet activation via two receptors: apolipoprotein E receptor 2′ and glycoprotein I bα. Front. Med. 10, 76–84 (2016).
Müller-Calleja, N. et al. Lipid presentation by the protein C receptor links coagulation with autoimmunity. Science 371, eabc0956 (2021).
Kim, K.-J., Baek, I.-W., Yoon, C.-H., Kim, W.-U. & Cho, C.-S. Elevated levels of soluble CD40 ligand are associated with antiphospholipid antibodies in patients with systemic lupus erythematosus. Clin. Exp. Rheumatol. 35, 823–830 (2017).
Patsouras, M. D. et al. Elevated expression of platelet-derived chemokines in patients with antiphospholipid syndrome. J. Autoimmun. 65, 30–37 (2015).
Provan, D. & Semple, J. W. Recent advances in the mechanisms and treatment of immune thrombocytopenia. eBioMedicine 76, 103820 (2022).
Al-Samkari, H. et al. A modern reassessment of glycoprotein-specific direct platelet autoantibody testing in immune thrombocytopenia. Blood Adv. 4, 9–18 (2019).
Kuwana, M., Kaburaki, J., Okazaki, Y., Miyazaki, H. & Ikeda, Y. Two types of autoantibody-mediated thrombocytopenia in patients with systemic lupus erythematosus. Rheumatology 45, 851–854 (2006).
Czirják, L. et al. Anti-platelet antibodies against gpIIb/IIIa in systemic sclerosis. Clin. Exp. Rheumatol. 12, 527–529 (1994).
Hally, K., Fauteux-Daniel, S., Hamzeh-Cognasse, H., Larsen, P. & Cognasse, F. Revisiting platelets and Toll-like receptors (TLRs): at the interface of vascular immunity and thrombosis. Int. J. Mol. Sci. 21, 6150 (2020).
Aslam, R. et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 107, 637–641 (2006).
Andonegui, G. et al. Platelets express functional Toll-like receptor-4. Blood 106, 2417–2423 (2005).
Ward, J. R. et al. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb. Haemost. 94, 831–838 (2005).
Vogel, S. et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Invest. 125, 4638–4654 (2015).
Koupenova, M. et al. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood 124, 791–802 (2014).
Brown, G. J. et al. TLR7 gain-of-function genetic variation causes human lupus. Nature 605, 349–356 (2022). This study shows that constitutive activation of TLR7 causes early and severe SLE in mice and humans.
Becker, Y. et al. Autoantibodies in systemic lupus erythematosus target mitochondrial RNA. Front. Immunol. 10, 1026 (2019).
Becker, Y. L., Julien, A.-S., Godbout, A., Boilard, É. & Fortin, P. R. Pilot study of anti-mitochondrial antibodies in antiphospholipid syndrome. Lupus 29, 1623–1629 (2020).
Thon, J. N. et al. The functional role of TLR9 in human platelets. Blood 118, 366 (2011).
Boudreau, L. H. et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124, 2173–2183 (2014).
Bjornevik, K. et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 375, 296–301 (2022).
Draborg, A. H. et al. Reduced response to Epstein-Barr virus antigens by T-cells in systemic lupus erythematosus patients. Lupus Sci. Med. 1, e000015 (2014).
Su, Y. et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 185, 881–895.e20 (2022).
Wood, R. A. et al. Serologic markers of Epstein-Barr virus reactivation are associated with increased disease activity, inflammation, and interferon pathway activation in patients with systemic lupus erythematosus. J. Transl. Autoimmun. 4, 100117 (2021).
Banerjee, M. et al. Platelets endocytose viral particles and are activated via TLR (Toll-like receptor) signaling. Arterioscler. Thromb. Vasc. Biol. 40, 1635–1650 (2020).
Ahmad, A. & Menezes, J. Binding of the Epstein-Barr virus to human platelets causes the release of transforming growth factor-beta. J. Immunol. 159, 3984–3988 (1997).
Swanson, K. V., Deng, M. & Ting, J. P.-Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Murthy, P. et al. The NLRP3 inflammasome and Bruton’s tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem. Biophys. Res. Commun. 483, 230–236 (2017).
Vogel, S. et al. The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2, 2672–2680 (2018).
Boilard, E. et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327, 580–583 (2010). This study shows that patients with rheumatoid arthritis have platelet-derived extracellular vesicles in the synovial fluid and inflamed joints, and that these vesicles promote inflammation through IL-1β release.
Chiang, T. M., Takayama, H. & Postlethwaite, A. E. Increase in platelet non-integrin type I collagen receptor in patients with systemic sclerosis. Thromb. Res. 117, 299–306 (2006).
Takahashi, H. & Moroi, M. Antibody against platelet membrane glycoprotein VI in a patient with systemic lupus erythematosus. Am. J. Hematol. 67, 262–267 (2001).
Hechler, B. & Gachet, C. Purinergic receptors in thrombosis and inflammation. Arterioscler. Thromb. Vasc. Biol. 35, 2307–2315 (2015).
Haas, C. B., Lovászi, M., Braganhol, E., Pacher, P. & Haskó, G. Ectonucleotidases in inflammation, immunity, and cancer. J. Immunol. 206, 1983–1990 (2021).
Chaurasia, S. N., Kushwaha, G., Pandey, A. & Dash, D. Human platelets express functional ectonucleotidases that restrict platelet activation signaling. Biochem. Biophys. Res. Commun. 527, 104–109 (2020).
Hesse, J. et al. Profound inhibition of CD73-dependent formation of anti-inflammatory adenosine in B cells of SLE patients. eBioMedicine 73, 103616 (2021).
Han, L. et al. Phenotypical analysis of ectoenzymes CD39/CD73 and adenosine receptor 2A in CD4+CD25highFoxp3+ regulatory T-cells in psoriasis. Australas. J. Dermatol. 59, e31–e38 (2018).
Fuentes, E., Moore-Carrasco, R., de Andrade Paes, A. M. & Trostchansky, A. Role of platelet activation and oxidative stress in the evolution of myocardial infarction. J. Cardiovasc. Pharmacol. Ther. 24, 509–520 (2019).
Praticò, D., Iuliano, L., Ghiselli, A., Alessandri, C. & Violi, F. Hydrogen peroxide as trigger of platelet aggregation. Haemostasis 21, 169–174 (1991).
Polidoro, L. et al. Platelet activation in patients with the Raynaud phenomenon. Intern. Med. J. 42, 531–535 (2012).
Pamuk, G. E. et al. Increased circulating platelet–leucocyte complexes in patients with primary Raynaudʼs phenomenon and Raynaudʼs phenomenon secondary to systemic sclerosis: a comparative study. Blood Coagul. Fibrinolysis 18, 297–302 (2007).
Estevez, B. & Du, X. New concepts and mechanisms of platelet activation signaling. Physiology 32, 162–177 (2017).
Petito, E. et al. A dichotomy in platelet activation: evidence of different functional platelet responses to inflammatory versus haemostatic stimuli. Thromb. Res. 172, 110–118 (2018).
Nagahama, M. et al. Platelet activation markers and soluble adhesion molecules in patients with systemic lupus erythematosus. Autoimmunity 33, 85–94 (2001).
Sheremata, W. A. et al. Evidence of platelet activation in multiple sclerosis. J. Neuroinflammation 5, 27 (2008).
Miao, D., Li, D.-Y., Chen, M. & Zhao, M.-H. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Res. Ther. 19, 252 (2017).
Collins, C. E., Cahill, M. R., Newland, A. C. & Rampton, D. S. Platelets circulate in an activated state in inflammatory bowel disease. Gastroenterology 106, 840–845 (1994).
Linge, P., Fortin, P. R., Lood, C., Bengtsson, A. A. & Boilard, E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat. Rev. Rheumatol. 14, 195–213 (2018).
Cloutier, N. et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes: immune complexes contain microparticles. EMBO Mol. Med. 5, 235–249 (2013).
Boilard, E., Blanco, P. & Nigrovic, P. A. Platelets: active players in the pathogenesis of arthritis and SLE. Nat. Rev. Rheumatol. 8, 534–542 (2012).
Nhek, S. et al. Activated platelets induce endothelial cell activation via an interleukin-1β pathway in systemic lupus erythematosus. Arterioscler. Thromb. Vasc. Biol. 37, 707–716 (2017).
Adnot, S., Houssaini, A., Abid, S., Marcos, E. & Amsellem, V. Serotonin transporter and serotonin receptors. Handb. Exp. Pharmacol. 218, 365–380 (2013).
Cloutier, N. et al. Platelets can enhance vascular permeability. Blood 120, 1334–1343 (2012).
Duerschmied, D. et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 121, 1008–1015 (2013).
Leon-Ponte, M., Ahern, G. P. & O’Connell, P. J. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood 109, 3139–3146 (2007).
Yin, J., Albert, R. H., Tretiakova, A. P. & Jameson, B. A. 5-HT1B receptors play a prominent role in the proliferation of T-lymphocytes. J. Neuroimmunol. 181, 68–81 (2006).
Bakogiannis, C., Sachse, M., Stamatelopoulos, K. & Stellos, K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine 122, 154157 (2019).
Du, Y. et al. Chemokines form nanoparticles with DNA and can superinduce TLR-driven immune inflammation. J. Exp. Med. 219, e20212142 (2022).
Krijgsveld, J. et al. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J. Biol. Chem. 275, 20374–20381 (2000).
Valle-Jiménez, X. et al. Human platelets and megakaryocytes express defensin alpha 1. Platelets 31, 344–354 (2020).
Speth, C. et al. Complement and platelets: mutual interference in the immune network. Mol. Immunol. 67, 108–118 (2015).
Zhang, Q. et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Mariani, A. et al. Serum calprotectin: review of its usefulness and validity in paediatric rheumatic diseases. Clin. Exp. Rheumatol. 33, 109–114 (2015).
Guo, L. et al. Increased platelet S100A8/S100A9 associated with vasculitis in granulomatosis with polyangiitis (GPA). Blood 138, 3142 (2021).
Lood, C. et al. Platelet-derived S100A8/A9 and cardiovascular disease in systemic lupus erythematosus. Arthritis Rheumatol. 68, 1970–1980 (2016).
Song, N.-P. et al. Plasma calprotectin was associated with platelet activation and no-reflow phenomenon in acute coronary syndrome. BMC Cardiovasc. Disord. 20, 443 (2020).
Guo, Q. et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe 29, 222–235.e4 (2021).
Colicchia, M. et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbα. Blood https://doi.org/10.1182/blood.2021014966 (2022).
Zhou, H. et al. Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv. 2, 638–648 (2018).
Maugeri, N. et al. Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity 45, 584–587 (2012).
Yang, B., Liu, X. & Mei, Q. HMGB1-positive platelet microparticles may be a biomarker of inflammatory bowel disease. Inflamm. Bowel Dis. 26, e10 (2020).
Stark, K. et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 128, 2435–2449 (2016).
Kwak, M. S. et al. Immunological significance of HMGB1 post-translational modification and redox biology. Front. Immunol. 11, 1189 (2020).
Taniguchi, N. et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 48, 971–981 (2003).
Feng, X. et al. HMGB1 protein promotes glomerular mesangial matrix deposition via TLR2 in lupus nephritis. J. Cell. Physiol. 235, 5111–5119 (2020).
Ek, M., Popovic, K., Harris, H. E., Nauclér, C. S. & Wahren-Herlenius, M. Increased extracellular levels of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in minor salivary glands of patients with Sjögren’s syndrome. Arthritis Rheum. 54, 2289–2294 (2006).
Kao, K., Cook, D. & Scornik, J. Quantitative analysis of platelet surface HLA by W6/32 anti-HLA monoclonal antibody. Blood 68, 627–632 (1986).
Chapman, L. M. et al. Platelets present antigen in the context of MHC class I. J. Immunol. 189, 916–923 (2012).
Marcoux, G. et al. Platelet EVs contain an active proteasome involved in protein processing for antigen presentation via MHC-I molecules. Blood 138, 2607–2620 (2021).
Zufferey, A. et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv. 1, 1773–1785 (2017).
Pariser, D. N. et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Invest. 131, e137377 (2021).
Maître, B. et al. Immature myeloid dendritic cells capture and remove activated platelets from preformed aggregates. J. Thromb. Haemost. 8, 2262–2272 (2010).
Coenen, D. M., Mastenbroek, T. G. & Cosemans, J. M. E. M. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood 130, 2819–2828 (2017).
Hottz, E. D. et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 122, 3405–3414 (2013).
Truchetet, M.-E. et al. Platelets induce thymic stromal lymphopoietin production by endothelial cells: contribution to fibrosis in human systemic sclerosis. Arthritis Rheumatol. 68, 2784–2794 (2016).
Maugeri, N. et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Transl. Med. 10, eaao3089 (2018).
Aslam, R., Speck, E. R., Kim, M., Freedman, J. & Semple, J. W. Transfusion-related immunomodulation by platelets is dependent on their expression of MHC class I molecules and is independent of white cells. Transfusion 48, 1778–1786 (2008).
Malý, P. et al. The α(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell 86, 643–653 (1996).
Polanowska-Grabowska, R. et al. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler. Thromb. Vasc. Biol. 30, 2392–2399 (2010).
Ehlers, R. et al. Targeting platelet–leukocyte interactions: identification of the integrin Mac-1 binding site for the platelet counter receptor glycoprotein Ibα. J. Exp. Med. 198, 1077–1088 (2003).
Li, N. et al. Platelet-leukocyte cross talk in whole blood. Arterioscler. Thromb. Vasc. Biol. 20, 2702–2708 (2000).
Schuhmann, M. K. et al. CD84 links T cell and platelet activity in cerebral thrombo-inflammation in acute stroke. Circ. Res. 127, 1023–1035 (2020).
Joseph, J. E., Harrison, P., Mackie, I. J., Isenberg, D. A. & Machin, S. J. Increased circulating platelet–leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 115, 451–459 (2001).
Irving, P. M. et al. Formation of platelet-leukocyte aggregates in inflammatory bowel disease. Inflamm. Bowel Dis. 10, 361–372 (2004).
Popp, S. K. et al. Circulating platelet-neutrophil aggregates characterize the development of type 1 diabetes in humans and NOD mice. JCI Insight 7, e153993 (2022).
Sellam, J. et al. Increased levels of circulating microparticles in primary Sjögren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res. Ther. 11, R156 (2009).
Sreeramkumar, V. et al. Neutrophils scan for activated platelets to initiate inflammation. Science 346, 1234–1238 (2014). This study shows that neutrophils scan the circulation for activated platelets through the P-selectin–PSGL1 axis and that this outside-in signalling promotes neutrophil migration and tissue injury in a mouse model of inflammation.
Yeo, E. L., Sheppard, J.-A. I. & Feuerstein, I. A. Role of P-selectin and leukocyte activation in polymorphonuclear cell adhesion to surface adherent activated platelets under physiologic shear conditions (an injury vessel wall model). Blood 83, 2498–2507 (1994).
Schmitt-Sody, M. et al. Platelet P-selectin is significantly involved in leukocyte-endothelial cell interaction in murine antigen-induced arthritis. Platelets 18, 365–372 (2007).
Elzey, B. D. et al. Platelet-mediated modulation of adaptive immunity: a communication link between innate and adaptive immune compartments. Immunity 19, 9–19 (2003).
Nakabo, S., Romo-Tena, J. & Kaplan, M. J. Neutrophils as drivers of immune dysregulation in autoimmune diseases with skin manifestations. J. Invest. Dermatol. 142, 823–833 (2022).
Wigerblad, G. & Kaplan, M. J. Neutrophil extracellular traps in systemic autoimmune and autoinflammatory diseases. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00787-0 (2022).
Li, P. et al. Glutathione peroxidase 4–regulated neutrophil ferroptosis induces systemic autoimmunity. Nat. Immunol. 22, 1107–1117 (2021).
Etulain, J. et al. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126, 242–246 (2015).
Panicker, S. R. et al. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood 130, 181–191 (2017).
Nayak, L. et al. A targetable pathway in neutrophils mitigates both arterial and venous thrombosis. Sci. Transl. Med. 14, eabj7465 (2022).
Gros, A. et al. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune-complex–mediated inflammation in mice. Blood 126, 1017–1026 (2015).
Sozzani, S., Del Prete, A. & Bosisio, D. Dendritic cell recruitment and activation in autoimmunity. J. Autoimmun. 85, 126–140 (2017).
Han, P. et al. Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci. Adv. 6, eaaz1580 (2020). This article shows that platelet P-selectin induces the maturation of monocytes to proinflammatory DCs with increased MHC class II and co-stimulatory molecule expression.
Li, T. et al. Platelets mediate inflammatory monocyte activation by SARS-CoV-2 spike protein. J. Clin. Invest. 132, e150101 (2021).
Saris, A. et al. Inhibition of dendritic cell activation and modulation of T cell polarization by the platelet secretome. Front. Immunol. 12, 631285 (2021).
Kral, J. B., Schrottmaier, W. C., Salzmann, M. & Assinger, A. Platelet interaction with innate immune cells. Transfus. Med. Hemother. 43, 78–88 (2016).
Semple, J. W. & Kapur, R. Platelet immunology from the inside out. ISBT Sci. Ser. 15, 315–319 (2020).
Chen, X. et al. The phosphatase PTEN links platelets with immune regulatory functions of mouse T follicular helper cells. Nat. Commun. 13, 2762 (2022).
Zamora, C. et al. Association of platelet binding to lymphocytes with B cell abnormalities and clinical manifestations in systemic lupus erythematosus. Mediators Inflamm. 2019, e2473164 (2019).
Cognasse, F. et al. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp. Hematol. 35, 1376–1387 (2007).
Viallard, J.-F. et al. Increased soluble and platelet-associated CD40 ligand in essential thrombocythemia and reactive thrombocytosis. Blood 99, 2612–2614 (2002).
Solanilla, A. Platelet-associated CD154 in immune thrombocytopenic purpura. Blood 105, 215–218 (2005).
Sonia D’Souza, C. et al. Platelets drive inflammation and target gray matter and the retina in autoimmune-mediated encephalomyelitis. J. Neuropathol. Exp. Neurol. 77, 567–576 (2018). This study reports that platelets are required for T cell infiltration and, ultimately, tissue damage in a mouse model of inflammatory CNS disease.
Schleicher, R. I. et al. Platelets induce apoptosis via membrane-bound FasL. Blood 126, 1483–1493 (2015).
Kocovski, P. et al. Platelet depletion is effective in ameliorating anxiety-like behavior and reducing the pro-inflammatory environment in the hippocampus in murine experimental autoimmune encephalomyelitis. J. Clin. Med. 8, 162 (2019).
Kronbichler, A. & Mayer, G. Renal involvement in autoimmune connective tissue diseases. BMC Med. 11, 95 (2013).
Ståhl, A. et al. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets via TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood 108, 167–176 (2006).
Kuligowski, M. P., Kitching, A. R. & Hickey, M. J. Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet-derived P-selectin in the absence of rolling. J. Immunol. 176, 6991–6999 (2006).
Finsterbusch, M., Norman, M. U., Hall, P., Kitching, A. R. & Hickey, M. J. Platelet retention in inflamed glomeruli occurs via selective prolongation of interactions with immune cells. Kidney Int. 95, 363–374 (2019).
Dewitte, A. et al. CD154 Induces interleukin-6 secretion by kidney tubular epithelial cells under hypoxic conditions: inhibition by chloroquine. Mediators Inflamm. 2020, e6357046 (2020).
Rigothier, C. et al. CD154 induces matrix metalloproteinase-9 secretion in human podocytes. J. Cell. Biochem. 117, 2737–2747 (2016).
Tanaka, T. Human platelets stimulate mesangial cells to produce monocyte chemoattractant protein-1 via the CD40/CD40 ligand pathway and may amplify glomerular injury. J. Am. Soc. Nephrol. 13, 2488–2496 (2002).
Yuan, Y. et al. Excessive activation of the TLR9/TGF-β1/PDGF-B pathway in the peripheral blood of patients with systemic lupus erythematosus. Arthritis Res. Ther. 19, 70 (2017).
Meng, X., Nikolic-Paterson, D. J. & Lan, H. Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 12, 325–338 (2016).
Christmann, R. B. et al. Association of interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 66, 714–725 (2014).
Solé, C., Gimenez-Barcons, M., Ferrer, B., Ordi-Ros, J. & Cortés-Hernández, J. Microarray study reveals a transforming growth factor-β-dependent mechanism of fibrosis in discoid lupus erythematosus. Br. J. Dermatol. 175, 302–313 (2016).
Sagonas, I. & Daoussis, D. Serotonin and systemic sclerosis. An emerging player in pathogenesis. Jt. Bone Spine 89, 105309 (2022).
Kowal-Bielecka, O. et al. Beta thromboglobulin and platelet factor 4 in bronchoalveolar lavage fluid of patients with systemic sclerosis. Ann. Rheum. Dis. 64, 484–486 (2005).
Rossaint, J. et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J. Exp. Med. 218, e20201353 (2021).
Bonaventura, A. et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 21, 319–329 (2021).
Berger, J. S. et al. Effect of P2Y12 inhibitors on survival free of organ support among non–critically ill hospitalized patients with COVID-19: a randomized clinical trial. JAMA 327, 227 (2022).
Hadjadj, J. et al. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. Science 369, 718–724 (2020).
Vial, G. et al. The impact of clopidogrel on plasma-soluble CD40 ligand levels in systemic lupus erythematosus patients: the CLOPUS phase I/II pilot study. Jt. Bone Spine 88, 105097 (2021).
Parker, W. A. E. et al. Aspirin, clopidogrel and prasugrel monotherapy in patients with type 2 diabetes mellitus: a double-blind randomised controlled trial of the effects on thrombotic markers and microRNA levels. Cardiovasc. Diabetol. 19, 3 (2020).
Avalos, I. et al. Aspirin therapy and thromboxane biosynthesis in systemic lupus erythematosus. Lupus 16, 981–986 (2007).
Shock, A., Humphreys, D. & Nimmerjahn, F. Dissecting the mechanism of action of intravenous immunoglobulin in human autoimmune disease: lessons from therapeutic modalities targeting Fcγ receptors. J. Allergy Clin. Immunol. 146, 492–500 (2020).
Arman, M. & Krauel, K. Human platelet IgG Fc receptor FcγRIIA in immunity and thrombosis. J. Thromb. Haemost. 13, 893–908 (2015).
Richez, C. et al. Practical management of patients on hydroxychloroquine. Jt. Bone Spine 88, 105316 (2021).
Cornwell, M. G. et al. Hydroxychloroquine is associated with lower platelet activity and improved vascular health in systemic lupus erythematosus. Lupus Sci. Med. 8, e000475 (2021).
Ruiz-Irastorza, G. et al. Effect of antimalarials on thrombosis and survival in patients with systemic lupus erythematosus. Lupus 15, 577–583 (2006).
Neumüller, J., Tohidast-Akrad, M., Fischer, M. & Mayr, W. R. Influence of chloroquine or acid treatment of human platelets on the antigenicity of HLA and the ‘thrombocyte-specific’ glycoproteins Ia/IIa, IIb, and IIb/IIIa. Vox Sang. 65, 223–231 (1993).
Morel, L. Immunometabolism in systemic lupus erythematosus. Nat. Rev. Rheumatol. 13, 280–290 (2017).
Yin, Y. et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 7, 274ra18 (2015).
Sun, F. et al. Safety and efficacy of metformin in systemic lupus erythematosus: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Rheumatol. 2, e210–e216 (2020).
Negrotto, L., Farez, M. F. & Correale, J. Immunologic effects of metformin and pioglitazone treatment on metabolic syndrome and multiple sclerosis. JAMA Neurol. 73, 520–528 (2016).
Xin, G. et al. Metformin uniquely prevents thrombosis by inhibiting platelet activation and mtDNA release. Sci. Rep. 6, 36222 (2016).
Lu, D.-Y. et al. Metformin use in patients with type 2 diabetes mellitus is associated with reduced risk of deep vein thrombosis: a non-randomized, pair-matched cohort study. BMC Cardiovasc. Disord. 14, 187 (2014).
Xu, H. et al. Colchicine for secondary prevention of coronary artery disease: a meta-analysis of randomised controlled trials. Heart Lung Circ. 31, 685–695 (2022).
Shah, B. et al. Effect of colchicine on platelet-platelet and platelet-leukocyte interactions: a pilot study in healthy subjects. Inflammation 39, 182–189 (2016).
Ito, M. et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat. Commun. 6, 7360 (2015).
Ataga, K. I. et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N. Engl. J. Med. 376, 429–439 (2017).
Karki, N. R., Saunders, K. & Kutlar, A. A critical evaluation of crizanlizumab for the treatment of sickle cell disease. Expert. Rev. Hematol. 15, 5–13 (2022).
Zhang, L. et al. P-selectin blockade ameliorates lupus nephritis in MRL/lpr mice through improving renal hypoxia and evaluation using BOLD-MRI. J. Transl. Med. 18, 116 (2020).
Wong, D. J. et al. A PSGL-1 glycomimetic reduces thrombus burden without affecting hemostasis. Blood 138, 1182–1193 (2021).
Furie, R. A. et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology 60, 5397–5407 (2021).
Jayne, D. et al. POS0687 a randomised dose ranging, placebo-controlled, phase II study assessing the efficacy and safety of BI 655064, an antagonistic anti-CD40 antibody, in patients with lupus nephritis. Ann. Rheum. Dis. 80, 589–590 (2021).
Andrianova, I. A. et al. In systemic lupus erythematosus anti-dsDNA antibodies can promote thrombosis through direct platelet activation. J. Autoimmun. 107, 102355 (2020).
Roberts, D. E., McNicol, A. & Bose, R. Mechanism of collagen activation in human platelets. J. Biol. Chem. 279, 19421–19430 (2004).
Eckly, A. et al. Megakaryocytes use in vivo podosome-like structures working collectively to penetrate the endothelial barrier of bone marrow sinusoids. J. Thromb. Haemost. 18, 2987–3001 (2020).
Lefrançais, E. et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544, 105–109 (2017).
Valet, C. et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J. Clin. Invest. 132, e153920 (2022).
Sun, S. et al. Single-cell analysis of ploidy and the transcriptome reveals functional and spatial divergency in murine megakaryopoiesis. Blood 138, 1211–1224 (2021).
Zhang, W. et al. Global characterization of megakaryocytes in bone marrow, peripheral blood, and cord blood by single-cell RNA sequencing. Cancer Gene Ther. 29, 1636–1647 (2022).
Lood, C. et al. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: up-regulation of the type I interferon system is strongly associated with vascular disease. Blood 116, 1951–1957 (2010).
Acknowledgements
M.S. is supported by academic sources including the Société Française de Rhumatologie, the Institut pour la Santé et la Recherche Médicale and the Bettencourt-Schueller Fondation. E.B. is recipient of an award from the Fonds de Recherche en Santé du Québec. P.B. is supported by the Fondation pour la Recherche Médicale, Société Française de Rhumatologie, Foundation for Research in Rheumatology (FOREUM) and Fondation Arthritis.
Author information
Authors and Affiliations
Contributions
M.S. wrote the first draft and conceptualized the figures. E.B., G.C.T., C.R. and P.B. edited the manuscript and suggested additions. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
C.R. has received consulting or speaker fees from AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Biogen, Eli Lilly, GlaxoSmithKline, Janssen Novartis and Pfizer and grants from Biogen, Eli Lilly and Nordic Pharma, all unrelated to this work. M.S. has received consulting fees from Sandoz, Amgen and Nordic Pharma, all unrelated to this work. The other authors report no competing interests.
Peer review
Peer review information
Nature Reviews Immunology thanks S. Massberg, J. Semple, S. Vogel and A. Zarbock for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Antineutrophil cytoplasmic antibody
-
A type of autoantibody that targets neutrophil components and is characteristic of antineutrophil cytoplasmic antibody-associated vasculitis.
- Antiphospholipid syndrome
-
(APS). An autoimmune disorder characterized by antiphospholipid antibodies responsible for arterial and venous thrombosis and obstetric issues.
- Clopidogrel
-
An anti-aggregation therapy that targets the platelet purine receptor P2Y12, which is widely prescribed for the prevention of cardiovascular disease in patients at high risk (such as patients with a history of myocardial infarction).
- Disseminated intravascular coagulation
-
A critical medical condition characterized by widespread and dysregulated clotting throughout the blood vessels.
- Extracellular vesicles
-
Small (10–1,000 nm) vesicles surrounded by a lipid bilayer that are released from a cell (often after activation) and that contain soluble factors and surface molecules from that cell.
- Ferroptosis
-
A form of programmed cell death that is dependent on intracellular iron, characterized by the intracellular accumulation of lipid peroxides.
- Immune thrombocytopenia
-
(ITP). An autoimmune disorder characterized by severe thrombocytopenia secondary to immune destruction of platelets that is responsible for increased bleeding risk.
- Inflammatory bowel disease
-
(IBD). A group of immune-mediated inflammatory diseases such as Crohn’s disease or ulcerative colitis characterized by inflammation of the bowel wall.
- Ischaemia–reperfusion injury
-
A condition in which tissue damage occurs after blood flow is restored to an ischaemic tissue owing to the local release of toxic factors after reperfusion.
- Mesangial cells
-
Mesenchymal cells of the kidney that are responsible for the architecture of the glomeruli.
- NETosis
-
A form of programmed cell death by which neutrophils release the contents of their cytoplasm and nucleus (including DNA and histones) into the extracellular milieu, often in the context of infection or autoimmunity.
- Primary haemostasis
-
The primary response to ensure blood vessel integrity upon injury, which involves platelets, subendothelial matrix tissue factor and circulating fibrinogen.
- Podocytes
-
Specialized epithelial cells that form the finely fenestrated lining of the kidney glomeruli and are responsible for blood filtration with minimal loss of protein in the urine.
- Psoriasis
-
A frequent skin inflammatory disease characterized by widespread scaly and itchy patches, which can be associated with inflammatory arthritis (psoriatic arthritis).
- Raynaud phenomenon
-
A condition in which the blood vessels in the extremities constrict upon exposure to cold, resulting in skin colour changes (typically white, blue then red) and pain. The condition may be benign (Raynaud disease) or associated with an autoimmune disease (secondary Raynaud phenomenon).
- Rheumatoid arthritis
-
A non-rare (prevalence of 0.5% in high-income countries) autoimmune disease characterized by widespread inflammation and destruction of joints, as well as involvement of other organs (lungs and eyes).
- Sjögren syndrome
-
A connective tissue disorder characterized by the involvement of exocrine glands, causing dry eyes and/or mouth, as well as systemic symptoms.
- Systemic lupus erythematosus
-
(SLE). A prototypical systemic autoimmune disease characterized by the production of antibodies to DNA and the involvement of a wide range of organs.
- Systemic sclerosis
-
A connective tissue disorder characterized by vasculopathy, autoimmunity and excessive fibrosis.
- von Willebrand factor
-
A multimeric blood glycoprotein involved in platelet adhesion to a wound and which is necessary for the initial steps of blood clotting.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Scherlinger, M., Richez, C., Tsokos, G.C. et al. The role of platelets in immune-mediated inflammatory diseases. Nat Rev Immunol 23, 495–510 (2023). https://doi.org/10.1038/s41577-023-00834-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-023-00834-4
This article is cited by
-
Mean platelet volume is associated with periodontitis: a cross-sectional study
BMC Oral Health (2024)
-
Proteomic profiling identifies SPP1 associated with rapidly progressive interstitial lung disease in anti-MDA5-positive dermatomyositis
Arthritis Research & Therapy (2024)
-
CaMK4 controls follicular helper T cell expansion and function during normal and autoimmune T-dependent B cell responses
Nature Communications (2024)
-
The day after intracerebral hemorrhage: platelet mass index as predictor of survival—a retrospective cohort study
The Egyptian Journal of Neurology, Psychiatry and Neurosurgery (2023)
