
Abstract
Eosinophilic granulomatosis with polyangiitis (EGPA) is a rare anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, characterized by asthma, eosinophilia and granulomatous or vasculitic involvement of several organs. The diagnosis and management of EGPA are often challenging and require an integrated, multidisciplinary approach. Current practice relies on recommendations and guidelines addressing the management of ANCA-associated vasculitis and not specifically developed for EGPA. Here, we present evidence-based, cross-discipline guidelines for the diagnosis and management of EGPA that reflect the substantial advances that have been made in the past few years in understanding the pathogenesis, clinical subphenotypes and differential diagnosis of the disease, as well as the availability of new treatment options. Developed by a panel of European experts on the basis of literature reviews and, where appropriate, expert opinion, the 16 statements and five overarching principles cover the diagnosis and staging, treatment, outcome and follow-up of EGPA. These recommendations are primarily intended to be used by healthcare professionals, pharmaceutical industries and drug regulatory authorities, to guide clinical practice and decision-making in EGPA. These guidelines are not intended to limit access to medications by healthcare agencies, nor to impose a fixed order on medication use.
Similar content being viewed by others
Introduction
Eosinophilic granulomatosis with polyangiitis (EGPA; formerly known as Churg–Strauss syndrome) is a rare small-vessel vasculitis that occurs in patients with asthma and eosinophilia and is histologically characterized by tissue eosinophilia, necrotizing vasculitis and eosinophil-rich granulomatous inflammation1,2. The incidence of EGPA ranges between 0.5 and 4.2 cases per million people per year and its prevalence between 10 and 14 cases per million inhabitants globally3,4,5. The frequency of the disease is comparable in men and women, and the mean age at diagnosis is ~50 years6. Paediatric cases are extremely rare7.
EGPA usually evolves through three different phases: a prodromic ‘allergic’ phase, which can last for several years and is marked by asthma and chronic rhinosinusitis; an eosinophilic phase, during which eosinophilia and end-organ involvement appear; and a vasculitic phase, characterized by clinical manifestations due to small-vessel vasculitis (for example, mononeuritis multiplex and glomerulonephritis). However, these phases often overlap, do not necessarily develop in the aforementioned sequence and some patients do not manifest vasculitic complications2,8.
The clinical phenotype of EGPA is quite heterogeneous and the diagnosis is not always straightforward. Anti-neutrophil cytoplasmic antibodies (ANCA), usually against myeloperoxidase (MPO), are detectable in ~40% of the cases and are associated with a different frequency of clinical manifestations: features of vasculitis, particularly glomerulonephritis, peripheral neuropathy and purpura, occur more often in ANCA-positive patients, whereas the so-called eosinophilic features such as cardiac involvement and gastroenteritis are more frequent in ANCA-negative patients6,9,10,11 (Fig. 1 and Table 1). Asthma and ear–nose–throat (ENT) disease, which occur in >90% and 60–80% of patients with EGPA, respectively, are equally distributed in the ANCA-positive and ANCA-negative groups. Histopathological evidence of vasculitis is more common in ANCA-positive than in ANCA-negative patients, although EGPA lesions usually include eosinophilic infiltrates (with or without granulomas) along with necrotizing vasculitis and are therefore difficult to categorize as vasculitic or eosinophilic12,13 (Fig. 2).

The clinical manifestations of eosinophilic granulomatosis with polyangiitis (EGPA) are quite heterogeneous and their frequencies differ on the basis of anti-neutrophil cytoplasmic antibody (ANCA) status. Specifically, vasculitic features (for example, glomerulonephritis, peripheral neuropathy and purpura) occur more often in ANCA-positive patients, whereas eosinophilic features (such as cardiac involvement and gastroenteritis) are more frequent in ANCA-negative patients. The vasculitic and eosinophilic phenotypes, however, are not clearly separated, as most patients manifest an overlap between vasculitic and eosinophilic features.

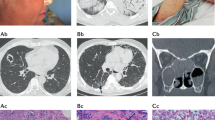
a, CT (coronal view) of the paranasal sinuses showing signs of diffuse rhinosinusitis (arrow). b, High-resolution CT (axial view) showing patchy bilateral lung infiltrates. c, Cardiac magnetic resonance phase-sensitive inversion recovery image showing a hypointense, small apical mass suggestive of intraventricular thrombus. d, Purpura of the lower limbs. e, Nasal polyp showing a dense, eosinophil-rich infiltrate within the submucosa (haematoxylin and eosin (H&E) stained; original magnification. ×20). f, Eosinophilic vasculitis in biopsy-obtained tissue of the airway mucosa (H&E stained; original magnification, ×20). g, Eosinophil-rich granuloma in airway mucosa (H&E stained; original magnification, ×20). h, Skin tissue from a patient with purpura showing perivascular inflammation of dermal vessels (arrows) (H&E stained; original magnification, ×10).
The pathogenesis of EGPA is driven by genetic and environmental factors14,15,16,17,18. Genetic studies have highlighted associations between HLA-DQ and MPO-ANCA-positive EGPA, whereas ANCA-negative EGPA is mainly associated with genetic variants involved in mucosal responses and eosinophil biology, such as GPA33 and IL5. Several other variants linked to asthma and eosinophil counts in the general population are associated with the whole EGPA spectrum14. Among environmental factors, exposure to silica, organic solvents and farming was associated with an increased risk of EGPA, whereas cigarette smoking was associated with a lower risk17. How genetics and environment interact to shape the susceptibility to and the phenotype of EGPA is still unclear.
Several cell types participate in the immunopathogenesis of the disease. Eosinophils are clearly central and are likely to mediate tissue damage, a concept supported by the evidence that targeting IL-5 (for example, using mepolizumab), a survival factor for eosinophils, is an effective therapy for EGPA19,20. CD4+ T cells orchestrate the adaptive immune response and are polarized towards a T helper 2 (TH2) phenotype, which enhances eosinophilic reactions; however, TH1 and TH17 cells might also have a role, especially in vasculitis and granuloma formation2,8,21. In a mouse model of eosinophilic vasculitis, type 2 innate lymphoid cells were important promoters of vascular permeability and secretion of eotaxins22, which in turn induce tissue influx of eosinophils23. Humoral and B cell responses are also dysregulated in EGPA: in addition to the production of ANCA, enhanced production of IgG4 is a common feature of EGPA and probably results from TH2-skewed immunity24. The pathogenic relevance of B cells is also underlined by the good response to B cell-depleting agents (such as rituximab) in a substantial proportion of patients25,26.
Given the rarity of EGPA, its heterogeneous clinical presentation and the clinical overlap with other vasculitic or eosinophilic disorders, the diagnosis of EGPA is often challenging. Multiple disciplines are involved in the care of patients, which dictates an integrated and collaborative approach. To date, no systematically developed, evidence-based guidelines have been specifically dedicated to the diagnosis and management of EGPA, and current practice is based mainly on the 2015 recommendations for EGPA published by a consensus task force12 and on the 2016 EULAR/European Renal Association (ERA)–European Dialysis and Transplant (EDTA) Association recommendations for ANCA-associated vasculitis (AAV)27. More recently, the 2021 ACR–Vasculitis Foundation guidelines for the management of AAV were developed28. The 2016 and 2021 guidelines, however, cover all forms of AAV and were not developed specifically for EGPA. In the past few years, considerable advances have been made in EGPA research, particularly in the differential diagnosis and in understanding of pathogenesis and clinical sub-phenotypes; additionally, new treatment options are available and long-term follow-up studies have enabled the definition of disease prognosis based on clinical presentation29. Here, we present comprehensive, evidence-based, cross-discipline guidelines for the diagnosis and management of EGPA, in order to contribute to the harmonization of patient care, improve quality of care and provide reliable instruments for patient education. These recommendations mainly address healthcare professionals, pharmaceutical industries and drug regulatory authorities, to guide clinical practice and decision-making in EGPA without limiting access to medications by healthcare agencies or imposing a fixed order on medication use. An update of the present guidelines would be periodically planned, based on the advances in the field.
Methods
Overview of the guideline project
This guideline follows the RIGHT (Reporting Items for Practice Guidelines in Healthcare) Statement for Practice Guidelines30. To generate this evidence-based guideline, a core committee and a voting panel were assembled. The core committee included specialists in immunology (G.E.), nephrology (A.V. and D.R.W.J.) and internal medicine (L.G.), as well as a methodologist (G.B.).
The voting committee included the core committee members and an additional 25 members with expertise in rheumatology, immunology, nephrology, internal medicine, pulmonology, cardiology, ENT surgery and pathology, as well as two project fellows, healthcare professionals and representatives of EGPA and vasculitis patient advocacy organizations.
A Delphi approach was used to identify questions to drive the literature search and the guideline statements. Voting group members were asked, by means of an e-questionnaire, to provide a level of agreement on the importance of a set of 21 questions that were proposed by the core committee and discussed during an online meeting among all voting members before the first vote (using a nine-point Likert scale, with 1–3 indicating ‘low importance’; 4–6, ‘uncertain importance’; and 7–9, ‘high importance’). After the first Delphi round, all questions were voted on again using the same scale in a second Delphi round, where some details were added to better explain unclear items. Only questions achieving positive consensus (that is, >75% of respondents providing a score of 7–9 points on the Likert scale) in the second round of voting were selected to drive the literature search (Supplementary Table 1).
Development of the PICO questions
The questions that achieved consensus were then converted by the core committee into PICO (population, intervention, comparator, outcomes) questions to be addressed in the literature search. Each PICO question represented the basis for a recommendation.
The population included patients with EGPA. With regard to interventions and comparators, evidence supporting the diagnostic (laboratory, imaging and procedures) and therapeutic interventions was retrieved on the basis of available literature studies. With regard to outcomes, not only disease-related but also treatment-related complications and comorbidities were considered. If no specific study was available for EGPA, recommendations were based on evidence derived from studies on other AAV, as well as on consensus reached among expert clinicians.
Literature search
The process of the literature search was conducted by two project fellows (A.Bet. and E.G.); the study selection is summarized in Supplementary Tables 1 and 2 and in Supplementary Fig. 1.
The PubMed, Embase and Cochrane library databases were systematically searched for literature published from 1980 until 6 September 2021. We considered all articles in English in humans, including prospective randomized controlled trials (RCTs), uncontrolled or observational studies, registries, reviews (published after 2000) and case series. The search strategy used for the PubMed database was “(EGPA OR Churg-Strauss OR “Eosinophilic Granulomatosis with Polyangiitis” OR “Churg-Strauss Syndrome”[Mesh])”; this strategy was adapted for the searches of the Embase and Cochrane library databases.
Of the articles retrieved after the systematic literature review, we selected only those relevant to the diagnosis and management of EGPA (the selection was made by two independent investigators (A.Bet and E.G.) and discrepancies in their choices were resolved by consensus of the convenor (A.V.) and the co-convenor (G.E.)); pertinent articles, identified by a manual search of the reference lists of the originally retrieved publications and by consultation with the convenor and the co-convenor, were also included. Case reports or case series including five or fewer patients were excluded. Abstracts were considered for inclusion only if they provided novel data supporting the statements and were not yet published as full-length articles.
After removal of duplicates, the systematic literature review retrieved 9,085 unique records. Of these publications, a total of 198 were finally considered for the development of this guideline (Supplementary Table 2). Further details of the article selection flow are given in Supplementary Fig. 1.
Grading of the recommendations
The convenor (A.V.), co-convenor (G.E.) and the two research fellows (A.Bet, E.G.) generated 16 recommendation statements and five overarching principles, which were further discussed through repeated circulation of the manuscript draft.
We adopted the grading system from the Oxford Centre for Evidence-Based Medicine31. The level of evidence was graded on the basis of the design and validity of the available studies, on a scale from 1a (systematic reviews of RCTs) to 5 (expert opinion); the recommendations were graded based on the total body of evidence using a letter scale from A (highest; consistent level 1 studies) to D (lowest; level 5 evidence or very inconsistent or inconclusive studies of any level). For each recommendation, members of the voting group were asked via e-questionnaire to rate their level of agreement with each statement on a 0–10 scale (with 0 indicating no agreement and 10 indicating full agreement), on the basis of both the available evidence from the literature and their own expertise. Members of the voting group could also provide their feedback on the wording of the statements.
Recommendations
The 16 statements are reported and discussed below and in Table 2, and the overarching principles are reported in Box 1. The level of evidence (L) and grade (G) are reported at the end of each statement.
Statement 1: The diagnosis of EGPA should be considered in patients with asthma, chronic rhinosinusitis and eosinophilia who develop end-organ involvement, particularly peripheral neuropathy, lung infiltrates, cardiomyopathy or other complications (for example, skin, gastrointestinal or kidney involvement). (L: 2b; G: B)
The vast majority (>90%) of patients with EGPA are affected by asthma, which usually arises in adulthood, rarely shows seasonal exacerbations and tends to worsen over time32. Asthma is often accompanied by ENT symptoms, which include chronic rhinosinusitis with nasal polyps (whereby polyps commonly recur after surgical excision) and other manifestations such as otitis media33,34. Eosinophilia (>10% or >1,500 cells per μl) is also observed in almost all patients with EGPA, although it can be masked by the use of systemic glucocorticoids21. The clinical suspicion of EGPA should be raised when patients with the above manifestations develop other complications. Lung infiltrates are common (40–50%); they are often multiple and migratory and respond to treatment with systemic glucocorticoids. Peripheral neuropathy occurs in 50–70% of patients35,36, has a mononeuritis multiplex pattern, is usually sensory but might also cause motor deficits, and has an axonal damage pattern on nerve conduction studies. Skin lesions are also frequent, but quite heterogeneous, with palpable purpura being the most vasculitis-specific lesion21,37.
Other organ manifestations that contribute to the clinical phenotype of EGPA include myocarditis and pericarditis, gastroenteritis, renal disease (revealed by proteinuria, haematuria and/or varying degrees of kidney failure) and systemic manifestations such as fatigue, weight loss, myalgia and arthralgia2.
Statement 2: There are no diagnostic criteria for EGPA. Classification criteria (including the 1990 ACR criteria and 2022 ACR–EULAR criteria) have established sensitivity and specificity, but should not be used as diagnostic criteria, as they were not developed for this purpose. Additional criteria (such as those used in the MIRRA trial) are based on expert opinion and require validation. A diagnosis of EGPA should be based on highly suggestive clinical features, objective evidence of vasculitis (for example, from histology) and ANCA. (L: 2b; G: B)
Several sets of criteria have been generated for EGPA, but none of them has been validated for diagnosis. In 1984, Lanham et al.38 proposed that asthma, eosinophilia and vasculitis involving two or more organs should be present to make a diagnosis of EGPA; these criteria are usually considered too stringent and were never validated. In 1990, the ACR defined classification criteria to distinguish the different vasculitic syndromes and identified six items for EGPA, namely, asthma, eosinophilia >10%, neuropathy, non-fixed lung infiltrates, paranasal sinus abnormalities and histological evidence of extravascular eosinophils. If four or more of these criteria are met, a patient with vasculitis can be classified as having EGPA with a sensitivity of 85% and a specificity of 99.7% (ref. 39). In 1993, the Chapel Hill Consensus Conference (CHCC) provided definitions for vasculitides, including EGPA, with a particular focus on histopathological aspects; in 2013, the revised CHCC nomenclature incorporated the concept that ANCA positivity is associated with renal involvement in EGPA1. The CHCC criteria, however, are descriptive statements based on expert opinion. In 2017, the MIRRA trial20 committee established eligibility criteria that could be used to define EGPA but they still require validation. These criteria included asthma, eosinophilia and at least two of the following: tissue evidence of eosinophilic vasculitis, perivascular eosinophilic infiltration or eosinophil-rich granulomatous inflammation; neuropathy; pulmonary infiltrates; sino-nasal abnormality; cardiomyopathy; glomerulonephritis; alveolar haemorrhage; palpable purpura; and ANCA positivity. The MIRRA criteria were therefore the first to include ANCA as a potentially diagnostic tool.
Finally, in 2022 the Diagnosis and Classification criteria in Vasculitis Study (DCVAS) defined ACR–EULAR-endorsed weighted criteria for the classification of small- and medium-sized vessel vasculitis, including EGPA40. These criteria comprise positively scored parameters, namely, a maximum eosinophil count ≥ 1 × 109/l (+5 points), obstructive airway disease (+3), nasal polyps (+3), extravascular eosinophilic-predominant inflammation (+2), and mononeuritis multiplex and/or motor neuropathy not due to radiculopathy (+1), all of which make the diagnosis of EGPA more likely. Other parameters make the likelihood of EGPA less probable and are therefore scored negatively; these parameters include a cytoplasmic ANCA (C-ANCA) pattern on immunofluorescence or anti-proteinase 3 (PR3)-ANCA positivity (−3) and haematuria (−1). If a cumulative score of 6 or more is reached, a patient with a diagnosis of small- or medium-sized vessel vasculitis can be classified as having EGPA with a sensitivity of 85% and a specificity of 99% (ref. 40).
Given the absence of diagnostic criteria, the diagnosis of EGPA — as for other small-vessel vasculitides — should be based on objective evidence of vasculitis, which should rely on histopathological findings. However, as a diagnostic biopsy is often lacking in EGPA patients, highly suggestive clinical features should be considered for the diagnosis. Examples of highly evocative clinical features are those included in the 1990 ACR or 2022 ACR–EULAR classification criteria (such as asthma, chronic rhinosinusitis with polyps, eosinophilia, neuropathy, lung infiltrates, eosinophilic cardiomyopathy or gastroenteritis, glomerulonephritis). ANCA status is also to be considered for the diagnosis of EGPA41.
Statement 3: The diagnostic evaluation of patients with suspected EGPA should always be multidisciplinary; it should rule out other eosinophilic and vasculitic disorders and investigate the main disease complications, particularly heart, respiratory, skin, renal and nervous system involvement, along with ANCA and eosinophilia. Biopsy is recommended when feasible, but is not essential to make the diagnosis of EGPA. (L: 3b; G: C)
Patients with suspected EGPA should undergo a multidisciplinary evaluation to confirm the diagnosis and investigate the involvement of the most common target organs. As shown in Fig. 3, diagnostic tests can be grouped into ‘baseline investigations’ and ‘investigations to be performed in selected cases’, which are clinically driven tests that can be ordered on the basis of specific disease manifestations and/or the positivity of baseline screening tests.

The figure shows the main investigations to be performed in patients with suspected eosinophilic granulomatosis with polyangiitis (EGPA). In the left-hand column, ‘baseline investigations’ indicate laboratory and imaging tests or procedures that are usually non-invasive and should be performed in all patients; the procedures listed in the right-hand column should be performed only in the presence of specific clinical manifestations. The investigations reported in parentheses are indicated only in selected cases. *Urinary protein excretion > 1 g per day, glomerular haematuria. ANCA, anti-neutrophil cytoplasmic antibodies; BAL, broncho-alveolar lavage; BNP, brain natriuretic peptide; CNS, central nervous system; CSF, cerebrospinal fluid; CV, cardiovascular; EMG–ENG, electromyography–electroneurography; ENT, ear–nose–throat; FESS, functional endoscopic sinus surgery; GI, gastrointestinal; HRCT, high-resolution CT; LDH, lactate dehydrogenase.
Biopsies of affected organs are encouraged because examination of tissues can contribute to the diagnostic evaluation, exclude differential diagnoses and in certain instances reflect the degree of activity/chronicity of the disease process42. Locations from which tissue samples are taken include the kidney, skin, ENT region, lung and gastrointestinal tract. Kidney tissues typically show crescentic necrotizing glomerulonephritis that can be accompanied by eosinophilic infiltrates, granulomatous changes and (eosinophil-rich) necrotizing vasculitis of arterioles and arteries. Atypical renal presentations with other glomerulopathies such as membranous nephropathy, in particular in ANCA-negative patients, can also occur43. Skin tissue in patients with EGPA who have palpable purpura invariably reveals necrotizing vasculitis of small arteries that can be accompanied by extravascular granulomas. Tissue eosinophils can be distributed in a vascular, perivascular or interstitial dermal pattern44. Biopsies of sino-nasal mucosa/polyps are often non-diagnostic33, despite attempts to use structured histopathological evaluations that have suggested that certain lesions, such as neutrophil aggregates, are more prevalent in EGPA than in chronic rhinosinusitis45. Examinations of lung and gastrointestinal tissue biopsies can reveal typical lesions13, but are seldom performed in clinical practice.
The differential diagnosis of EGPA mainly includes other small-vessel vasculitides and eosinophilic disorders. The differential diagnosis with other small-vessel vasculitides such as granulomatosis with polyangiitis (GPA) and microscopic polyangiitis is often straightforward owing to differences in phenotypes and histology, although granulomatosis with polyangiitis can sometimes present with peripheral or tissue eosinophilia and also a small proportion of patients with EGPA present with PR3-ANCA and an associated granulomatous and eosinophilic phenotype46. Other small-vessel vasculitides (such as IgA vasculitis and cryoglobulinaemia) typically show immune deposits, which are absent in EGPA and other AAV. Eosinophilic disorders are numerous and have different aetiologies, including allergic forms, haematological conditions (for example, lymphocytic and myeloproliferative hypereosinophilic syndromes, the latter of which is characterized by FIP1L1 fusion genes), parasitic infections and hypersensitivity disorders such as allergic broncho-pulmonary aspergillosis. Other conditions that only occasionally present with eosinophilia but can have overlapping features with EGPA (such as HIV infection or IgG4-related disease) should also be considered2.
Statement 4: ANCA testing should be performed in all patients with suspected EGPA. ANCA are detectable in 30–40% of patients with EGPA, most of whom test positive for MPO-ANCA. MPO-ANCA-positive patients frequently show vasculitis features, such as glomerulonephritis, neuropathy and purpura, whereas ANCA-negative patients more frequently manifest cardiomyopathy and lung involvement. (L: 2a; G: B)
ANCA can be detected by indirect immunofluorescence, which essentially shows cytoplasmic and perinuclear patterns (C-ANCA and P-ANCA, respectively), but the reference test for AAV is enzyme-linked immunosorbent assay for PR3-ANCA or MPO-ANCA. ANCA positivity is detectable in 30–40% of patients with EGPA and most of these patients test positive for P-ANCA and MPO-ANCA47. In patients with a compatible clinical phenotype (asthma, eosinophilia, rhinosinusitis and lung infiltrates), ANCA positivity supports the diagnosis of EGPA, with MPO-ANCA being considered more specific than P-ANCA for the diagnosis of vasculitis. In fact, isolated P-ANCA positivity (with negativity for MPO-ANCA) can be found in other inflammatory, non-vasculitic conditions (for example, inflammatory bowel disease). ANCA are usually not present in primary eosinophilic disorders29.
MPO-ANCA positivity is associated with clinical manifestations such as peripheral neuropathy, renal involvement and purpura, whereas it confers a lower risk of having pulmonary infiltrates and cardiac manifestations14,48. However, when considering the ANCA-positive and ANCA-negative phenotypes, the possibility of a substantial overlap between the two should be taken into consideration and the clinical value of ANCA positivity should not be overestimated47. Patients with PR3-ANCA-positive EGPA are rare and differ from MPO-ANCA-positive or ANCA-negative patients, as they more frequently have lung nodules and skin manifestations, and less frequently have active asthma, peripheral neuropathy and hypereosinophilia46. Their phenotype seems, therefore, closer to that of GPA.
ANCA status could have prognostic implications: overall survival seems to be worse in ANCA-negative patients6,9, probably attributable to the higher frequency of cardiac involvement, whereas relapses tend to be more frequent in ANCA-positive patients, although some controversies still exist6,49,50. ANCA status itself is not useful in the choice of treatment51.
Statement 5: EGPA remission is defined as the absence of clinical signs or symptoms attributable to active disease, including asthma and ENT manifestations. The daily dose of glucocorticoids should also be considered for the definition of remission, and a maximum daily dose of 7.5 mg of prednisone can be chosen as the cut-off. (L: 5; G: D)
According to the EULAR/ERA–EDTA recommendations, EGPA remission is defined as the absence of clinical signs or symptoms attributable to active disease, with a Birmingham Vasculitis Activity Score (BVAS) of zero on a maximum prednisone (or equivalent) dose of 7.5 mg per day52. This definition is currently used to assess efficacy outcomes in most observational studies and clinical trials of EGPA20,53,54,55,56, although more stringent definitions have also been adopted (that is, BVAS of zero on a maximum prednisone dose of 4 mg per day)20.
On the basis of current evidence20,28,52, we also recommend defining remission as a BVAS of zero, with or without concomitant glucocorticoid and/or immunosuppressive therapy. In the case of concomitant glucocorticoid treatment, the definition of remission could include a maximum prednisone (or equivalent) dose of 7.5 mg per day. This dose is arbitrarily fixed; considering the availability of new agents (such as anti-IL-5 biologics), which can also enable corticosteroid sparing in patients with refractory respiratory manifestations, we conclude that a more stringent definition of remission, including a maximum prednisone dose of 4 mg per day, might be adopted. Future treatment strategies should definitely be aimed at further minimization or withdrawal of glucocorticoids; therefore, the definition of remission might entail steroid-free therapy.
We also recommend including the control of asthma and/or ENT manifestations in the definition of remission. Although it is commonly agreed that ENT manifestations and/or asthma flares do not necessarily reflect vasculitis activity, we believe that current evidence is insufficient to exclude these manifestations from the definition of EGPA remission. However, the BVAS has important limitations in the assessment of asthma and ENT disease: a BVAS of zero does not preclude abnormal lung function tests57, whereas normal lung function is an important objective in asthma treatment and contributes to the definition of asthma control. Therefore, disease scores that specifically address asthma and ENT disease, such as the Asthma Control Questionnaire58 or the 22-item Sino-Nasal Outcome Test59, could be combined with the BVAS for a more comprehensive disease assessment in patients with EGPA.
Statement 6: Remission-induction treatment should be tailored on the basis of clinical manifestations with prognostic relevance. Organ-threatening manifestations included in the Five-Factor Score (renal insufficiency, proteinuria, cardiomyopathy, gastrointestinal tract and central nervous system involvement) as well as peripheral neuropathy and other rare manifestations (for example, alveolar haemorrhage) should be considered when choosing remission-induction strategies. (L: 2b; G: B)
The Five-Factor Score (FFS) predicts the risk of mortality in patients with an established diagnosis of EGPA, as well as polyarteritis nodosa, microscopic polyangiitis or GPA. It includes five factors associated with shortened overall survival, namely, renal insufficiency (serum creatinine >1.58 mg/dl), proteinuria >1 g per day, cardiomyopathy, gastrointestinal involvement and central nervous system (CNS) involvement60. The FFS considers clinical manifestations only at the time of diagnosis; hence, the appearance of new manifestations during follow-up should also be taken into account when choosing remission-induction regimens for disease flares60.
The original FFS was subsequently revised61 to include age >65 years as a poor prognostic factor and ENT involvement as a favourable prognostic factor, whereas CNS involvement was no longer included in the score. However, most studies considering the FFS for treatment decisions refer to its original version60.
In addition to the items included in the FFS, other disease features influence remission-induction therapy. Peripheral neuropathy has also necessitated immunosuppression in large observational studies and should thus be considered62,63,64. Evidence regarding the treatment of rare but severe complications such as alveolar haemorrhage or some forms of eye involvement (such as central retinal artery or vein occlusion, ischaemic optic neuropathy, orbital myositis and vasculitis, infarcts or oedema of the retina)65 is scarce, but the clinical experience derived from the other AAV suggests that these complications should also be treated aggressively66,67.
Statement 7: For remission induction in patients with new-onset, active EGPA, glucocorticoids should be administered as initial therapy. In patients with severe disease (unfavourable prognostic factors discussed in Statement 6) cyclophosphamide or, as an alternative, rituximab, should be added. In patients with non-severe disease, glucocorticoids alone should be used. (L: 2b; G: B)
Remission-induction treatment should be stratified on the basis of disease severity, whereby severe disease is defined according to the presence of at least one adverse prognostic factor (that is, the factors included in the FFS and those considered as manifestations of severe disease, such as peripheral neuropathy, alveolar haemorrhage, mesenteric ischaemia, limb digital ischaemia and eye disease). Patients with severe disease should be treated with pulsed intravenous glucocorticoids (usually daily methylprednisolone pulses of 500–1,000 mg each over 3 days, for a maximum total dose of 3 g) followed by high-dose oral glucocorticoids (for example, 0.75–1 mg/kg per day) (Fig. 4). Cyclophosphamide should be added to glucocorticoids for remission induction in patients with severe disease. The evidence for the use of cyclophosphamide is supported by an RCT performed in patients with FFS ≥ 1, which showed that relapse-free survival was longer after 12 cyclophosphamide pulses than after six cyclophosphamide pulses (administered every 2 weeks for 1 month, then every 4 weeks thereafter, at a dose of 0.6 g/m2 per pulse)68. However, the optimal duration of cyclophosphamide induction therapy in severe EGPA remains to be established. In routine clinical practice, we recommend that cyclophosphamide induction be conducted until remission is achieved, usually within 6 months; longer induction periods (up to 9–12 months) can be reserved for patients who improve slowly but do not reach complete remission by month 6.

This algorithm is based on the evidence-based statements and differentiates the treatment of patients with new-onset, active disease from that of patients with relapsing disease. The treatment approaches are also tailored on the basis of disease severity. EGPA, eosinophilic granulomatosis with polyangiitis; FFS, five-factor score.
Observational studies have initially highlighted the potential role of rituximab for remission induction26,69,70. The REOVAS RCT, published in abstract form in 2021, showed that rituximab (1-gram pulses 2 weeks apart) is comparable with cyclophosphamide (nine intravenous pulses over 5 months) for induction of remission (defined as BVAS of zero and a prednisone dose ≤ 7.5 mg per day) in patients with FFS ≥ 1. Adverse events and cumulative prednisone exposure were comparable in the two groups71. Unlike in previous observational studies, no significant differences in response to rituximab were found between ANCA-positive and ANCA-negative patients; likewise, no differences were found between patients with new-onset and relapsing disease.
In patients with non-severe disease, glucocorticoids alone are usually sufficient to induce remission. In a prospective trial involving 72 patients with an FFS of zero, the remission rate after glucocorticoid monotherapy was 93% (ref. 72). However, a considerable proportion of patients who responded to treatment experienced early relapses (35% within the first year of treatment), mostly respiratory, and thus received treatment with immunosuppressants such as cyclophosphamide and azathioprine. Although the evidence supporting the use of traditional immunosuppressants for remission maintenance in non-severe EGPA is scarce, these agents are often used in routine clinical practice.
The MIRRA RCT tested the efficacy and safety of mepolizumab versus placebo in achieving remission (BVAS of zero and prednisolone dose ≤ 4 mg per day) in patients with relapsing or refractory EGPA without organ- or life-threatening manifestations. Mepolizumab proved significantly more efficacious than placebo and had comparable toxicity. ANCA status did not influence response, although the proportion of ANCA-positive patients included in the trial was low (10%)20. Therefore, the combination of mepolizumab and glucocorticoids for remission induction in non-severe EGPA should be considered19,73. Further details on the MIRRA trial, the indications for mepolizumab in EGPA and the suggested dosage are discussed in Statement 13.
Overall, in both severe and non-severe disease, remission induction is centred on the use of high-dose glucocorticoids, which certainly contributes to short-term and long-term treatment-related toxic effects. Treatment strategies (such as mepolizumab) are already heading towards glucocorticoid sparing, as demonstrated by the MIRRA trial, which, however, enrolled patients without organ- or life-threatening manifestations. It is advisable that remission induction in patients with severe disease has the same goal, as demonstrated in other forms of AAV in trials published in the past few years (for example, PEXIVAS)74.
Statement 8: For remission maintenance, in patients with severe EGPA, we recommend using rituximab, mepolizumab or traditional DMARDs in combination with glucocorticoids. In patients with non-severe EGPA, we suggest glucocorticoids, alone or in combination with mepolizumab. Glucocorticoids should be tapered to the minimum effective dosage to reduce toxicity. (L: 2b; G: B)
After remission induction, a maintenance treatment should be considered to reduce the risk of toxicity and of relapse. Glucocorticoid-related toxicity is particularly relevant in patients with EGPA as they are often exposed to high cumulative doses of glucocorticoids and only a small proportion of them can be weaned off glucocorticoids. Therefore, several efforts are being made to reduce glucocorticoid exposure without putting patients at risk of relapse. The available evidence on remission-maintenance therapies in EGPA is limited. We recommend adopting different remission-maintenance strategies based on the presence of unfavourable prognostic factors (as defined in Statement 6). In patients with severe disease, the maintenance approach is uncertain. Observational studies have reported the use of glucocorticoids combined with azathioprine, methotrexate and leflunomide to maintain remission55,75, but none of these approaches has been demonstrated to prolong relapse-free survival (as compared with glucocorticoid monotherapy). Despite the absence of evidence from the literature, DMARDs are routinely used in clinical practice for remission maintenance12,28,76,77.
Rituximab has been proposed as an induction therapy for EGPA, but also seems to be effective for remission maintenance: in an observational study, scheduled rituximab maintenance therapy (500 mg every 6 months) reduced the relapse rate as compared with unscheduled treatment (that is, a single 1-gram infusion, administered only in the case of relapse)25. In particular, all patients receiving scheduled rituximab were able to maintain remission throughout the follow-up period. In a retrospective study published in 2020, rituximab maintenance also showed efficacy in reducing the median glucocorticoid dose for the control of asthma and systemic manifestations70. We recommend rituximab maintenance in patients with severe disease, particularly in those who achieved remission on rituximab.
Mepolizumab is commonly used during remission maintenance, mainly for the control of asthma and to reduce glucocorticoid exposure. However, some observational studies19,73 suggest that it might also be effective in the treatment of major organ manifestations (such as neuropathy and cardiomyopathy); therefore, its use for remission maintenance in patients with severe manifestations can be considered. In patients with non-severe disease, glucocorticoids combined with mepolizumab are often effective in maintaining remission, as shown in the MIRRA trial in patients with relapsing or refractory disease20 and in observational studies19,73.
Statement 9: EGPA relapse is defined as the recurrence of clinical signs or symptoms attributable to active disease following a period of remission. The need for an increase in the glucocorticoid dosage or the initiation of or an increase in an immunosuppressant should also be considered a relapse. The relapse or new onset of systemic vasculitis (systemic relapse) should be differentiated from the isolated exacerbation of asthma and ENT manifestations (respiratory relapse). (L: 5; G: D)
EGPA relapse can be defined as the recurrence of clinical signs or symptoms attributable to active disease following a period of remission12,28,78. In line with latest trials20, we recommend considering as disease relapse the need for an increase in the daily glucocorticoid dosage or the initiation of or an increase in an immunosuppressive therapy. When defining relapse, we recommend distinguishing the relapse of systemic vasculitis (systemic relapse) from the isolated exacerbation of asthma and ENT manifestations (respiratory relapse). An increase in the eosinophil count without accompanying clinical manifestations should not be considered a relapse.
Systemic relapses can be differentiated into severe and non-severe, the former presenting either with manifestations included in the FFS or with life- or organ-threatening manifestations (Statement 6)68. For example, relapsing peripheral neuropathy, glomerulonephritis, cardiomyopathy or gastroenteritis are usually considered severe relapses, whereas skin manifestations (for example, urticaria), arthralgia or systemic symptoms (such as fatigue or weight loss) are usually considered non-severe.
Statement 10: Relapses should be treated according to type (systemic versus respiratory) and severity. For severe systemic relapses, we recommend using rituximab or cyclophosphamide with glucocorticoids. For non-severe systemic and respiratory relapses, we recommend increasing the dose of glucocorticoids and/or adding mepolizumab. (L: 2b; G: C)
The treatment of relapses depends primarily on their type (systemic versus respiratory relapses) and severity (severe versus non-severe, for systemic relapses), but should also take into account previous treatments and the burden of chronic damage. For severe systemic relapses, rituximab or cyclophosphamide can be considered the main remission-induction agents. Rituximab can be preferred over cyclophosphamide, especially when re-treatment with cyclophosphamide is to be avoided, as in patients who previously achieved remission on rituximab or failed to respond to cyclophosphamide. Cyclophosphamide can be considered in recurrent and severe cardiac disease, in other severe or life-threatening complications and/or in patients who previously failed to respond to rituximab. These recommendations are essentially based on the results of observational studies25,26,70,77, as none of the published trials enrolled patients with severely relapsing disease. The REOVAS trial included patients with relapsing disease, as well as those with new-onset disease, but the results on these two subgroups are still unavailable71.
For patients with non-severe systemic relapses, several options are available, and must be chosen on a patient-by-patient basis. Some minor relapses can be managed with optimization of glucocorticoid therapy; mepolizumab can also be used in addition to glucocorticoids to treat minor relapses. For respiratory relapses, a stepwise approach should be followed. First, topical therapies (for example, bronchodilators) should be optimized (Statement 14). Second, the dose of oral glucocorticoids can be increased and short courses of high-dose glucocorticoids (0.5–1 mg/kg per day for 5–7 days) can be given and stopped without tapering. Third, mepolizumab can be added. Functional endoscopic sinus surgery can be considered for relapsing ENT disease that does not adequately respond to the above approach.
Statement 11: Refractory EGPA is defined as unchanged or increased disease activity after 4 weeks of appropriate remission-induction therapy. The persistence or worsening of systemic manifestations should be distinguished from that of respiratory manifestations. (L: 5; G: D)
Refractory EGPA denotes persisting or worsening disease despite appropriate remission-induction therapy28,52,69. Refractory EGPA with severe manifestations is rare if patients are treated with cyclophosphamide as the remission-induction regimen68. The minimum duration of remission induction to define refractoriness has not been established, but 4 weeks can be considered a reasonable time frame, analogous to the other AAV52.
EGPA can be defined as refractory only after addressing the following issues52: the primary diagnosis should be re-evaluated, and refractory manifestations being attributable to other aetiologies such as infections or malignancies must be excluded; the appropriateness of the remission-induction treatment (Statement 7) should be checked; the patient’s compliance with the remission-induction regimen should be assessed; and persistently active manifestations should be distinguished from irreversible damage (a supporting tool is the Vasculitis Damage Index).
Once refractoriness has been established, it must be ascertained whether it is attributable to persistence or worsening of systemic manifestations or of asthma and/or ENT disease, or both. For patients with refractory systemic EGPA despite remission-induction treatment with high-dose glucocorticoids plus cyclophosphamide, the use of rituximab is recommended, whereas cyclophosphamide should be used for patients in whom remission induction with rituximab fails69. For patients with refractory asthma/ENT disease (without systemic manifestations) despite use of high-dose glucocorticoids and optimized inhaled therapy, the addition of mepolizumab is recommended20. In patients whose disease does not respond to these approaches, different therapeutic options can be considered, including other anti-IL-5 agents (Statement 13), plasma exchange and intravenous immunoglobulin therapy; anti-IgE agents have also been tried but with unsatisfactory results73,79,80,81,82. In selected patients, the use of IFNα83 or mycophenolate mofetil can also be considered for remission induction84. However, no solid evidence supports their use as maintenance therapy.
Statement 12: The IL-5 inhibitor mepolizumab in combination with glucocorticoids is recommended to induce remission in patients with relapsing-refractory EGPA without organ- or life-threatening manifestations. Mepolizumab can also be used for remission maintenance, particularly in patients requiring a daily prednisone dose ≥7.5 mg for the control of their respiratory manifestations. (L: 2b; G: B)
IL-5 is a key cytokine for eosinophil maturation, differentiation and survival. In the past decade, interest around the use of therapies targeting IL-5 or IL-5 receptor in EGPA has been growing. Among them, the monoclonal antibody mepolizumab was tested in observational studies85,86,87 and subsequently in the phase III MIRRA RCT20, which included 136 patients with relapsing or refractory EGPA and without life- or organ-threatening manifestations. The results of this trial indicate that mepolizumab (300 mg every 4 weeks) is effective at inducing and maintaining remission, while improving lung function and allowing glucocorticoid sparing88.
However, cohort studies have shown that a lower mepolizumab dosage (100 mg every 4 weeks) is also effective for EGPA, especially for the control of respiratory manifestations19,73. In the largest of these studies19, the efficacy of 100 mg every 4 weeks and 300 mg every 4 weeks was comparable, although these findings resulted from a retrospective analysis.
We recommend consideration of mepolizumab for induction therapy in patients with relapsing-refractory disease without organ- or life-threatening manifestations. Mepolizumab should also be considered for remission maintenance, mainly for the control of asthma and to reduce glucocorticoid exposure. The approved dosage for EGPA is 300 mg every 4 weeks. However, a lower initial dosage (100 mg every 4 weeks) can be considered, particularly in patients with limited respiratory manifestations; this dosage can subsequently be titrated up to 300 mg every 4 weeks in patients with an unsatisfactory response to treatment19. The efficacy of other IL-5 or IL-5 receptor inhibitors (such as benralizumab and reslizumab) has been reported in case reports and case series89,90; their use can therefore be considered in patients with disease refractory to mepolizumab therapy.
Statement 13: In patients with EGPA who have active asthma or ENT involvement, topical and/or inhaled therapy must be optimized. The approach to the management of these disease manifestations must involve specialists such as pulmonologists and otolaryngologists. (L: 5; G: D)
Asthma and ENT manifestations negatively affect the quality of life of patients with EGPA. Moreover, respiratory involvement is among the most frequently relapsing manifestations in EGPA, with a course mostly independent of systemic disease involvement75.
Although the use of systemic therapies (such as glucocorticoids and mepolizumab) is the mainstay for the control of respiratory EGPA manifestations, combination with inhaled therapies should be considered as a supportive treatment for asthma control91. In particular, in patients with asthmatic manifestations the combination of high-dose inhaled glucocorticoids and long-acting β2-agonists seems to be a valid option92. However, consultation with a pulmonologist is strongly recommended.
Patients with ENT involvement might also benefit from nasal rinses and other topical therapies (for example, antibiotics or lubricants) for the long-term control of these symptoms. Consultation with an otolaryngologist is strongly encouraged for these patients.
Statement 14: Treatment decisions should be modified as necessary in special populations of patients such as children, elderly patients, women of child-bearing age and those with comorbidities. There is still no evidence that different phenotypes (such as ANCA-positive versus ANCA-negative) necessitate different approaches. (L: 5; G: D)
Special populations should also be considered when choosing the treatment approach.
EGPA is extremely rare in children7; therefore, there is no guidance for treatment of this special population. Glucocorticoids and other traditional immunosuppressants remain the mainstay of therapy. However, as cyclophosphamide reduces the ovarian reserve and can affect male fertility, rituximab could be the preferred option for young patients. Also, mepolizumab can be considered an optimal glucocorticoid-sparing therapy, and is approved for use in EGPA in patients >6 years old93.
In all patients with EGPA, we strongly recommend tapering of glucocorticoids to the minimum effective dosage in order to reduce long-term toxicity. Also, a reduction in the dose of immunosuppressants should be considered to limit the risk of complications, especially infections. These recommendations particularly apply to the elderly population (aged >65 years), considering their intrinsic fragility and increased burden of comorbidities. An open-label trial including 104 patients with systemic necrotizing vasculitis (of whom 14 had EGPA) aged >65 years indicated that a reduction of cyclophosphamide dose (from 500 mg/m2 to a fixed dose of 500 mg) and a reduction in the duration of glucocorticoid treatment (from 26 to 9 months) is useful to lower the risk of adverse events and does not affect remission rates94.
Pregnant women should not discontinue treatment, as disease flare could have a negative effect on pregnancy outcomes; however, only glucocorticoids, intravenous immunoglobulins and azathioprine are considered to be safe for use during pregnancy95. Cyclophosphamide, mycophenolate mofetil and methotrexate are also contraindicated during pregnancy and should be stopped 3–6 months before conception. Rituximab and mepolizumab should also be avoided during pregnancy owing to a lack of safety data93,96. Considering that pregnancy loss can occur in up to 20% of patients with EGPA, dedicated management by obstetric specialists is advocated95.
Patients with EGPA can be subclassified according to the ANCA status (ANCA-positive versus ANCA-negative). Preliminary evidence, mainly from observational studies, has suggested that ANCA-positive and ANCA-negative patients have different sensitivity to treatments; in particular, ANCA-positive patients seemed to be more responsive to rituximab therapy than ANCA-negative patients26,97. This view has been challenged by the results of the REOVAS trial, which did not reveal significant differences in the rates of response to rituximab between ANCA-positive and ANCA-negative patients56. The MIRRA trial did not reveal any significant difference in response to mepolizumab between the two subgroups either, although the ANCA-positive subgroup accounted for only 10% of the enrolled patients20. These results support the recommendation put forth in 2020 that ANCA status should not influence treatment decisions51, even though it denotes differences in clinical phenotype and genetic backgrounds.
Statement 15: Although some laboratory parameters (for example, eosinophil count or ANCA) are commonly monitored, there are no reliable biomarkers to measure disease activity in EGPA. Disease activity should therefore be assessed on follow-up only using validated clinical tools. (L: 5; G: D)
During follow-up, EGPA is usually monitored clinically, by detecting signs and symptoms of active disease and by means of appropriate imaging or functional studies (such as pulmonary function tests, electromyography–electroneurography and echocardiography), and with routine laboratory tests. However, no biomarker reliably correlates with disease activity or predicts relapse. The eosinophil count is routinely assessed in patients with EGPA as it is thought to mirror disease activity; however, despite eosinophil counts being markedly high in patients at diagnosis and decreased during remission, relapses can also occur without an increase in the eosinophil count98. In a cohort study of 141 patients, the eosinophil count — as well as erythrocyte sedimentation rate and serum C-reactive protein and IgE concentrations — showed weak or no association with disease activity and disease flares99. Therefore, the role of these parameters as longitudinal biomarkers seems limited. Concentrations of other biomarkers of eosinophil biology such as eosinophil cationic protein100, CCL26 (eotaxin-3)23 and CCL17 (thymus and activation-regulated chemokine)101 are high in patients at the time of diagnosis but do not correlate with disease activity during follow-up and therefore are not used in clinical practice.
Although its use is still limited, monitoring of serum IgG4 concentration might have some value for the assessment of disease activity. In an observational study including 72 patients with AAV (of whom 46 had EGPA), 25 with atopic asthma and 20 healthy individuals, serum IgG4 concentrations were found to be markedly increased in patients with active EGPA and correlated positively with BVAS and number of organs involved24. Nevertheless, these data are not yet confirmed and the use of IgG4 as a biomarker of disease activity is controversial.
The value of ANCA monitoring in EGPA is also debated, as ANCA positivity or titres are not clearly associated with disease activity or response to treatment19. However, serum ANCA monitoring is advisable in patients with MPO-ANCA positivity at disease onset, because persistence, rise or reappearance of ANCA might justify more frequent clinical assessment51.
Statement 16: Routine monitoring of EGPA-related manifestations, with particular reference to lung function, cardiovascular events and neurological complications, is recommended. Long-term monitoring of comorbidities (such as cancer, infections and osteoporosis) is also recommended. (L: 2b; G: B)
EGPA is associated with a consistent burden of morbidity and mortality. Among the most frequent complications, persistent asthma negatively affects quality of life and life expectancy. Close monitoring of lung function is recommended, particularly in patients who are overweight, those presenting with pulmonary infiltrates, in cases of uncontrolled or severe asthma at diagnosis and in patients with rhinosinusitis, as these features have been associated with a more severe asthma course32,91.
Major vascular events102,103 and cardiac involvement104 are frequent in EGPA and seem to be associated with a poorer survival105,106,107. Periodic echocardiography and electrocardiography is recommended in all patients108 for early detection of asymptomatic cardiac involvement. Cardiac magnetic resonance monitoring is recommended only in patients with overt cardiomyopathy, whereas its routine use in asymptomatic patients seems limited109,110.
Another severe complication of EGPA is related to sequelae of neuropathy. Although neuropathy is not life threatening, we strongly recommend appropriate management of this complication given the risk of disability due to muscle atrophy and neuropathic pain35,64,111. Consultation with a neurologist and a physiotherapist is strongly encouraged in the management of these patients.
Some other complications should also be assessed and prevented. Patients with EGPA seem to be at an increased risk of infections, which is related to the disease and also to immunosuppressive therapy108. We advocate prophylaxis against Pneumocystis jirovecii infection with sulfamethoxazole–trimethoprim (800 mg–160 mg on alternate days or 400 mg–80 mg daily) in all patients treated with cyclophosphamide and/or rituximab27,112. Screening for major chronic infections (such as hepatitis B virus and HIV) is also strongly recommended before initiating treatment with cyclophosphamide or rituximab. Therapy with cyclophosphamide and rituximab has a negative effect on the humoral vaccine response and can lead to clinically relevant secondary hypogammaglobulinaemia. Accordingly, timely vaccination according to current recommendations, passive immunization if necessary and monitoring of quantitative IgG serum concentrations are recommended.
The risk of cancer should be carefully considered, especially in patients treated with cyclophosphamide113,114,115. All patients should undergo age-appropriate cancer screening; cyclophosphamide-treated patients should also be regularly screened for bladder cancer (for example, urine cytology examination), myeloid leukaemia (evaluation of peripheral blood cell counts and/or haematological examination), and skin cancer (dermatological surveillance)113,116,117.
The risk of osteoporosis should also be assessed, particularly in patients receiving prolonged glucocorticoid treatment118. Periodic assessment of bone density is recommended in all patients with EGPA, especially those with a high cumulative glucocorticoid dose and in those with concomitant traditional risk factors for osteoporosis.
Although only a subgroup of patients with EGPA have allergies (30–40%)119,120, testing allergies, particularly perennial ones, through a prick test and/or a radioallergosorbent test, is encouraged in EGPA patients, and appropriate anti-histaminic treatment should be considered in allergic patients, also to control ENT symptoms119.
Conclusions
EGPA is a rare form of vasculitis and has a complex phenotype. Clinicians face several challenges in the diagnosis and management of this condition, given the absence of diagnostic biomarkers and the paucity of controlled clinical trials. The management of the disease requires a multidisciplinary approach and is based on the use of glucocorticoids, traditional immunosuppressants and novel biologic agents. The evidence-based guideline defined in this article provides guidance for diagnosis and the best possible management strategies.
Future research concerning EGPA will have to address several issues (Box 2), such as improving understanding of its pathogenesis and the role of genetics. Defining diagnostic criteria and exploring biomarkers that can assist with the differential diagnosis and the assessment of disease activity are also of utmost importance. Management of comorbidities or disease-related complications such as cardiovascular disease is warranted. Finally, the indications for new treatment options need to be better defined.
References
Jennette, J. C. et al. 2012 Revised International Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 65, 1–11 (2013).
Vaglio, A., Buzio, C. & Zwerina, J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy 68, 261–273 (2013).
Mahr, A., Guillevin, L., Poissonnet, M. & Ayme, S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: a capture-recapture estimate. Arthritis Rheum. 51, 92–99 (2004).
Mohammad, A. J., Jacobsson, L. T., Westman, K. W., Sturfelt, G. & Segelmark, M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology 48, 1560–1565 (2009).
Watts, R. A., Lane, S. & Scott, D. G. What is known about the epidemiology of the vasculitides? Best. Pract. Res. Clin. Rheumatol. 19, 191–207 (2005).
Comarmond, C. et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum. 65, 270–281 (2013).
Zwerina, J., Eger, G., Englbrecht, M., Manger, B. & Schett, G. Churg-Strauss syndrome in childhood: a systematic literature review and clinical comparison with adult patients. Semin. Arthritis Rheum. 39, 108–115 (2009).
Mahr, A. et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Curr. Opin. Rheumatol. 26, 16–23 (2014).
Healy, B. et al. Antineutrophil cytoplasmic autoantibodies and myeloperoxidase autoantibodies in clinical expression of Churg-Strauss syndrome. J. Allergy Clin. Immunol. 131, 571–576.e6 (2013).
Sablé-Fourtassou, R. et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann. Intern. Med. 143, 632–638 (2005).
Sinico, R. A. et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 52, 2926–2935 (2005).
Groh, M. et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) consensus task force recommendations for evaluation and management. Nephron 129, 158 (2015).
Vaglio, A., Corradi, D., Ronda, N., Garini, G. & Buzio, C. Large bowel obstruction heralding Churg-Strauss syndrome. Am. J. Gastroenterol. 99, 562–563 (2004).
Lyons, P. A. et al. Genome-wide association study of eosinophilic granulomatosis with polyangiitis reveals genomic loci stratified by ANCA status. Nat. Commun. 10, 5120 (2019).
Martorana, D. et al. Fcγ-receptor 3B (FCGR3B) copy number variations in patients with eosinophilic granulomatosis with polyangiitis. J. Allergy Clin. Immunol. 137, 1597–1599.e8 (2016).
Vaglio, A. et al. HLA-DRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum. 56, 3159–3166 (2007).
Maritati, F. et al. Occupational exposures and smoking in eosinophilic granulomatosis with polyangiitis: a case–control study. Arthritis Rheumatol. 73, 1694–1702 (2021).
Fagni, F., Bello, F. & Emmi, G. Eosinophilic granulomatosis with polyangiitis: dissecting the pathophysiology. Front. Med. 8, 627776 (2021).
Bettiol, A. et al. Mepolizumab for eosinophilic granulomatosis with polyangiitis (EGPA): a European multicenter observational study. Arthritis Rheumatol. 74, 295–306 (2022).
Wechsler, M. E. et al. Mepolizumab or placebo for eosinophilic granulomatosis with polyangiitis. N. Engl. J. Med. 376, 1921–1932 (2017).
Vaglio, A. et al. Churg-Strauss syndrome. Kidney Int. 76, 1006–1011 (2009).
Kotas, M. E. et al. A role for IL-33-activated ILC2s in eosinophilic vasculitis. JCI Insight 6, e143366 (2021).
Zwerina, J. et al. Eotaxin-3 in Churg-Strauss syndrome: a clinical and immunogenetic study. Rheumatology 50, 1823–1827 (2011).
Vaglio, A. et al. IgG4 immune response in Churg-Strauss syndrome. Ann. Rheum. Dis. 71, 390–393 (2012).
Emmi, G. et al. Scheduled rituximab maintenance reduces relapse rate in eosinophilic granulomatosis with polyangiitis. Ann. Rheum. Dis. 77, 952–954 (2018).
Mohammad, A. J. et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Ann. Rheum. Dis. 75, 396–401 (2016).
Yates, M. et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann. Rheum. Dis. 75, 1583–1594 (2016).
Chung, S. A. et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Care Res. 73, 1088–1105 (2021).
Trivioli, G., Terrier, B. & Vaglio, A. Eosinophilic granulomatosis with polyangiitis: understanding the disease and its management. Rheumatology 59 (Suppl. 3), iii84–iii94 (2020).
Chen, Y. et al. A reporting tool for practice guidelines in health care: the RIGHT statement. Ann. Intern. Med. 166, 128–132 (2017).
CEBM. Oxford Centre for Evidence-Based Medicine: Levels of Evidence (March 2009) https://www.cebm.ox.ac.uk/resources/levels-of-evidence/oxford-centre-for-evidence-based-medicine-levels-of-evidence-march-2009 (2009).
Cottin, V. et al. Respiratory manifestations of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Eur. Respir. J. 48, 1429–1441 (2016).
Bacciu, A. et al. Ear, nose and throat manifestations of Churg-Strauss syndrome. Acta Otolaryngol. 126, 503–509 (2006).
Bacciu, A. et al. Nasal polyposis in Churg-Strauss syndrome. Laryngoscope 118, 325–329 (2008).
Cho, H. J. et al. Clinical characteristics and treatment response of peripheral neuropathy in the presence of eosinophilic granulomatosis with polyangiitis (Churg-Strauss Syndrome): experience at a Single Tertiary Center. J. Clin. Neurol. 13, 77–83 (2017).
Padoan, R. et al. Overall disability sum score for clinical assessment of neurological involvement in eosinophilic granulomatosis with polyangiitis. J. Clin. Rheumatol. 24, 197–202 (2018).
Micheletti, R. G. et al. Cutaneous manifestations of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. 72, 1741–1747 (2020).
Lanham, J. G., Elkon, K. B., Pusey, C. D. & Hughes, G. R. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine 63, 65–81 (1984).
Masi, A. T. et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 33, 1094–1100 (1990).
Grayson, P. C. et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology Classification Criteria for Eosinophilic granulomatosis with polyangiitis. Arthritis Rheumatol. 74, 386–392 (2022).
Watts, R. et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann. Rheum. Dis. 66, 222–227 (2007).
Cottin, V. et al. Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): a study of 157 patients by the Groupe d’Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun. Rev. 16, 1–9 (2017).
Durel, C. et al. Renal involvement in eosinophilic granulomatosis with polyangiitis (EGPA): a multicentric retrospective study of 63 biopsy-proven cases. Rheumatology 60, 359–365 (2021).
Tabb, E. S., Duncan, L. M. & Nazarian, R. M. Eosinophilic granulomatosis with polyangiitis: cutaneous clinical and histopathologic differential diagnosis. J. Cutan. Pathol. 48, 1379–1386 (2021).
Contro, G. et al. Neutrophil infiltrates and eosinophil aggregates in chronic rhinosinusitis with nasal polyps and EGPA. Clin. Rheumatol. 40, 1949–1957 (2021).
Papo, M. et al. Significance of PR3-ANCA positivity in eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Rheumatology 60, 4355–4360 (2021).
Moiseev, S. et al. Association of venous thromboembolic events with skin, pulmonary and kidney involvement in ANCA-associated vasculitis: a multinational study. Rheumatology 60, 4654–4661 (2021).
Chang, H. C., Chou, P. C., Lai, C. Y. & Tsai, H. H. Antineutrophil cytoplasmic antibodies and organ-specific manifestations in eosinophilic granulomatosis with polyangiitis: a systematic review and meta-analysis. J. Allergy Clin. Immunol. Pract. 9, 445–452.e6 (2021).
Samson, M. et al. Long-term outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. J. Autoimmun. 43, 60–69 (2013).
Sokolowska, B. M. et al. ANCA-positive and ANCA-negative phenotypes of eosinophilic granulomatosis with polyangiitis (EGPA): outcome and long-term follow-up of 50 patients from a single Polish center. Clin. Exp. Rheumatol. 32, S41–S47 (2014).
Moiseev, S. et al. International consensus on antineutrophil cytoplasm antibodies testing in eosinophilic granulomatosis with polyangiitis. Am. J. Respir. Crit. Care Med. 202, 1360–1372 (2020).
Yates M. et al. Validation of the EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis by disease content experts. RMD Open 3, e000449 (2017).
French Vasculitis Study Group. MAINRITSEG trial https://clinicaltrials.gov/ct2/show/NCT03164473 (2022).
AstraZeneca. MANDARA trial https://clinicaltrials.gov/ct2/show/NCT04157348 (2023).
Moosig, F. et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): monocentric experiences in 150 patients. Ann. Rheum. Dis. 72, 1011–1017 (2013).
Vega Villanueva, K. L. & Espinoza, L. R. Eosinophilic vasculitis. Curr. Rheumatol. Rep. 22, 5 (2020).
Suppiah, R. et al. A cross-sectional study of the Birmingham Vasculitis Activity Score version 3 in systemic vasculitis. Rheumatology 50, 899–905 (2011).
Santino, T. A., Chaves, G. S., Freitas, D. A., Fregonezi, G. A. & Mendonca, K. M. Breathing exercises for adults with asthma. Cochrane Database Syst. Rev. 3, CD001277 (2020).
Cai, S., Xu, S., Lou, H. & Zhang, L. Comparison of different biologics for treating chronic rhinosinusitis with nasal polyps: a network analysis. J. Allergy Clin. Immunol. Pract. 10, 1876–1886.e7 (2022).
Guillevin, L. et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome: a prospective study in 342 patients. Medicine 75, 17–28 (1996).
Guillevin, L. et al. The five-factor score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine 90, 19–27 (2011).
Samson, M. et al. Mononeuritis multiplex predicts the need for immunosuppressive or immunomodulatory drugs for EGPA, PAN and MPA patients without poor-prognosis factors. Autoimmun. Rev. 13, 945–953 (2014).
Hattori, N. et al. Clinicopathological features of Churg-Strauss syndrome-associated neuropathy. Brain 122, 427–439 (1999).
Hattori, N. et al. Mortality and morbidity in peripheral neuropathy associated Churg-Strauss syndrome and microscopic polyangiitis. J. Rheumatol. 29, 1408–1414 (2002).
Akella, S. S., Schlachter, D. M., Black, E. H. & Barmettler, A. Ophthalmic eosinophilic granulomatosis with polyangiitis (Churg-Strauss Syndrome): a systematic review of the literature. Ophthalmic Plast. Reconstr. Surg. 35, 7–16 (2019).
Perez-Jacoiste Asin, M. A. et al. Ocular involvement in granulomatosis with polyangiitis: a single-center cohort study on 63 patients. Autoimmun. Rev. 18, 493–500 (2019).
Abe, Y., Kusaoi, M., Tada, K., Yamaji, K. & Tamura, N. Efficacy of plasma exchange therapy for diffuse alveolar hemorrhage in patients with microscopic polyangiitis. Ther. Apher. Dial. 26, 515–521 (2022).
Cohen, P. et al. Churg-Strauss syndrome with poor-prognosis factors: a prospective multicenter trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in forty-eight patients. Arthritis Care Res. 57, 686–693 (2007).
Thiel, J. et al. Rituximab as induction therapy in eosinophilic granulomatosis with polyangiitis refractory to conventional immunosuppressive treatment: a 36-month follow-up analysis. J. Allergy Clin. Immunol. Pract. 5, 1556–1563 (2017).
Casal Moura, M. et al. Asthma control in eosinophilic granulomatosis with polyangiitis treated with rituximab. Clin. Rheumatol. 39, 1581–1590 (2020).
Terrier, B. P. G. et al. Rituximab versus conventional therapeutic strategy for remission induction in eosinophilic granulomatosis with polyangiitis: a double-blind, randomized, controlled trial [abstract]. Abstract No. L21. Arthritis Rheumatol. https://acrabstracts.org/abstract/rituximab-versus-conventional-therapeutic-strategy-for-remission-induction-in-eosinophilic-granulomatosis-with-polyangiitis-a-double-blind-randomized-controlled-trial/ (2021).
Ribi, C. et al. Treatment of Churg-Strauss syndrome without poor-prognosis factors: a multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. 58, 586–594 (2008).
Canzian, A. et al. Use of biologics to treat relapsing and/or refractory eosinophilic granulomatosis with polyangiitis: data from a European collaborative study. Arthritis Rheumatol. 73, 498–503 (2021).
Walsh, M. et al. Plasma exchange and glucocorticoids in severe ANCA-associated vasculitis. N. Engl. J. Med. 382, 622–631 (2020).
Puéchal, X. et al. Non-severe eosinophilic granulomatosis with polyangiitis: long-term outcomes after remission-induction trial. Rheumatology 58, 2107–2116 (2019).
Terrier, B. et al. ANCA-associated vasculitides: recommendations of the French Vasculitis Study Group on the use of immunosuppressants and biotherapies for remission induction and maintenance. Presse Med. 49, 104031 (2020).
Maritati, F. et al. Methotrexate versus cyclophosphamide for remission maintenance in ANCA-associated vasculitis: a randomised trial. PloS ONE 12, e0185880 (2017).
Terrier, B. et al. French recommendations for the management of systemic necrotizing vasculitides (polyarteritis nodosa and ANCA-associated vasculitides). Orphanet J. Rare Dis. 15, 351 (2020).
Celebi Sozener, Z. et al. Omalizumab in the treatment of eosinophilic granulomatosis with polyangiitis (EGPA): single-center experience in 18 cases. Allergy: Eur. J. Allergy Clin. Immunol. 74, 450 (2019).
Jachiet, M. et al. Anti-IgE monoclonal antibody (omalizumab) in refractory and relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss): data on seventeen patients. Arthritis Rheumatol. 68, 2274–2282 (2016).
Yamada, Y. et al. Efficacy of plasma exchange for antineutrophil cytoplasmic antibody-associated systemic vasculitis: a systematic review and meta-analysis. Arthritis Res. Ther. 23, 28 (2021).
Guillevin, L. et al. Corticosteroids plus pulse cyclophosphamide and plasma exchanges versus corticosteroids plus pulse cyclophosphamide alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome patients with factors predicting poor prognosis. A prospective, randomized trial in sixty-two patients. Arthritis Rheum. 38, 1638–1645 (1995).
Metzler, C., Schnabel, A., Gross, W. L. & Hellmich, B. A phase II study of interferon-alpha for the treatment of refractory Churg-Strauss syndrome. Clin. Exp. Rheumatol. 26, S35–S40 (2008).
Philobos, M. et al. A real-world assessment of mycophenolate mofetil for remission induction in eosinophilic granulomatosis with polyangiitis. Rheumatol. Int. 41, 1811–1814 (2021).
Kahn, J. E. et al. Sustained response to mepolizumab in refractory Churg-Strauss syndrome. J. Allergy Clin. Immunol. 125, 267–270 (2010).
Kim, S., Marigowda, G., Oren, E., Israel, E. & Wechsler, M. E. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J. Allergy Clin. Immunol. 125, 1336–1343 (2010).
Moosig, F., Gross, W. L., Herrmann, K., Bremer, J. P. & Hellmich, B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann. Intern. Med. 155, 341–343 (2011).
Steinfeld, J. et al. Evaluation of clinical benefit from treatment with mepolizumab for patients with eosinophilic granulomatosis with polyangiitis. J. Allergy Clin. Immunol. 143, 2170–2177 (2019).
Manka, L. A. et al. Efficacy and safety of reslizumab in the treatment of eosinophilic granulomatosis with polyangiitis. Ann. Allergy Asthma Immunol. 126, 696–701.e1 (2021).
Koga, Y. et al. Perspectives on the efficacy of benralizumab for treatment of eosinophilic granulomatosis with polyangiitis. Front. Pharmacol. 13, 865318 (2022).
Berti, A. et al. Eosinophilic granulomatosis with polyangiitis: clinical predictors of long-term asthma severity. Chest 157, 1086–1099 (2020).
Latorre, M. et al. Asthma control and airway inflammation in patients with eosinophilic granulomatosis with polyangiitis. J. Allergy Clin. Immunol. Pract. 4, 512–519 (2016).
European Medicines Agency. Nucala. https://www.ema.europa.eu/en/medicines/human/EPAR/nucala (2015).
Pagnoux, C. et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol. 67, 1117–1127 (2015).
Pagnoux, C., Mahendira, D. & Laskin, C. A. Fertility and pregnancy in vasculitis. Best. Pract. Res. Clin. Rheumatol. 27, 79–94 (2013).
EMA. MABTHERA-EMA. https://www.ema.europa.eu/en/medicines/human/EPAR/mabthera (2018).
Teixeira, V., Mohammad, A. J., Jones, R. B., Smith, R. & Jayne, D. Efficacy and safety of rituximab in the treatment of eosinophilic granulomatosis with polyangiitis. RMD Open 5, e000905 (2019).
Dejaco, C. et al. Serum biomarkers in patients with relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss). PLoS ONE 10, e0121737 (2015).
Grayson, P. C. et al. Value of commonly measured laboratory tests as biomarkers of disease activity and predictors of relapse in eosinophilic granulomatosis with polyangiitis. Rheumatology 54, 1351–1359 (2015).
Guilpain, P. et al. Serum eosinophil cationic protein: a marker of disease activity in Churg-Strauss syndrome. Ann. N. Y. Acad. Sci. 1107, 392–399 (2007).
Dallos, T. et al. CCL17/thymus and activation-related chemokine in Churg-Strauss syndrome. Arthritis Rheum. 62, 3496–3503 (2010).
Bettiol, A. et al. Risk of acute arterial and venous thromboembolic events in eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome). Eur. Respir. J. 57, 2004158 (2021).
Allenbach, Y. et al. High frequency of venous thromboembolic events in Churg-Strauss syndrome, Wegener’s granulomatosis and microscopic polyangiitis but not polyarteritis nodosa: a systematic retrospective study on 1130 patients. Ann. Rheum. Dis. 68, 564–567 (2009).
Cereda, A. F., Pedrotti, P., De Capitani, L., Giannattasio, C. & Roghi, A. Comprehensive evaluation of cardiac involvement in eosinophilic granulomatosis with polyangiitis (EGPA) with cardiac magnetic resonance. Eur. J. Intern. Med. 39, 51–56 (2017).
Bourgarit, A. et al. Deaths occurring during the first year after treatment onset for polyarteritis nodosa, microscopic polyangiitis, and Churg-Strauss syndrome: a retrospective analysis of causes and factors predictive of mortality based on 595 patients. Medicine 84, 323–330 (2005).
Hazebroek, M. R. et al. Prevalence and prognostic relevance of cardiac involvement in ANCA-associated vasculitis: eosinophilic granulomatosis with polyangiitis and granulomatosis with polyangiitis. Int. J. Cardiol. 199, 170–179 (2015).
Hasegawa, W. et al. Factors that predict in-hospital mortality in eosinophilic granulomatosis with polyangiitis. Allergy 70, 585–590 (2015).
Garcia-Vives, E., Segarra-Medrano, A., Martinez-Valle, F., Agraz, I. & Solans-Laque, R. Prevalence and risk factors for major infections in patients with antineutrophil cytoplasmic antibody-associated vasculitis: influence on the disease outcome. J. Rheumatol. 47, 407–414 (2020).
Dunogué, B. et al. Impact of cardiac magnetic resonance imaging on eosinophilic granulomatosis with polyangiitis outcomes: a long-term retrospective study on 42 patients. Autoimmun. Rev. 14, 774–780 (2015).
Drosos, G. C. et al. EULAR recommendations for cardiovascular risk management in rheumatic and musculoskeletal diseases, including systemic lupus erythematosus and antiphospholipid syndrome. Ann. Rheum. Dis. 81, 768–779 (2022).
Cattaneo, L. et al. Peripheral neuropathy in Wegener’s granulomatosis, Churg-Strauss syndrome and microscopic polyangiitis. J. Neurol. Neurosurg. Psychiatry. 78, 1119–1123 (2007).
Kronbichler, A. et al. Trimethoprim-sulfamethoxazole prophylaxis prevents severe/life-threatening infections following rituximab in antineutrophil cytoplasm antibody-associated vasculitis. Ann. Rheum. Dis. 77, 1440–1447 (2018).
Choi, S. T., Ahn, S. V., Lee, P. H. & Moon, C. M. The cancer risk according to three subtypes of ANCA-associated vasculitis: a propensity score-matched analysis of a nationwide study: cancer risk in AAV three subtypes. Semin. Arthritis Rheum. 51, 692–699 (2021).
Ahn, S. S. et al. Risk of cancers in antineutrophil cytoplasmic antibody-associated vasculitis: results from the Korea national health insurance claims database 2010–2018. J. Clin. Med. 8, 1871 (2019).
Yoo, J. et al. Cancer development in Korean patients with ANCA-associated vasculitis: a single centre study. Clin. Exp. Rheumatol. 36, S73–S77 (2018).
Calatroni, M., Buzio, C. & Vaglio, A. The evolving paradigm of cancer risk related to cyclophosphamide therapy in granulomatosis with polyangiitis. Rheumatology 54, 1339–1341 (2015).
Faurschou, M. et al. Prolonged risk of specific malignancies following cyclophosphamide therapy among patients with granulomatosis with polyangiitis. Rheumatology 54, 1345–1350 (2015).
Box, C. D., Cronin, O. & Hauser, B. The impact of high dose glucocorticoids on bone health and fracture risk in systemic vasculitides. Front. Endocrinol. 13, 806361 (2022).
Bottero, P. et al. The common allergens in the Churg-Strauss syndrome. Allergy 62, 1288–1294 (2007).
Bello, F. et al. Microarray evaluation of allergen-specific IgE in eosinophilic granulomatosis with polyangiitis. Ann. Rheum. Dis. 80, 1247–1248 (2021).
Acknowledgements
The authors thank M. Tesi for preparing Fig. 1, and F. Bello, A. Biscarini and M. Zampieri for helping to source CT and MR images. The authors also thank the European EGPA study group members who participated in the 4th EESG meeting at the 20th International Vasculitis and ANCA Workshop (Dublin, April 2022) and contributed to the discussion of the current guideline.
Author information
Authors and Affiliations
Contributions
G.E., A.Bet., E.G., G.B., L.G., D.R.W.J. and A.V. researched data for the article. G.E., A.Bet. and A.V. wrote the article. All authors contributed substantially to discussion of the content and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
N.V. has received speaking and consulting fees from GSK. B.S. has received advisory and/or speaker fees from Boehringer Ingelheim and GSK. A.V. has received speaking and consulting fees from GSK, Otsuka and Vifor. G.E. has received speaking and consulting fees from AstraZeneca, Boehring Ingelheim, GSK, Janssen, Novartis, Otsuka, Roche and Sobi, and R.A.S. has received advisory and consulting fees from GSK, Otsuka and Roche. A. Berti has received speaking and consulting fees from GSK. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Rheumatology thanks P. Gatenby and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Emmi, G., Bettiol, A., Gelain, E. et al. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol 19, 378–393 (2023). https://doi.org/10.1038/s41584-023-00958-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41584-023-00958-w
This article is cited by
-
Benralizumab noninferior to mepolizumab for EGPA
Nature Reviews Rheumatology (2024)
-
Cardiovascular Disease in Anti-neutrophil Cytoplasm Antibody-Associated Vasculitis
Current Rheumatology Reports (2024)
-
Asthma bronchiale und allergische Rhinitis – die Hautprobe offenbart eine schwerwiegende Systemerkrankung
Die Dermatologie (2024)
-
ANCA-negative eosinophilic granulomatosis with polyangiitis in a girl misdiagnosed as asthma and pulmonary tuberculosis
Pediatric Rheumatology (2023)
