
Propionic Acidemia
Disclaimer
Metabolic crises in infants and children with organic acid disorders are complex medical emergencies and must be treated as such to avoid death or serious brain injury. These materials are only a guideline and should not be used for definitive treatment without metabolic consultation. It is essential to call or page the on-call genetics/metabolism fellow, or failing this, the on-call metabolic attending at your hospital or nearest pediatric tertiary care center, as rapidly as possible. Please read our Terms of Use.
About
These acute illness materials are a guideline for health care professionals treating the sick infant or child who is known to have propionic acidemia. The materials were developed at Boston Children’s Hospital under the direction of Dr. Harvey Levy, Senior Physician in Medicine/Genetics and Dr. Jonathan Picker, Fragile X Program Director, and was updated by Dr. Patroula Smpokou, Clinical Genetics Fellow.
Introduction
Propionic Acidemia (PA) is an autosomal recessively inherited organic acid disorder characterized by a defect in the mitochondrial biotin-dependent propionyl-coenzyme A carboxylase (propionyl-CoA carboxylase).
Pathophysiology
In normal metabolism, the essential amino acids isoleucine, valine, methionine, and threonine as well as odd-chain fatty acids and cholesterol are degraded to propionyl-CoA at which point propionyl-CoA carboxylase would normally catalyze further conversion to methylmalonyl-CoA. In propionic acidemia, deficient activity of propionyl-CoA carboxylase prevents this conversion (see figure below).
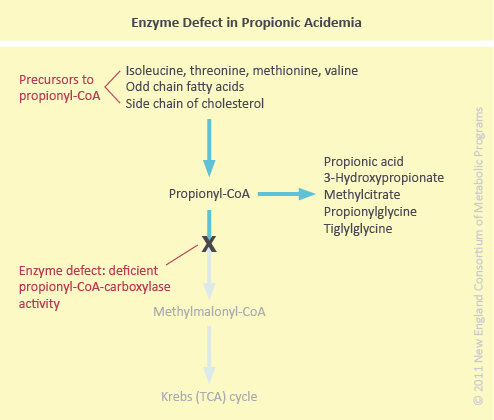
When acute illness occurs in PA, propionic acid accumulates leading to biochemical features including profound metabolic acidosis (due to ketone body production and organic acid accumulation), hypoglycemia, and hyperammonemia (see figure below).

Catabolic stress such as normal perinatal catabolism, or an acute illness (e.g. in the setting of infection, injury, surgery, febrile illness) produces endogenous protein breakdown leading to increase in the involved amino acids (isoleucine, valine, methionine, threonine). When excessive protein is ingested, a similar increase in available amino acids occurs.
Hence, the constellation of laboratory findings in PA is the following:
Metabolic acidosis with anion gap
Ketonuria
Hypoglycemia
Hyperammonemia
Hyperglycinemia
The ketoacidosis, hyperammonemia and hypoglycemia can explain the lethargy and obtundation that is sometimes seen in PA patients in acute crisis. The ketoacidosis also produces vomiting. Mobilization of free fatty acids from stores in the liver in response to the hypoglycemia produces a fatty liver. Prolonged metabolic decompensation can lead to bone marrow suppression with resulting neutropenia.
Acute Presentation
Lethargy, altered mental status
Nausea, vomiting
Hepatomegaly
Laboratory Findings
Hypoglycemia
Metabolic acidosis with anion gap
Hyperammonemia
There are two types of presentation, depending on the severity of the metabolic defect. The neonatal form presents within the first days of life with a life-threatening picture of severe lethargy progressing to obtundation. The infantile or late-onset form has a more insidious presentation with failure to thrive, developmental delay, and perhaps other neurologic features such as seizures and spasticity. These children can decompensate acutely during catabolic stress, usually brought on by infection.
Parents of children with diagnosed metabolic disorders know the signs of decompensation in THEIR children.
It is important to listen to the parents’ insight into their child’s illness.
Immediate Assessment
dextrose stick for blood glucose
vital signs, cardiovascular stability
hydration status
presence of fever; signs of infection
hepatomegaly
neurologic status; evidence of increased intracranial pressure
Laboratory Evaluation
Blood
venous blood gas for blood pH
electrolytes, measured CO2, glucose
blood ammonia (NOTE: draw WITHOUT tourniquet, from free-flowing blood, place on ice and transport STAT to lab to be run immediately)
AST, ALT, AlkPO4, PT, PTT, INR
amylase, lipase
plasma amino acids
plasma total and free carnitine
CBC, differential WBC count, platelet count
blood culture (if indicated)
Urine
urinalysis for specific gravity and ketones
urine for organic acids
urine culture (if indicated)
NOTE: organic acids and ammonia are toxic to the brain and accumulations of these may result in cerebral edema. Caution should be exercised when considering the need for a lumbar puncture.
MaNagement
Specific management guidelines are listed here, with details below:
Stop all protein intake
Provide hydration with high caloric supplementation
Correct biochemical abnormalities
Eliminate toxic metabolites
Treat precipitating factor(s)
Provide cofactor supplementation
Prevent associated sequelae
1. Protein intake
All protein intake should be halted once it is determined that a patient with PA is having metabolic decompensation severe enough to need treatment and acute metabolic management.
2. Hydration/caloric supplementation
In a metabolic crisis, a patient with PA should receive high rate intravenous fluids (either via a peripheral venous catheter or a central venous catheter) for rehydration purposes and also for provision of calories. High dextrose-containing fluids (10% glucose) should be administered with the addition of electrolytes (sodium as half- or full-normal saline solution, and also potassium if urine output is adequate and renal function is sufficient) at a high rate of 1.5 times the maintenance rate for the particular patient. Intravenous fluids should ideally be maintained until the patient is able to tolerate oral fluids or nutrition or until rehydration goals are reached.
Any patient with PA in metabolic crisis should receive high caloric supplementation to reverse the catabolism precipitated by any stressor and achieve anabolism. An intravenous lipid infusion (e.g. intralipid) should be considered to provide the additional calories that may be required. Protein can be added to the nutritional regimen at a slow rate once the patient is able to tolerate protein depending on a combination of different factors, including clinical status, mental status, laboratory derangements and improvement of those.
3. Correct Biochemical abnormalities
a. Metabolic acidosis: this should slowly correct with rehydration and high caloric intake; if bicarbonate level is less than 15 the administration of intravenous or oral bicarbonate in the form of sodium bicarbonate or sodium acetate should be considered.
b. Hypoglycemia: if blood glucose is below 50mg/dL, give 5-10 mL/kg bolus of D10 intravenously (alternatively may give: 1-2 mL/kg of D50 or 2-4 mL/kg of D25). This should be followed by a continuous infusion of D10 with electrolytes (potassium should be added after patient is able to void and is not hyperkalemic) at a rate of 1.5 times the maintenance rate for the particular patient (based on current weight).
c. Hyperammonemia: if ammonia levels are extremely elevated, there are two modes of treatment: medical treatment (ammonul infusion which provides “scavenger” medications to reduce ammonia level) and hemodialysis.
4. Eliminate toxic metabolites
a. Hemodialysis: this is indicated in cases of intractable metabolic acidosis, severe hyperammonemia (blood ammonia level > 600 umol/L) unresponsive to other treatment), coma, and severe electrolyte disturbances. This mode of intervention should be instituted in consultation with a pediatric nephrology service.
b. L-carnitine (oral or intravenous) to conjugate and detoxify propionyl-CoA (forming propionylcarnitine), thus reducing propionic acid and related toxic metabolites. In addition, free carnitine levels are generally low in PA due to increased esterification of organic acid metabolites. Thus, carnitine supplementation may be required for repletion. While carnitine supplementation is controversial, case reports have shown that it is helpful during acute metabolic decompensations. Carnitine should be mixed in 10% glucose solution and run as an infusion to provide 100 mg/kg/day divided every 8 to 12 hours (maximum dose 5 grams/day). When patient is able to tolerate oral intake, the carnitine can be given orally at a dose of 100 mg/kg/day divided over 8 or 12 hours.
c. Antibiotics: Gut bacteria are a significant source of propionic acid; eradicating this source of organic acid production with short, intermittent courses of antibiotics may help to decrease the propionic acid load.
5. Precipitating factors
An acute metabolic decompensation in a patient with propionic acidemia is almost always precipitated by a stressor, such as infection, injury, surgery, hormonal changes, or significant dietary changes (involving increased protein intake). It is of utmost importance to identify and address the precipitating factor for the patient’s metabolic decompensation as treatment of the stressor will facilitate treatment of the metabolic derangements. In the case of infection, antibiotics should be provided to treat the particular infection.
In the case of surgery, prevention of a metabolic decompensation in association with the stress of surgery should be undertaken prior to surgery by providing adequate hydration before and after surgery, avoiding prolonged fasting to the extent possible, addressing pain issues, and providing adequate calories to promote fast healing.
In the case of hormonal changes, the particular patient situation needs to be assessed and possible dietary changes should be made in accordance with the patient’s hormonal status (e.g. puberty, growth spurt, menarche, thyroid disorder).
A change in the patient’s diet, with excessive protein intake, should be assessed; this would be an easy situation to address by adjusting the protein intake.
6. Cofactor supplementation
Biotin, the cofactor for propionyl CoA carboxylase, at 10 mg/day may be useful in cases of vitamin-responsive enzyme defects. In children with an established diagnosis of PA, parents will often know whether or not their child’s condition is biotin-responsive.
Clinical sequelae associated with an acute crisis
Acute pancreatitis: one should be aware of this potential sequela and measure serum amylase and lipase if there are any signs that indicate the possibility of acute pancreatitis. These include vomiting, abdominal pain, and feeding intolerance.
Stroke (particularly involving the basal ganglia): any neurologic deterioration in a patient with PA in metabolic crisis should be addressed emergently as the risk of neurologic stroke is higher in this patient population. A brain CT or MRI should be performed in the presence of any neurologic signs that suggest the possibility of a stroke. Management of increased intracranial pressure is similar to that for any other neurologic disorder causing increased intracranial pressure, including elevation of the head of the bed, hyperventilation (if patient is mechanically ventilated), mannitol, and other diuretics.
Monitoring the Patient
Clinical parameters
Mental status
Hydration status/fluid balance/oral intake
Evidence of bleeding (if thrombocytopenic)
Symptoms of infection (if neutropenic)
Monitor for signs/symptoms of LVH/cardiomyopathy
Biochemical parameters
Electrolytes, measured CO2, glucose, ammonia, blood gases (q 4-6 hours or as indicated)
CBC with differential, platelets
Urine for ketones and specific gravity with every void
Recovery
The patient should be kept NPO until his/her mental status is improved. Anorexia and nausea/vomiting during the acute crisis period makes a significant oral intake unlikely. If the patient is not significantly compromised neurologically, consideration should be given to providing the patient by PO or NG a modified formula preparation containing all but the offending amino acids (see the section above titled “Hydration/caloric supplementation”).
When the infant/child is able to take fluids orally or per NG/gastrostomy tube, a metabolic fellow/staff or the metabolic nutritionist should be contacted since each patient has a unique, modified diet. Each day, the nurses caring for the patient should review the menu with the parents or nutritionist to avoid dietary mistakes; these do happen and can be disastrous in the peri-crisis period.