导读
半月龄男婴第一次「呕吐咖啡渣样物」,胃镜检查提示胃多发溃疡。改用氨基酸配方奶后仍反复呕吐咖啡渣样物,复查胃镜溃疡改善不明显,腹部 B 超见该患儿胃壁逐渐增厚,幽门逐渐狭窄……
这个小婴儿为何反复呕吐咖啡渣样物?小小年纪为何多发胃溃疡?胃壁逐渐增厚,难道是肿瘤?亦或是自身免疫性胃病?
反复呕吐咖啡渣样物,排黑便
患儿男,2月龄,因「反复呕吐咖啡渣样物1月余」入院。
患儿出生后第 14 天开始无明显诱因呕吐咖啡色样物, 多发生于喂奶后半小时内,并排黑色大便,曾停母乳改氨基酸配方奶喂养后仍症状反复。
外院全麻胃镜检查提示胃多发性溃疡(活动期)、幽门狭窄,电子结肠镜检查提示结肠炎, 腹部 B 超检查怀疑肥厚性幽门狭窄。我院门诊 B 超检查提示幽门区未见明显肿块回声。
入院体检:身长 53 cm,体重 3.7 kg,营养不良貌,全身浅表淋巴结未及肿大,心肺无异常,腹软,未及包块,肝脾未及肿大。
实验室检查:WBC 10.3 × 109/L,血红蛋白 81 g/L,嗜酸细胞 5%,生化凝血功能正常。
上消化道造影提示幽门不全性梗阻。
入院后予氨基酸奶粉喂养、静脉用奥美拉唑 4 mg 每 12 小时 1 次抑酸、口服胃肠黏膜保护剂,仍反复呕吐咖啡渣样物。
住院 21 天,复查胃镜、B 超,
正规治疗,病情却持续恶化
入院第 6 天行第 1 次胃肠镜检查:胃窦、小弯、胃底见大片溃疡灶,幽门口狭窄变形(图 1A、1B)。
黏膜病理提示胃窦、胃体、胃角黏膜上皮完整,腺体无萎缩,固有层少许淋巴细胞浸润,散在个别嗜酸性粒细胞浸润(胃窦、胃体、胃角分别达 1、2、10 个/HFP),未见幽门螺杆菌(HP),未见肠化(图 2);结肠镜下所见大致正常。
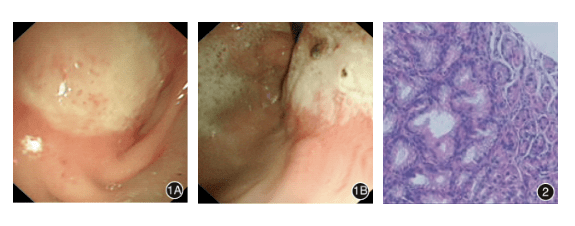
入院后第 1 次内镜检查所见 1A:胃窦小弯侧见大片状溃疡,覆厚白苔,幽门口狭窄变形,直径约 0.5 cm;1B:胃角大片溃疡延伸至胃体、胃底。图 2:黏膜上皮完整,腺体无萎缩,固有层少许淋巴细胞浸润,散在个别嗜酸性粒细胞浸润(1~10 个/HPF),未见幽门螺旋杆菌,未见肠化 HE × 200
入院第 21 天行第 2 次胃镜检查,镜下见溃疡较上一次检查时改善不明显(图 3)。
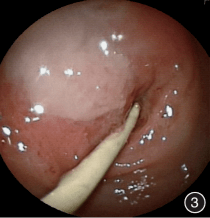
图 3 入院后第 2 次内镜检查:
胃窦溃疡较前一次检查时无明显改善,幽门狭窄变形
行胃镜下空肠置管未成功,改行腹腔镜下空肠置管,氨基酸配方奶持续空肠喂养,由 5 ml/H 起渐增加至 50 ml/H ;持续胃肠减压,胃管引流量 50~150 ml/d,胃液 PH 值波动于 4~6,当 PH = 4 时反复出现咖啡渣样物。
复查 B 超提示:胃窦部及幽门部胃壁不规则增厚,较厚处约 13.0 mm。
正规治疗后效果差,考虑什么?
考虑患儿正规治疗后效果差,不排除胃泌素瘤、自身免疫性胃病、特殊病原体感染等少见病。
胃泌素检查结果正常,肿瘤筛查结果仅糖类抗原⁃199 稍高(39.06 U/ml,正常值< 37 U/ml)免疫相关血清检查均阴性。免疫 六项中 IgG、IgA、IgM、IgE、C4 正常,C3 稍低(0.43 g/L,正常值 0.74~1.38 g/L),CMV-DNA 定量、EBV-IgM 阴性,大便 HP 抗原阴性,胃黏膜病理未证实 HP 感染。
入院第 72 天行第 3 次胃镜检查,仍有幽门狭窄和巨大溃疡。B 超检查发现胃窦部全层胃壁弥漫性增厚,较厚处约 12.0 mm(图 4)。胃肠 MRI 检查发现胃小弯肿物(图 5)。
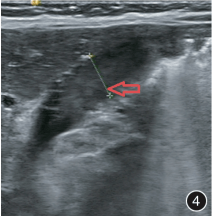
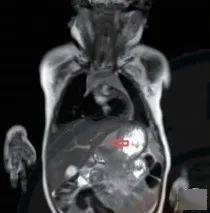
图 4:B 超检查见胃窦部全层胃壁弥漫性增厚,较厚处约 12.0 mm(胃大弯侧),浆膜层回声增强增厚;幽门肌增厚,较厚处约 3.5 mm;图 5:腹部 MRI 检查见胃小弯侧胃壁全层增厚,向下延伸至幽门处,最厚处约 1.8 cm;胃小弯浆膜层内可见一异常信号影,边界清,大小约 2.3 cm × 0.9 cm × 3.4 cm,T2WI 呈高信号,T1WI 呈等信号
住院 3 个月,最终确诊 IMT
全身 PET-CT 检查提示胃小弯侧(近幽门)团块状高代谢病灶,不除外淋巴瘤。
外科剖腹探查见胃小弯侧胃壁一肿物,厚约 1.0 cm,长约 6.0 cm,宽约 2.0 cm,上延至贲门,下延至十二指肠降部中段。质硬,较固定,与肝门部、胰腺紧密粘连,无法行根治术,行肿瘤活检和胃空肠吻合术(图 6)。
术中冰冻切片提示炎性肌纤维母细胞瘤(IMT),肿物病理可见梭形肿瘤细胞增生,细胞轻度异型,胶原丰富,间质大量炎性细胞浸润(图 7)。
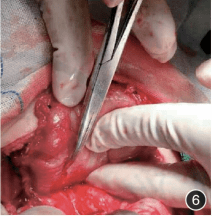
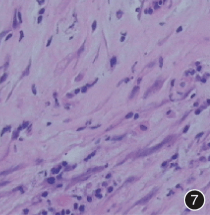
图 6:术中发现肿物质硬,较固定,与肝门部、胰腺紧密粘连;图 7:肿物光镜病理,可见梭形肿瘤细胞增生,细胞轻度异型,胶原丰富,间质大量炎性细胞浸润,可见浆细胞 HE x 400
术后拔除空肠营养管,恢复经口进食,未再出现呕吐咖啡渣样物。术后 1 个月体重持续增加(约 40 g/d),达到追赶生长,复查 B 超提示肿瘤体积较前稍缩小,未见肿瘤转移。
IMT 是什么?
IMT 是一种少见而独特的间叶性肿瘤,常有低度恶性的表现或交界性肿瘤的特点[1]。
WHO 软组织肿瘤分类(2002)将 IMT 定义为 「 由分化的肌纤维母细胞性梭形细胞组成的, 常伴大量浆细胞和(或)淋巴细胞的一 种肿瘤 」[2]。
WHO(2010)消化系统肿瘤病理学和遗传学分类首次将 IMT 列入胃肿瘤组织学分类中[3]。
IMT 最常见于肺,也可见于腹腔、肠系膜、后腹膜、盆腔、肝、胰、泌尿道、四肢及躯干、上呼吸道、中枢神经系统、皮肤及软组织等。发生在腹腔的 IMT 称为 AIMT,常与周围脏器或组织粘连,具有与腹腔内其他肿瘤的影像学、临床表现甚至病理学形态相似的特征,故常常造成诊断上的困难。
免疫组化标记 Vim、MSA 和 SMA 特异性高[4],是 AIMT 与腹腔脏器其他类型肿瘤鉴别的重要依据。
IMT 确切病因仍不清楚,相关因素有手术、创伤、炎症、 异常修复、EB 病毒或特殊细菌感染,也有学者认为与基因有关。
有研究证实,IMT 在遗传学上存在异质性,50%~70% 的儿童或年轻患者常有克隆性细胞遗传学重排。受累的染色体是 2p23,该片段上的 ALK 基因与多种伙伴基因(TPM3、TPM4、CLTC、RANBP2、ATIC 等)[5]。
这种单克隆核型基因组的重排和肿瘤特异性融合基因的产生,为 IMT 的单克隆性及肿瘤性起源提供了有力证据。文献报道 50%~60% 的患者 ALK 免疫组化阳性,其中 30%~67% 的 ALK 阳性患者存在 ALK 基因突变[6]。本病例患儿免疫组化 ALK 阴性,所以未进行基因学测定。
Coffin 等[7]根据病理学表现把 IMT 分成 3 型,黏液样型或血管型、梭形细胞紧密型、细胞减少纤维增生型。目前 IMT 的治疗以手术为主,可达到治愈目的,术后无需放化疗等进一步治疗。
大多数 IMT 在不完全性切除下可稳定或消退,但仍存在复发、转移的可能,复发率达 10%~25%[8]。Montgomery 等[9]报道转移率达 5%,肿瘤囊实性改变、细胞核轻微非典型改变、核仁突出、有丝分裂象增多、DNA 非整倍体者易发生转移,肿瘤大或未切净者易复发。Coffin 等[10]认为,远处转移与特定的 ALK 融合伙伴基因(如 RANBP2)以及圆形细胞的组织类型有关,ALK 阳性患者一般预后较好,ALK 阴性患者发生转移的危险性更高。
复盘与随访
本例肿瘤呈慢性进展过程,患儿 2 个月大时 B 超未发现胃肠肿物,多次复查 B 超示胃窦部及幽门部胃壁不规则增厚,初期考虑为巨大溃疡导致黏膜水肿增厚和幽门不全梗阻,忽视了少见的胃部肿瘤侵犯浆膜面导致的消化道溃疡。
住院第 3 个月才通过胃肠 MRI 和 PET-CT 发现胃部肿物,经过 HE 染色及免疫组化检测 Vim 和 SMA 阳性确诊 IMT。
本患儿 ALK 阴性, 随访时间短,需要更长时间密切随访。由于术前明确诊断较为困难,有必要术前活检或术中冰冻切片明确诊断,以免误行根治性手术。
本例患儿术中发现肿物与肝门部、胰腺紧密粘连,无法行根治术,遂行胃空肠吻合术解决不全幽门梗阻症状,保证经口摄入足够营养,术后可顺利经口喂养。
患儿体重增加满意,至 6 个月龄时体重达到 6.9 kg,并且术后 1 个月复查腹部 B 超提示胃部肿瘤较前缩小,未见转移灶,目前仍在随访之中。
本文作者:广州市妇女儿童医疗中心消化科 许朝晖 主任医师
参考文献:(上下滑动可查看完整内容)
[1] 朱玉春, 周伟, 王建良, 等. 后腹膜炎性肌纤维母细胞瘤一例 [J]. 中华放射学杂志, 2008, 042(008):848.
[2] Fletcher CD,Unni KK,Mertens F.World Health Organization classification of tumors.Pathology and genetics of tumors of soft tissue and bone[M].IARC Press,2002:48
[3] Bosman FT, Carneiro F, Hruban RH, et al.World Health Organization classification of tumors Pathology and genetics of tumors of digestive system[M].Lyon:IARC Press,2010:74.
[4] 黄晓赤, 罗克枢, 赵剑萍, 等. 腹腔脏器炎性肌纤维母细胞瘤的临床病理观察 [J]. 临床与实验病理学杂志, 2006(01):45-48.
[5] Mariño-Enríquez A, Wang W L, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK[J]. The American journal of surgical pathology, 2011, 35(1): 135-144.
[6] Teoh J Y C, Chan N H, Cheung H Y, et al. Inflammatory myofibroblastic tumors of the urinary bladder: a systematic review[J]. Urology, 2014, 84(3): 503-508.
[7] Coffin C M, Watterson J, Priest J R, et al. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases[J]. The American journal of surgical pathology, 1995, 19(8): 859-872.
[8] Pradhan M R, Ranjan P, Rao R N, et al. Inflammatory myofibroblastic tumor of the urinary bladder managed by laparoscopic partial cystectomy[J]. Korean journal of urology, 2013, 54(11): 797-800.
[9] Montgomery E A, Shuster D D, Burkart A L, et al. Inflammatory myofibroblastic tumors of the urinary tract: a clinicopathologic study of 46 cases, including a malignant example inflammatory fibrosarcoma and a subset associated with high-grade urothelial carcinoma[J]. The American journal of surgical pathology, 2006, 30(12): 1502-1512.
[10] Coffin C M, Hornick J L, Fletcher C D M. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases[J]. The American journal of surgical pathology, 2007, 31(4): 509-520.
策划:春花
投稿邮箱:wangliming1@dxy.cn
本文首发于丁香园旗下专业平台:儿科时间返回搜狐,查看更多
责任编辑: