- +1
【重磅综述】从机制到治疗——深度解析细胞衰老与个体衰老
以下文章来源于老顽童说 ,作者老顽童说

老顽童说
公众号致力于传播衰老相关的前沿科研进展和趣味科普,帮助大家更深入地了解衰老背后的科学故事~

关注我们,获取更多相关资讯
翻译 By 曹天玲 张彬 杨远涵 胡健立
引言
俗话说千里之堤,溃于蚁穴。如果把个体衰老比作千里之堤,那细胞衰老就是那防不胜防的蚁穴。清除蚁穴可以暂且保全石堤,那清除衰老的细胞是否可以延缓个体衰老呢?今天小编就给大家分享一篇来自San Raffaele Telethon基因治疗研究所Raffaella Di Micco实验室和FIRC分子肿瘤学研究所的Fabrizio d’Adda di Fagagna实验室于2020年12月16日合作发表在Nature Reviews Molecular Cell Biology上发表的一篇标题为“Cellular senescence in ageing: from mechanisms to therapeutic opportunities”的综述,该文章总结了衰老细胞的特性以及细胞衰老的机制并探讨了通过清除衰老细胞进而延缓衰老的方法,小编这里将为您详细揭秘细胞衰老与个体衰老之间的“爱恨情仇”。
通讯作者:Raffaella Di Micco(左) Fabrizio d’Adda di Fagagna(右)
(图片来源于网络)
摘要
细胞衰老一词最早于1961年提出,随着研究的日益深入,目前已经成为很多生物技术公司用于改善人类各种疾病的重要靶标。细胞衰老以永久性的增殖停滞为特征,在响应包括端粒功能障碍、癌基因激活和持续性DNA损伤等内源或外源刺激时发生。此外,细胞衰老还可以在多种生物发生过程中发生,例如早期胚胎发育等。衰老细胞的外源活性,主要和衰老相关分泌表型(senescence-associated secretory phenotype,SASP)相关,它不仅扩大了衰老细胞增长停滞的影响,还会阻碍组织再生并导致衰老相关的慢性疾病和个体衰老的发生。本篇综述主要讨论了细胞衰老相关分泌表型的机制和调节因子,并概述了以衰老细胞为靶标,通过senolytic和senomorphic疗法进行衰老和衰老相关疾病干预的手段的潜在价值。
前言
细胞衰老在响应多种诱发因素时产生,这些诱发因素主要包括DNA损伤、端粒功能障碍、癌基因激活和细胞器应激等。此外,细胞衰老还与肿瘤抑制、组织修复、胚胎发生和个体衰老等过程相关。1961年,Hayflick和Moorhead证明:正常培养的人类成纤维细胞在进入不可逆的生长停滞(称为复制衰老)之前会表现出有限的细胞分裂能力。于是,科学家们提出了这样的假设:细胞增殖能力的逐渐丧失最终会导致组织的衰老,这可能是因为细胞增殖对于受损细胞的替换至关重要。但是,开发能够证明该假设的工具就已花费科学家们数十年之久。
确定衰老细胞的特异标志是在活组织中检测衰老细胞的第一个难题。衰老相关的β-半乳糖苷酶(senescence-associated-β-galactosidase, SA-β-gal)活性是鉴定衰老细胞的最早生物标记之一,该标记有效证明了衰老细胞会在多种哺乳动物的衰老相关疾病病灶和衰老组织中逐渐积累。衰老细胞的另一个显著特征是细胞周期抑制蛋白(统称为细胞周期依赖性激酶抑制剂)的表达增加,这会导致衰老细胞的停滞状态的维持,进而导致衰老细胞的不断积累,其中p16INK4A(以下称为p16)是最主要的细胞周期抑制蛋白。有实验表明p16缺乏的小鼠容易自发形成肿瘤。20世纪90年代末,发现过度的致癌信号或肿瘤抑制作用的丧失都会过早地诱发细胞衰老。后来科学家们又发现异常的DNA复制和DNA损伤积累会也会诱发细胞衰老。但是,这些特点都不是衰老细胞普遍存在的,因此,同时测试多个生物标志物以定义衰老状态非常重要。
为了从进化的角度调和衰老细胞拥有的看似对立的促衰老和抗癌作用,衰老细胞被认为符合衰老的拮抗多效性理论,该理论假设自然选择有利于促进早期生殖健康的基因,这可能伴随着未经选择的后果,并在生命后期产生负面影响,尽管这还没有得到证实。或者,可以假设衰老与进化成本和收益理论有关。该理论暗示衰老细胞在整个生命中都具有有益作用(例如,限制组织损伤和抑制肿瘤发生),但是老年时这些作用的代价超过了益处。最近有几种方法证实了衰老细胞在许多疾病中都起到了致病作用。这些方法包括INK-ATTAC11和p16-3MR12转基因小鼠模型的建立(可选择性消除表达p16的细胞),以及senolytic和senomorphic药物制剂。senolytics主要是通过消除衰老的细胞来发挥功效,而senomorphics的功能实现则是通过调节衰老细胞的特性而非消除它们。
在这篇综述中,作者首先描述了衰老细胞的特性以及导致细胞衰老的机制。随后,作者讨论了衰老细胞在各种生物学过程中的作用,以及如何通过将其去除或减弱其特性来进行疾病干预和健康寿命的延长。
写在前面|名词解释
Sirtuins蛋白(Sirtuins)
烟酰胺二核苷酸依赖(NAD+)的脱酰酶,调节DNA修复、炎症、代谢和衰老等多种细胞过程。
线粒体功能障碍相关衰老(Mitochondrial dysfunction-associated senescence,MiDAS)
线粒体损伤引发的衰老具有明显的分泌表型,即缺乏IL-1 (Interleukin-1,IL-1)依赖型炎症反应。
外泌体(Exosomes)
胞内小室产生的细胞外囊泡参与细胞间通讯。
HMGB蛋白(HMGB proteins)
结合DNA并影响染色质压缩的非组蛋白分子。
骨髓偏斜(Myeloid skewing)
骨髓和血液中,与衰老相关的骨髓细胞比例增加,但其他谱系的细胞有所减少。
tau
神经元中发现的一种蛋白质,对于维持轴突的微管结构非常重要。在多种神经退行性疾病(包括阿尔茨海默症)中发现了突变体和高磷酸化形式的tau。
纤维化(Fibrosis)
细胞外基质在疾病组织中的积累限制了正常的组织功能并导致长期的组织损伤。
低密度脂蛋白受体(LDL receptor)
介导低密度脂蛋白进入细胞的受体。编码该受体的基因突变容易导致动脉粥样硬化的发展。
白内障(Cataracts)
眼睛中的晶状体混浊导致视力下降。在老年人中,通过手术更换患病的晶状体的是白内障常见的治疗手段。
脊柱后凸(Lordokyphosis)
在实验室小鼠和人类中均观察到脊柱向后弯曲的异常状态。
脂肪代谢障碍(Lipodystrophy)
脂肪组织在体内的异常分布,包括过多沉积或不足。
INK-ATTAC
在衰老细胞中少量活跃的p16启动子的控制下,带有药物诱导的胱天蛋白酶8的转基因小鼠模型可以选择性消除表达p16的衰老细胞
p16-3MR
在p16启动子的控制下表达红色荧光蛋白、萤光素酶和单纯疱疹病毒胸苷激酶的三峰报告基因的转基因小鼠模型,可以追踪和消除表达p16的衰老细胞。
细胞衰老的诱因和特征
细胞衰老是一种稳定的、生长停滞的终末状态,在这种状态下即使细胞在最佳生长条件和有丝分裂刺激下仍无法增殖 (框 1,2;图 1)。在衰老细胞中细胞存活通路往往被上调(包括抗凋亡蛋白BCL-2家族),这使得衰老细胞即使在外源应激下也具有较强的抗凋亡能力。而衰老细胞这种生存能力的增强是细胞在抗凋亡过程中适者生存的结果还是伴随衰老过程而产生的结果,还有待确定。细胞最终发生凋亡还是衰老的分子机制目前尚不清楚,但这可能取决于初始刺激的强度和持续时间,以及损伤的性质和细胞类型。因为衰老和凋亡的关键调节因子类似,例如p53通路的激活,因此衰老细胞的抗凋亡能力可能依赖于p53的水平和活性。因为衰老和凋亡的程序在关键成分上趋同,包括p53通路的激活,衰老细胞对凋亡的抵抗可能依赖于p53的水平和活性。虽然衰老被认为是一个永久的细胞周期阻滞的状态,但最近的证据表明,至少在肿瘤形成和抗癌治疗方面,细胞衰老的形成可能涉及表观遗传机制,表观遗传能以细胞自主的方式重编程癌细胞,使其获得一定程度的干细胞特性。值得注意的是,衰老的形成是一个动态的过程,从退出细胞周期到衰老后期的不同阶段中,参与其中的分子途径有所重叠但不相同。
框1 | 衰老的生物标志物
衰老领域的一个主要限制是缺乏单一的、通用的或模型特异性的生物标志物来识别培养中的衰老细胞或组织样本。目前,对衰老细胞的鉴定依赖于多种标记的组合,当这些标记同时存在时,可以区分稳定停滞的衰老细胞和对应的静止或分化的细胞。
在培养细胞和新鲜组织样本中检测衰老细胞,第一个也是最广泛使用的生物指标是一种名为“衰老相关-β-半乳糖苷酶”(senescence-associated-β-galactosidase,SA-β-gal)的溶酶体酶的积累。该标记物在大多数衰老细胞中可通过组织化学染色检测到,而在未衰老的,静息的或永生的和转化的细胞中一般不存在,但是SA-β-gal在组织培养中也可在血清饥饿或过度融合的细胞中积累,并可能标记体内巨噬细胞的一个特定亚群作为免疫刺激可逆反应的一部分。脂褐素积累是衰老细胞的另一个特征。最近开发的一种基于生物素连接的苏丹黑B类似物的方法正在成为一种可靠的,可在各种细胞和组织类型中追踪衰老细胞的检测系统。
衰老细胞的另一个特点是形态异常增大、扁平,胞质核比不成比例地增加。虽然这种体积庞大的细胞质最初被描述为伴随细胞衰老形成的特征,但最近的一项研究表明,细胞大小的增加可能在衰老相关的生长停滞中起着重要作用。此外,在单细胞水平上,SA-β-gal阳性的衰老细胞与SA-β-gal阴性细胞相比,细胞体积增加。衰老细胞的另一个明显标志是缺乏DNA复制,这通常可以通过加入核苷类似物(例如,5-溴脱氧尿苷或[3H]胸腺嘧啶)或增殖标志物免疫染色鉴定(如PCNA和Ki-67)。这些标记物无法将衰老细胞与静止细胞或者有丝分裂后分化的细胞区分开来。
p21和p16是两种周期蛋白依赖性的激酶抑制剂,是p53和RB控制的肿瘤抑制通路的组成部分,通常在衰老细胞中积累。因为p21和p16的表达水平足以建立和维持与衰老相关的生长阻滞,它们被用于识别组织和培养细胞中的衰老细胞。特别是p16,它被用作代替衰老标记物使用,已用于衰老细胞选择性根除的工程小鼠模型的构建。然而,并不是所有的衰老细胞类型都表达p16,因为它也可以被某些肿瘤细胞表达,特别是那些失去RB功能的细胞。
细胞核衰老相关异染色质聚集(senescence-associated heterochromatin foci,SAHF)也被用于识别衰老细胞,但它们似乎对由激活的致癌基因和DNA复制应激源诱导的衰老程序具有特异性。持续性DNA损伤反应因子在损伤部位积累是细胞学上可检测的核病灶,也被用作衰老细胞的标记,当在端粒序列积累时,端粒相关病灶是衰老状态的一个强有力的标记。
最后,衰老相关分泌表型的成分,主要是促炎细胞因子白细胞介素-6 (interleukin-6,IL-6)和IL-8,可以在转录和蛋白水平上用于评估一般的组织或细胞培养衰老。然而,单独的SASP不能作为一个可靠的衰老生物标志物。事实上,由p16过表达引发的衰老并不会引起SASP转录程序的改变。总之,衰老表型具有高度异质性,可能因初始触发因子和所研究的细胞类型而不同,这一证据不断挑战着对普遍衰老生物标志物的寻找。因此,转录组学和蛋白质组学在相关细胞和组织类型的单细胞水平上的研究对于找到独特或共同的衰老状态标记至关重要,包括可以从衰老和病变组织中分离衰老细胞的细胞表面分子。最近,科学界对创新的成像工具和荧光示踪剂的开发非常感兴趣,这可能代表了基于衰老的转化医学应用的一个转折点。这些工具和荧光示踪剂可以实时监测衰老负荷,并监测senotherapies在临床样本中的治疗效果。
框2 | 衰老和自噬
功能障碍的细胞器,如线粒体和溶酶体,通常通过激活细胞内名为“自噬”的降解系统而被降解。然而,自噬是一个促进衰老的诱发因素,还是一种在衰老过程中失去的生存机制,仍然是一个需要严密研究的科学研究问题。事实上,据报道,一种通过哺乳动物的雷帕霉素靶点(mammalian target of rapamycin,mTOR)激活的选择性自噬途径,有助于维持许多衰老相关分泌表型因子蛋白质的合成,这些因子主要存在于癌基因诱导的衰老细胞中,其中几种自噬调节因子的下调可延缓癌基因诱导衰老的发生。最近,一种类泛素的自噬蛋白LC3B,被发现与核纤层蛋白Lamin B1有关,并有助于其在癌基因诱导的衰老细胞的溶酶体中降解。重要的是,核纤层相关的染色质结构域也通过同样的机制从细胞核转运到溶酶体中,并促进了衰老细胞中胞浆染色质片段的积累。抑制自噬可防止Lamin B1降解,并确保衰老细胞的核膜完整性。
在治疗癌细胞诱导的衰老背景下,也有研究表明,自噬是由衰老触发的,以应对有毒大分子积累导致的负荷增加,自噬的药理靶向性导致衰老细胞被消除。然而,自噬也被认为通过促进受损细胞器和其他细胞成分的降解来抑制衰老,一些研究支持了这一观点。在成人肌肉干细胞中,基础的自噬通过抑制衰老来维持干性。衰老过程中,肌肉干细胞(卫星细胞)的自噬活性随着干细胞再生能力的降低而下降,从而导致老年小鼠衰老卫星细胞的积累。恢复衰老的卫星细胞的自噬可以防止衰老并恢复其再生能力。同样,自噬对氧化应激引起的衰老也有保护作用。在过度氧化应激下,通过mTOR的抑制作用增强自噬活性可以延缓细胞衰老,并在功能上恢复线粒体和溶酶体功能。进一步证明自噬在预防衰老中的作用,最近一项旨在鉴定缓解复制性衰老化合物的高通量筛选显示,共济失调血管扩张症突变(ataxia telangiectasia mutated,ATM)抑制剂KU-60019通过阻断空泡蛋白ATP6V1G1的磷酸化,功能性恢复溶酶体活性,从而增强自噬能力。ATM抑制剂也能恢复线粒体功能,减轻衰老表型。这些看似相反的作用共同反映了自噬和细胞衰老间复杂的相互调节,自噬与衰老可以被几个衰老的触发条件、不同的细胞类型和在衰老调控网络中作用自噬程序的时空特异性的激活联系在一起。
细胞衰老和DNA损伤应答
多种应激因素可诱导细胞的衰老,其中核DNA损伤是最常见的一种,它主要表现为可激活DNA损伤应答(DNA damage response,DDR)的DNA双链断裂(DNA double-strand breaks,DSBs)的积累。(图 1)。DDR发挥检查点功能来阻断细胞周期,并防止错误的遗传信息遗传给子细胞。一些DDR因子在DNA损伤部位积累,形成由扩展染色质修饰事件(如组蛋白H2AX磷酸化)和与其相关的蛋白质(包括MDC1、53BP1和毛细血管扩张性共济失调突变(ataxia telangiectasia mutated,ATM)激酶的激活形式)组成的细胞学可检测的核位点。这些位点标记DNA损伤的部位,有助于损伤被修复前,检查点功能的执行和细胞周期的阻滞。如果DNA损伤持续存在,它会导致DDR信号的延长和以细胞衰老为形式的持久性增殖阻滞。最近的研究证明,在培养的衰老细胞中观察到的持久性DDR位点含有未修复的DSBs,这支持了细胞衰老类似于延长检查点激活时间的观点。抑制DDR信号激酶(ATM、ATR、CHK1和CHK2)使衰老细胞重新进入细胞周期。在DDR级联效应的后期,肿瘤抑制物p53被激活(p53是ATM及其旁系同源物ATR的靶点)并刺激细胞周期素依赖性激酶抑制剂p21的表达(p21是衰老相关细胞周期阻滞的重要介质)。p16是CDK4和CDK6的抑制剂,也是几种衰老类型的关键酶;p21在衰老早期被激活,p16在衰老后期被激活,可能是为了维持衰老表型。除了DDR级联效应被激活外,肿瘤抑制因子ARF还稳定p53,从而诱发衰老。目前很多工作主要集中在研究DDR和ARF这两条主要途径在p53依赖型衰老的发生(特别是为了应对致癌挑战)过程中的作用。最初的观点主要是基于小鼠的研究,即DDR和ARF发挥拮抗作用,因为ARF在肿瘤发生过程中以一种与DDR无关的方式被转录激活。最近,一项研究报道了人类癌症模型中紧密的调控网络,在这种网络中,ATM抑制ARF水平,当ATM被灭活时,ARF作为癌症进展的次要障碍。与这种时间规律相一致,DDR先于ARF参与,ARF激活在癌症进展的后期才被检测到并且比DDR更少。
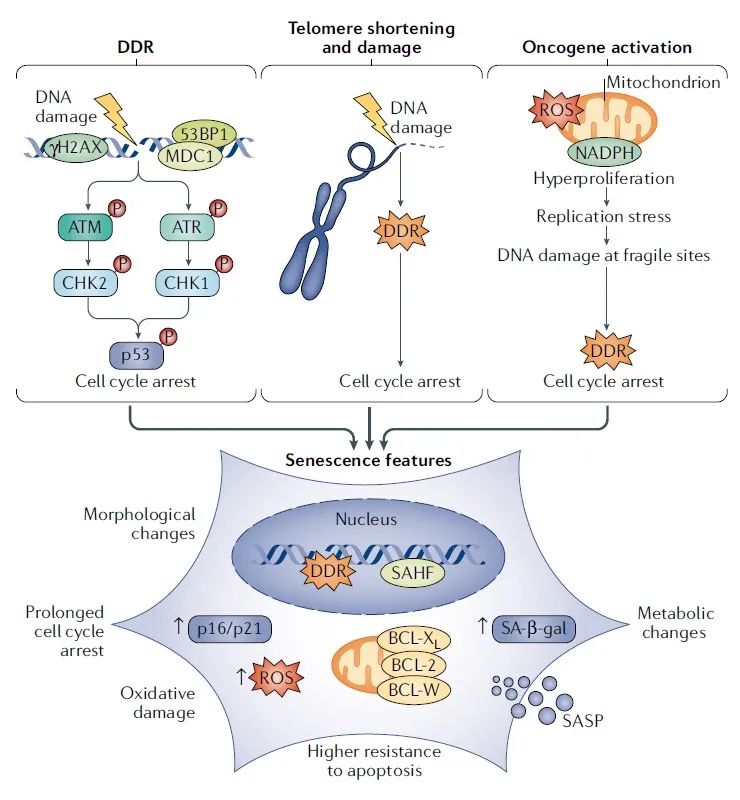
图 1 | 衰老的驱动因素和表型。 核DNA损伤与衰老的发生往往是因果关系。DNA损伤激活一个信号级联,定义为DNA损伤应答(DNA damage response,DDR),其特征是磷酸化组蛋白H2AX(γH2AX)、53BP1和MDC1、顶部的激酶毛细血管扩张性共济失调突变(ataxia telangiectasia mutated,ATM)和ATR以及下游激酶CHK2和CHK1。信号最终汇聚于p53激活,这反过来又导致细胞周期阻滞。延长的DDR激活会触发衰老。在染色体末端的一个或几个DDR信号端粒,足以触发复制性细胞衰老。癌基因激活也是一个强大的衰老触发因子。具体来说,大多数活化的癌基因,部分是通过活性氧(ROS)的产生,导致过度增殖和改变DNA复制模式,最终导致复制应激和DNA损伤积累在脆性位点,包括端粒。除了延长DDR活化外,衰老特征还包括细胞周期阻滞(通过上调p21和p16细胞周期抑制剂)、氧化损伤(通过增加ROS水平检测到)、上调抗凋亡蛋白BCL-2家族,从而促进细胞凋亡的抗性、代谢变化(包括衰老相关β-半乳糖苷酶(SA-β-gal)积累)、衰老相关异染色质聚集(senescence-associated heterochromatin foci,SAHF)和衰老相关分泌表型(senescence-associated secretory phenotype,SASP)。
端粒缩短和损伤
细胞衰老发生的第一个也是最显著的特征机制之一是端粒缩短。由于标准的DNA复制程序无法完全复制染色体DNA末端,在没有端粒维持机制的情况下,如端粒酶的表达或端粒之间的重组,端粒随着每一轮DNA复制而缩短。在一定长度以下,端粒封闭因子或保护结构的丢失使端粒非常短,类似于单端DSBs,从而触发与DNA DSBs触发的DDR非常相似的DDR(图 1)。一个或几个DDR信号端粒足以触发复制性细胞衰老,端粒酶的强制表达可防止细胞衰老,促进细胞无限增殖。
持续的DDR激活也发生在端粒未严重缩短的情况下,因为当DSBs位于端粒时,暴露于外源性基因毒性处理的不分裂细胞和不分裂的衰老细胞中的修复效率要低得多。由于端粒DSBs的持续存在,细胞衰老会发生并被维持。因此,端粒上DDR的持续激活是细胞衰老的一个触发条件,它既可以发生在增殖细胞的端粒缩短时,也可以发生在不增殖细胞(静止或终末分化)的端粒DNA损伤时,而不受端粒长度的影响。
癌基因诱导的衰老
癌基因活化是细胞衰老的一个强力诱因。癌基因的表达触发了一个初始的高增殖期,这一时期与DNA复制的改变密切相关,这种改变最终会参与DDR途径并导致衰老。这一过程被称为癌基因诱导衰老(oncogene-induced senescence,OIS)。肿瘤抑制因子表达的丧失也会导致增殖停滞,如PTEN丢失引起的细胞衰老(PTEN loss-induced cellular senescence,PICS)。虽然最初PTEN丢失诱导的细胞衰老被认为与DDR的激活无关,但后来又发现在体内PTEN的丢失与过度增殖、DDR的参与和细胞衰老均有联系。值得注意的是,不同于致癌的RAS或BRAF,研究表明,PI3K - AKT通路的激活会促进p53依赖性衰老,这一过程通常发生在没有明显的过度增殖和强烈的DNA损伤积累的情况下,这暗示了不同的潜在机制。
端粒对DNA复制应激异常敏感,包括癌基因导致的端粒功能障碍和癌基因积累引起的端粒功能障碍,而且已经在人类增生性癌症病变中观察到DDR。活性氧(Reactive oxygen species,ROS)在肿瘤中积累,除了公认的作为DNA破坏因子外,它们还可以作为信号分子,介导并促进癌基因的表达。 最近由于意外发现癌基因诱导的活性氧主要通过NADPH氧化酶产生,并可以改变DNA复制过程,造成DNA损伤积累来促进初始增生期从而诱导细胞衰老,ROS在促进细胞增殖和促进与衰老相关的DNA损伤方面自相矛盾的作用被部分解决了。
线粒体功能障碍和细胞衰老
衰老细胞氧化应激的增加与功能失调的线粒体的积累有关。事实上,衰老细胞的特征是线粒体质量、膜电位和线粒体形态的改变。线粒体功能失调可能在衰老过程中发挥重要作用,因为线粒体去乙酰化酶(一组调节不同物种衰老的进化保守的蛋白)的缺失以及线粒体功能的选择性化学抑制一般会触发衰老。有证据支持核DNA损伤与线粒体功能障碍之间的相互影响。值得注意的是,线粒体功能障碍相关衰老(mitochondrial dysfunction-associated senescence,MiDAS)以一种独特的表型为特征,表现出一种独特的细胞非自主程序,这可能是衰老动物体内代谢改变和脂肪细胞分化异常的原因。
衰老细胞中的染色质变化
大多数衰老细胞表现出表观基因组和染色质组装的深刻变化。这些变化与衰老相关增殖阻滞的细胞自主性和旁分泌(即对周围细胞的影响)有关。衰老相关异染色质聚集(senescence-associated heterochromatin foci,SAHF)是异染色质的空间领域,可以检测到富含抑制性染色质标记和蛋白的致密的4’,6-二氨基-2-苯吲哚(4′,6-diamidino-2-phenylindole,DAPI)阳性核结构,包括三甲基化组蛋白H3 Lys9(trimethylated histone H3 Lys9,H3K9me3),异染色质蛋白1 (heterochromatin protein 1,HP1),高迁移率基团蛋白A (high mobility group protein A,HMGA)因子,组蛋白变体macroH2A和组蛋白分子伴侣HIRA和ASF1A。
虽然SAHF在癌基因激活的时候会通过DNA复制以及依赖于ATR途径的方式产生,然而其并不是普遍的衰老标志物。 SAHF最初被认为能够抑制促细胞周期相关基因的表达,但后来发现相反的是,SAHF加强了耐DDR的异染色质结构,从而抑制了DDR信号传递。使用能够诱导染色质松弛的蛋白去乙酰化酶(histone deacetylase,HDAC)抑制剂处理,会通过细胞凋亡途径增强DDR信号,最终导致细胞死亡。这种治疗可能是首次报道的成功的senolytic方法的例子,这一点后来被报道的HDAC抑制剂panobinostat的sensenolytic 活性所支持。 HDAC抑制剂也可诱导正常人成纤维细胞的细胞衰老,这可能与其对DDR的影响有关。
衰老细胞的另一个染色质特征是异染色质结构域的展开,这种展开主要是中心粒卫星序列的扩张,在不同的物种中观察到这些现象,并遵循不同的衰老诱导模式。染色质结构的这些变化与选择性去除抑制性组蛋白标记无关,但与核结构蛋白的变化有关,包括核纤层的破裂。在衰老细胞的胞质中,核纤层的缺失可导致胞质染色质片段(cytosolic chromatin fragments,CCFs)的释放。虽然尚未证实,在癌基因诱导的DNA复制的背景下,难以复制的基因组区域,如脆性位点,端粒序列和重复的DNA可能与CCFs有关。目前尚不清楚CCF是否仅在深度衰老的细胞中形成。重要的是,CCFs通过激活环GMP-AMP合成酶(cyclic GMP–AMP synthase,cGAS)和干扰素基因刺激因子(stimulator of interferon genes,STING)受体通路(稍后讨论)来调控与衰老相关的旁分泌功能。据报道,低剂量的HDAC抑制剂可以降低CCFs并抑制衰老相关的分泌表型(senescence-associated secretory phenotype,SASP)。
最近,在染色质修饰的全基因组图谱方面,研究人员取得了技术进步,推动了衰老发生及其过程中分子图谱的建立。在复制衰老过程中,复制后期、基因贫乏的区域表现出广泛的DNA低甲基化,而局灶性高甲基化则见于肿瘤抑制基因。这些发现催生了衰老细胞可能是表观遗传的恶性转化的假设。但是这个假说最近受到了一项观察的挑战,该观察发现,与OIS逃逸的细胞相比,OIS细胞中甲基化模式的变化有限,这表明肿瘤相关的甲基组变化可能是随机发生的,并独立于衰老状态。与DNA甲基化变化的情况相比,癌基因诱导的衰老细胞和晚期复制衰老的成纤维细胞在核小体水平上显示出染色质可及性的显著增加,大多数开放染色质区域映射到调控元件和重复序列。基因组重复位点的染色质松动导致转座因子表达增加,在非应激细胞中转座因子通常是表观遗传沉默和休眠的。
尽管转座元件在通过转座触发基因组不稳定中的作用被广泛接受,但转座元件的再激活也有助于介导衰老细胞的非细胞自主功能,详见下文。H3K4me3的全基因组分析,衰老细胞中的H3K27me3和H3k27ac也揭示了大规模染色质结构域的动态获取和耗竭,这些结构域已被认为调节衰老下游关键效应因子的表达。
SASP的组成和调节
通过转录激活SASP也是衰老细胞发挥其多种生物学功能的一种潜在机制,衰老细胞可通过分泌细胞因子、趋化因子、生长因子和细胞外基质(extracellular matrix,ECM)蛋白酶加重自身衰老进程或影响衰老细胞的局部组织微环境甚至个体(图 2)。SASP激活是一个伴随衰老发生的动态过程。SASP最初被定义为一个重要的分泌过程,包括几十个甚至数百个生物活性因子。
SASP的组成因细胞类型和初始刺激的性质而不同,与复制或辐射诱导的衰老相比,致癌因素放大了蛋白质的分泌。尽管SASP在不同组织和衰老模型中存在一些定性和定量差异,但在所有类型的体外衰老细胞中,都报道了IL-6、CXC 趋化因子配体8(CXC chemokine ligand 8,CXCL8,下称IL-8) 和单核细胞趋化蛋白1(monocyte chemoattractant protein 1,MCP1;又称CCL2),这三者组成了SASP的核心。SASP不仅包括促炎分子,还包括参与ECM重塑的酶,如基质金属蛋白酶(matrix metalloproteinases,MMPs),丝氨酸/半胱氨酸蛋白酶抑制剂(serine/cysteine proteinase inhibitors,SERPINs)和金属蛋白酶组织抑制剂(tissue inhibitors of metalloproteinases,TIMPs)。最近对SASP进行的全面无偏定量蛋白质组学分析鉴定出了更多不同的SASP效应物,它们能作为可溶分子或外泌体被释放出去。外泌体中含有一系列成分,这些成分在先前报道中会富集在衰老和衰老相关疾病人群的血浆里。外泌体最近被确定为SASP旁分泌衰老效应以及外泌体促癌属性的关键媒介。
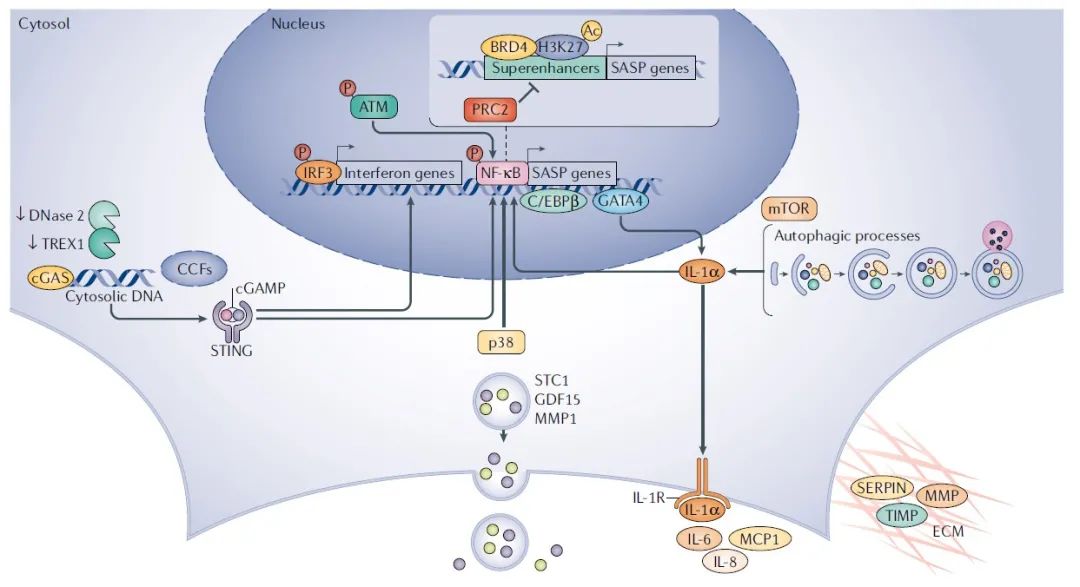
图 2 | SASP调节。衰老相关分泌表型(senescence-associated secretory phenotype,SASP)激活是一个动态的过程,由衰老触发,伴随着细胞周期的退出。一个核心的SASP程序主要包括以IL-1依赖方式调节的促炎白细胞介素-6(interleukin-6,IL-6)、IL-8和单核细胞趋化蛋白1(monocyte chemoattractant protein 1,MCP1),以及参与细胞外基质(extracellular matrix,ECM)重塑的酶,如基质金属蛋白酶(matrix metalloproteinases,MMPs)、丝氨酸/半胱氨酸蛋白酶抑制剂(serine/cysteine proteinase Inhibitors,SERPINs)和组织金属蛋白酶抑制剂(tissue inhibitors of metalloproteinases,TIMPs)。最近,发现了其他作为可溶性分子或外泌体释放的SASP核心效应物, 包括GDF15、STC1和MMP1。DNA损伤反应因子,包括上游DNA损伤反应激酶,通过核因子-κB(nuclear factor-κB,NF-κB)诱导SASP基因。促分裂原活化蛋白激酶p38也通过增加NF-κB的活性来诱导SASP基因。几种转录因子和染色质调节因子的激活与SASP的激活和调控有关。NF-κB转录因子CCAAT/增强子结合蛋白β(CCAAT/enhancer-binding protein-β,C/EBPβ)结合SASP基因的启动子并调节其激活。GATA4通过产生IL-1间接调控NF-κB和SASP基因。哺乳动物类雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)途径的靶点促进SASP的产生,通过增加mRNA亚群的翻译,包括编码IL-1α。与转录因子协同作用,表观遗传阅读器含溴域蛋白4(bromodomain-containing protein 4,BRD4)是一种参与肿瘤发生的乙酰化组蛋白结合蛋白,被招募到SASP基因附近作为超级增强子,从而有助于细胞衰老的正确执行。BRD4结合乙酰化组蛋白H3Lys27(acetylated histone H3 Lys27,H3K27),从而与多梳抑制复合物2(polycomb repressor complex 2,PRC2)竞争,后者甲基化相同的组蛋白残基(给予三甲基化H3K27)进行转录抑制。与此相一致,PRC2抑制衰老细胞中的SASP 基因。最近,据报道,DNA感受器环GMP-AMP 合成酶(ccyclic GMP–AMP Synthase,GAS)和干扰素刺激基因(stimulator of interferon genes,STING)受体是SASP程序在物种和衰老模式中的主要调节因子,可能是通过激活NF-κB和干扰素反应因子IR F3 来识别胞浆DNA 和胞浆染色质片段(cytosolic chromatin fragments,CCFs)。cGAS-STING通路的异常激活可能与DNA酶的下调有关(例如,DNase 2和TREX1),这些酶通常参与细胞质DNA降解。ATM,毛细血管扩张性共济失调突变;cGAMP,环GMP-AMP;IL-1R,白介素-1受体。
DDR和SASP之间的交互作用
诱导p16可抑制正常细胞的增殖,促进细胞衰老,但不足以诱导完整的SASP。 值得注意的是,持续的DDR信号通常需要激活炎症细胞因子的分泌。与上游DDR元件在促进衰老细胞的细胞自主性和旁分泌功能方面的作用相一致,ATM、NBS1和CHK2是SASP主要的被激活基因,因为在应对遗传毒性应激的反应时,这些DDR上游调节因子的缺失抑制了细胞因子的产生。抑制p53则有相反的作用,因为它在衰老诱导的损伤后进一步增强SASP,这可能有助于产生易于衰老逃逸和恶性转化的炎症微环境。
最近,ATM被发现通过介导SASP基因中组蛋白变体macroH2A1.1的去除来间接调控SASP基因的表达,以响应DNA损伤和致癌应激。然而,由于DDR的激活是一个快速的反应,而SASP的建立是缓慢的,必须有额外的通路控制SASP。事实上,在没有DNA损伤的情况下,应激诱导的MAPK p38的激活被证明是触发生长停滞和SASP的充分必要条件。与ATM一样,p38通过增加核因子-κB(nuclear factor-κB,NF-κB)的活性来诱导SASP转录本的表达,表明虽然DDR和p38激活是独立的,但它们可以汇合于SASP的激活。
在增殖和非增殖细胞中衰老是DDR增加相关的因素之一,年龄的增长会显著导致衰老细胞的积累。不完全的DNA修复可能进一步有助于DNA病变的积累和DDR激活,以及在衰老过程中在不同细胞类型和生物体水平上广泛的染色质变化。此外,DDR是代谢重编程的驱动因素,可以增强SASP。因此,DDR通过多种途径调节SASP可能是DDR驱动年龄相关炎症反应的方式之一。
SASP的转录与表观遗传调控
多种转录因子和染色质调控因子在转录水平上参与SASP的调控。NF-κB和转录因子CCAAT/增强子结合蛋白-β(CCAAT/enhancer-binding protein-β,C/EBPβ)结合在SASP基因的启动子上,并调控其激活。此外,转录因子GATA4激活大量参与免疫应答和炎症反应的基因,包括编码IL-6、IL-8、CXCL1(也称为GROα)、粒细胞-巨噬细胞集落刺激因子、ECM蛋白酶及其抑制剂。虽然GATA4对NF-κB通路的调控是通过增加NF-κB上游调控因子IL-1α的表达和分泌而间接进行的,但是在DDR激活的细胞及衰老细胞中GATA4表达水平仍然升高,因此有研究人员提出将GATA4作为DDR信号和随后NF-κB激活之间的分子连接,以完善SASP激活的整体过程。
在PTEN缺失导致的细胞衰老模型中,JAK2-STAT3通路激活了一组具有免疫抑制特性的SASP因子。因此,JAK/STAT抑制剂可以有效地重编程SASP,以增强化疗和T细胞介导的癌细胞与衰老细胞的清除。此外,JAK抑制剂还能够缓解老年小鼠的身体机能衰退。SASP基因的表达是动态变化的。据报道,衰老过程中NOTCH1随时间的活性变化能够调节SASP的成分。早期NOTCH1水平升高会激活转化生长因子-β及其效应因子,同时通过抑制其正调控因子C/EBPβ来调控SASP级联的促炎通路。在后期,严重衰老的细胞中,NOTCH1表达水平降低,诱导SASP促炎细胞因子IL-1、IL-6和IL-8的表达。然而,目前还不清楚NOTCH1的这种功能是否与最近报道的其直接抑制ATM的作用有关。
表观遗传阅读器——含溴结构域蛋白4(bromodomain-containing protein 4,BRD4)是一种乙酰化的组蛋白结合蛋白,被招募到OIS中SASP基因附近的超增强子中,参与肿瘤发生过程。BRD4有助于细胞衰老过程的进行,并出人意料地发挥了肿瘤抑制作用。事实上,通过药物或基因编辑使BRD4失活会导致SASP减少,限制了OIS免疫介导的细胞清除,因此这些方法可能无法清除易于衰老逃逸的受损细胞。然而,最近的一项药物筛选发现了一种降解BRD4的小分子,并具有清除衰老细胞的活性。此外,在小鼠和人类的细胞中BRD4都能促进端粒的延长。因此,BRD4抑制剂可能会抑制SASP的激活,但同时也会通过促进端粒缩短进而导致细胞衰老。BRD4与乙酰化的组蛋白H3 Lys27结合,从而竞争能够甲基化相同的组蛋白残基(产生三甲基化的H3Lys27)的多梳抑制复合体2(Polycomb repressor complex 2,PRC2)。与BRD4和EZH2(PRC2的催化核心亚基)竞争相同残基并具有拮抗作用相一致的,过表达EZH2通过多种机制阻止OIS,包括DDR调控和抑制SASP基因表达。还有研究人员发现,转录相关的组蛋白甲基转移酶和癌蛋白MLL1对于SASP激活是必不可少的。但是,这主要是通过癌基因诱导的过度复制和DDR参与,而不是通过SASP基因的直接转录调控的。
HMGB蛋白也参与SASP的调控。HMGB2直接结合并特异性调节癌基因诱导的衰老细胞中的SASP基因表达。并且,HMGB2的耗竭会导致SASP的减少,而不会影响衰老的生长停滞。HMGB1(也被称为alarmins)主要作为一种损伤相关的分子释放在细胞外,来诱导SASP介导的旁分泌衰老并刺激免疫系统。此外,OIS过程中核孔密度的增加(导致SAHF的关键)通过介导异染色质重组调节SASP的表达。
SASP和先天免疫
据报道,DNA传感器cGAS和适配器蛋白STING是跨物种的SASP主要调控因子,可能是通过激活NF-κB和干扰素反应因子IRF3调控SASP。cGAS-STING激活主要是通过识别衰老细胞胞浆中的细胞自身的双链DNA或染色质片段。在体内衰老模型中,cGAS-STING基因的缺失导致了促炎症SASP和衰老免疫监测的下降。据报道,cGAS-STING途径的异常激活可能与通常用于细胞质DNA降解的DNase(例如DNase 2和TREX1)的下调有关。DNase的下调导致了衰老过程中胞质DNA的积累,增加了SASP调控的复杂性。虽然在衰老细胞中引起胞质染色质释放的机制仍在研究中,但是这些发现表明STING抑制剂将可能用于治疗衰老相关的慢性炎症。
cGAS-STING并不是参与SASP启动和执行的唯一先天免疫途径。炎症小体也参与其中。炎症小体是一种多蛋白复合物,包含半胱天冬酶1和抗病原体防御机制的关键调节剂,以及调节炎症小体的Toll样受体,可促进OIS期间SASP因子的成熟和分泌。衰老细胞,特别是在体外培养或体内持续时间较长时,会表现出I型干扰素反应和下游靶点的深度激活。转座子的重新激活和cGAS-STING激活在一定程度上导致了这种独特的I型干扰素的深度激活。使用核苷逆转录酶抑制剂治疗,可以抑制转座子的逆转录,减少SASP与衰老相关的有害影响,并改善老年动物的慢性炎症。
干细胞衰老
干细胞和祖细胞是维持组织在生理转换和组织损伤过程中稳态的关键。随着年龄的增长,干细胞在体内的数量不一定会有所减少,但是其功能会有所下降。在正常和病理性衰老过程中,不同组织和物种的各种干细胞类型中都会表现出DNA损伤和DDR激活标记。这表明干细胞也不能避免DNA损伤积累和DDR激活。
尽管DDR通路的激活有利于基因组稳定性和干性,但有证据表明,DNA损伤后,DDR调控将导致永久性细胞周期停滞,并使细胞具有细胞衰老和细胞分化的特征。事实上,小鼠暴露于电离辐射下会导致毛皮变灰,这是由于一轮细胞分裂后受损的毛发黑色素干细胞的分化。虽然没有检测到一些细胞衰老标志物的变化,如p16和SA-β-gal,但黑色素干细胞的分化与持续的DDR激活有关,并且在Atm敲除小鼠中增强。相似地,端粒功能障碍或外源DNA损伤,会诱导淋巴样分化,继而抑制造血干细胞(haematopoietic stem cells, HSC)的自我更新。人类HSC的单细胞转录组学分析揭示了对DSB衰老样程序的剂量依赖性激活。其特征在于p53的激活和促炎程序的诱导,导致了细胞成瘤潜能,迁移能力和谱系输出的降低。相似地,老年小鼠HSC中DNA复制应力的积累与HSC衰老、造血分化不平衡和髓样偏斜有关。但是,特别是在人类中,老年人的骨髓造血障碍是由髓样引发的HSC分化增加或淋巴细胞分化受损所导致的仍有待进一步明确。
DNA损伤,例如电离辐射引起的DNA损伤能促进小鼠神经干细胞的分化和诱导细胞衰老。DNA损伤导致了干性基因表达的缺失,以及向星形胶质细胞分化的转录谱特征。这一细胞分化程序由ATM和可溶性因子控制,特别是通过BMP2信号。辐射小鼠体内的谱系追踪实验证实了诱导分化标记物在通常由神经干细胞组成的脑室下区表达。因此,DNA损伤诱导的细胞衰老可能与细胞分化相一致。
值得注意的是,在小鼠胚胎干细胞中,p53诱导的程序与分化过程的转录激活和多能干细胞基因的抑制有关。与上述结果一致,衰老以程序化方式进行,并有助于哺乳动物胚胎发育和组织发生,但主要是通过诱导p21,p15和SASP等介导因子的表达,而不是通过DDR信号转导。
总的来说,在黑色素细胞干细胞、造血干细胞、神经干细胞、胚胎干细胞和整个胚胎中的这些实验表明,干细胞中持续存在的遗传毒性应激,且可能在祖细胞和分化程度较低的细胞中更为广泛,可导致伴随有细胞分化特征的细胞衰老。尽管细胞衰老通常不被认为是细胞分化的一种形式,因为它通常是大分子损伤的结果,而细胞分化却不是,但共同点是惊人的:它们都涉及细胞周期停滞,并具有通常由可溶性因子控制的独特转录程序。从最开始,衰老研究就普遍使用分化程度较高的细胞(最典型的是成纤维细胞)。这可能导致细胞衰老未被作为应激诱导分化程序的发现,也许在不同的研究中,细胞衰老也可以被认为是DNA损伤诱导的细胞分化的一种形式。
有丝分裂后的细胞会变得“衰老”吗?
在衰老过程中,终末分化的细胞中会积累持续的DNA损伤和DDR标志物。这引出了一个问题——即持续的DNA损伤信号传导是否导致细胞周期抑制因子的表达,并最终导致细胞衰老,从而使细胞从非分裂的生理状态转换为非分裂的病理状态。虽然目前仅主要在神经元中进行了研究,但越来越多的证据表明,细胞衰老可能与终末分化的细胞有关。在衰老小鼠不同类型的神经元中均检测到DDR信号转导、异染色质诱导和SASP激活的标志物,包括IL-6的分泌和SA-β-gal的积累。此外,短期的饮食限制则能够减少它们的积累。这些表型在端粒酶失活的小鼠中会加重,但是p21的缺失会导致这些标记物的减少。相反地,小鼠肥胖则与大脑特定部位神经元中衰老标志物的表达有关。在阿尔茨海默病小鼠模型中,对含有tau蛋白的的神经元进行转录组分析表明,这类细胞的表达谱与衰老细胞一致。还有研究发现在视网膜病变中,视网膜神经节细胞(视网膜中的神经细胞)中会发生DDR的激活以及SA-β-gal、p16和p21这些标志物的积累。
在摄入过多热量的小鼠模型中成熟的有丝分裂后脂肪细胞以p53依赖的途径表现出较多SA-β-gal和SASP的积累,但目前尚无DNA损伤积累等其他标记的报道。骨细胞是调控骨稳态的有丝分裂后分化细胞。在老年小鼠中,骨细胞表现出端粒功能障碍标志物的表达以及p16的积累。研究表明,骨细胞中的SASP激活可促进破骨细胞活性,从而导致骨强度的下降。在这种情况下,清除衰老细胞的药物减少了骨质流失。
此外,心肌细胞中持续的端粒DNA损伤会导致衰老表型,其特征包括p16和p21的表达增加,以及导致心脏肥大和纤维化的非经典SASP途径。事实上,通过遗传或药物的方法清除表达p16的衰老细胞均可以改善老年小鼠的心脏功能。确定衰老细胞对某种疾病作用,最有力的方式是通过遗传操作或药物的方式将其清除。然而,由于目前没有工具可以选择性地靶向分化的衰老细胞亚群,所以它们在衰老相关过程中的作用仍不清楚。
细胞衰老的有益作用
细胞衰老是一种进化过程中发挥一些基本功能及有益功能的应激反应(图3)。在胚胎发育中,衰老细胞表现出了明显的有益作用。细胞衰老的一种独特形式发生在哺乳动物发育中,调控其胚胎和胎盘的生长发育。同样,在两栖动物中,细胞衰老发生在发育的特定阶段以塑造身体的生长。因此,生命早期的细胞衰老对正常的发育和形态发生非常重要;而在生命后期,细胞衰老则对组织修复和抑制肿瘤生长起重要作用。虽然肿瘤抑制活性主要由细胞自主性细胞周期停滞介导,但大多数其他衰老功能均涉及SASP。随着时间的积累,尽管SASP有利于组织的正常发育与修复,及免疫细胞的募集;但其持久的SASP作用可能会引发慢性炎症,并导致衰老相关疾病,甚至相反地引起癌症。
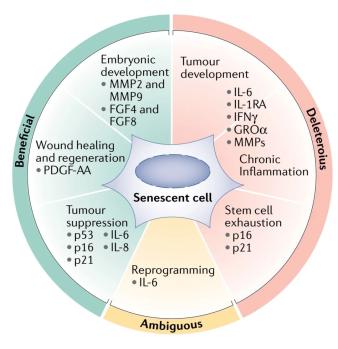
图3 | 细胞衰老的生物学结果。衰老细胞执行不同的生物学功能,这些功能取决于上下游的途径可能产生有害或有益的结果。作为有益的功能,衰老细胞通过分泌FGF4和FGF8来引导组织再生和胚胎发育,并通过基质金属蛋白酶2和9(metalloproteinase 2 and 9, MMP2 and MMP9)塑造胎盘的结构和功能。衰老细胞还通过限制细胞过度增殖来抑制组织损伤,并通过分泌PDGF-AA部分地促进伤口愈合。此外,衰老最显著的功能之一是抑制肿瘤。衰老细胞通过上调p53,p16和p21的细胞自主阻断细胞周期进程来限制肿瘤的发展,并通过分泌白介素6(IL-6)和IL-8促进邻近细胞的衰老,从而以细胞非自主的方式限制肿瘤的发展。作为有害功能,衰老细胞可以诱导促炎性微环境,并通过多种SASP成分促进肿瘤的发展。同样,衰老细胞在衰老期间及多种衰老相关疾病中会促进慢性炎症。SASP因子,包括IL-6,IL-1受体拮抗剂(IL-1 receptor antagonist,IL-1RA),GROα和干扰素-γ(interferon-γ,IFNγ)是上述效应的主要介导因子。其他SASP因子,包括MMP,也可能会进一步破坏组织结构并促进炎症和肿瘤发生。当干细胞或祖细胞由于细胞周期抑制蛋白(例如p16和p21)的上调而进入衰老时,它们不再能够通过提供新生细胞来发挥其促进组织再生的功能,从而限制了组织的再生潜力。衰老细胞至少还可以通过IL-6促进的重编程过程部分重编程为胚胎状态。 一方面,重编程可以促进组织再生,但是另一方面,也会促进肿瘤的发展。
细胞衰老有助于维持损伤后组织结构和功能的稳定。例如,肝纤维化是与肝脏瘢痕形成和功能降低有关的疾病,衰老限制了ECM的产生和肝星状细胞的活化与增殖。这种反应抑制了肝损伤后肝纤维化的发展。这些细胞中的SASP会募集自然杀伤(natural killer,NK)细胞,从而清除肝脏中的衰老细胞,从而恢复肝脏的稳态。细胞通讯网络因子1(cellular communication network factor 1,CCN1,也称为CYR61),是一种在肝脏中介导细胞衰老的ECM蛋白,在皮肤伤口愈合期间促进成纤维细胞的衰老,从而限制了皮肤纤维化。在伤口愈合过程中,SASP组分PDGF-AA则会加速伤口的愈合。此外,衰老诱导过程也限制了胰腺纤维化的进程。
细胞衰老也与其他系统的组织修复有关。在斑马鱼中,衰老受损会阻止截肢后鳍的再生;而在蝾螈中,衰老细胞也与肢体再生有关。总而言之,这些结果表明细胞衰老已经发展为了一种抑制生物体中的组织损伤反应的程序,并能够促进组织修复和重塑,迅速恢复组织的功能状态。
一旦执行了有益的功能,衰老细胞的稳态功能就取决于免疫系统对它们的清除作用。特异的SASP趋化因子能够吸引免疫细胞的不同亚群,包括NK细胞、嗜中性粒细胞、树突状细胞、单核细胞/巨噬细胞、B细胞和T细胞。在这些细胞类型中,NK细胞、T细胞和巨噬细胞可以在病理及生理条件下与衰老细胞发生物理性的相互作用。这种反应是由SASP成分以及免疫细胞和衰老细胞之间的直接相互作用介导的。免疫系统对衰老细胞的监视和清除是必要的,这样才能抑制恶性病变和癌症治疗后的肿瘤发生。同样,衰老免疫监视对于限制病理性纤维化病和衰老也是必不可少的。相反地,在衰老细胞积累相关的癌症发展过程中,SASP可以募集未成熟的髓样细胞以旁分泌方式促进肿瘤发生。另外,衰老细胞能够通过SASP促进癌症的发展和转移,并使治疗失败和复发风险的增加。因此,SASP是衰老表型的一个组成部分,并且是衰老细胞向免疫系统发出的信号。此外,SASP还可促进衰老细胞的清除。但是当衰老细胞持续存在时,其产生的SASP则会产生有害作用。
衰老的有害影响
衰老细胞能够通过多种机制促进机体的衰老。随着年龄的增长,个体往往会表现出高循环水平炎症分子的状态(即炎症)。炎性衰老是各种衰老相关的慢性疾病的一个风险因素,包括心血管疾病、一些癌症和神经退行性疾病,并且还与过早死亡相关。此外,老年人血液中的炎症分子还与体重减轻、肌肉萎缩、身体虚弱、慢性炎症和抑郁相关(这些症状被认为是身体机能下降的表现)。在探究衰老相关慢性表型的共同基因组变异时,研究人员揭示了细胞衰老、炎症和身体机能下降之间的分子联系。最近,全基因组关联研究揭示了编码p15INK4B、p16和ARF的INK4/ARF基因位点是衰老生长停滞的关键效应因子。该位点是癌症、糖尿病、心血管疾病以及老年人的生理功能障碍等几种衰老相关疾病易发的基因组热点。此外,在衰老过程中,端粒长度缩短与代谢和心脏功能障碍有关。SASP可能导致了多种衰老器官的功能障碍。事实上,IL-6、IL-1、IL-1RA和肿瘤坏死因子(tumour necrosis factor,TNF)受体等所有重要的SASP效应因子在血液中浓度升高均可作为预测老年人慢性疾病的依据。在小鼠体内移植相对较少的衰老细胞,即会导致组织功能障碍和寿命缩短。这可以证明细胞衰老是导致身体机能下降和衰老相关疾病的原因。
最近,研究人员在LDL受体缺失的动脉粥样硬化斑块小鼠模型中,发现了大量SA-β-gal的积累和p16阳性的内皮细胞、血管平滑肌细胞和巨噬细胞。研究人员通过遗传操作方法和senolytics清除p16-3MR和INK-ATTAC转基因小鼠中的p16阳性细胞,减少了斑块形成和增大,同时抑制了SASP的表达。与小鼠中的这些数据一致,虽然p16主要由炎性巨噬细胞表达,但人动脉粥样硬化斑块中出现了p16阳性细胞的高度富集。此外,研究人员也不能排除在易患动脉粥样硬化的小鼠中,senolysis的积极作用是消除炎症性巨噬细胞的结果。研究人员在患有组织细胞病的病人中,以及伴有多器官弥散高度炎症和p16阳性髓系细胞的癌基因激活相关的血液肿瘤中,也发现了具有衰老样特征的巨噬细胞。
如前所述,除了通过SASP在机体水平上对炎症和慢性疾病的影响外,衰老细胞还可能通过抑制干细胞和祖细胞的增殖潜能来影响组织再生。积累了DNA和分子损伤,并在上调p16的刺激下进入衰老状态的肌肉祖细胞并不能促进损伤后的肌肉再生。综上所述,衰老以细胞自主的方式抑制了干细胞和祖细胞的增殖。另外,最近有报道称,当HSC暴露于衰老的基质细胞产生的SASP因子时,其克隆形成特性会受到损害。这表明衰老还可能以旁分泌方式影响再生。
虽然研究人员已经充分明确了衰老对成体干细胞功能的有害影响,但在体细胞重编程为胚胎样状态的过程中,细胞衰老则有待进一步研究。体外研究表明细胞衰老是细胞自身阻碍OCT4,SOX2,KLF4和MYC(通常称为OSKM)进行转录因子介导重编程的屏障,其作用类似于抑制肿瘤。但是,这些因子在体内的表达会诱导衰老和SASP产生,从而促进旁分泌衰老以及非细胞自主的方式在非衰老细胞中进行重编程。在这些相同的模型中,驱动细胞衰老的外源性组织损伤促进了重新编程。诱导衰老对于细胞重编程是必要的,因为SASP因子的产生以旁分泌方式促进了重编程为诱导性多能干细胞的过程。SASP中的IL-6对于诱导性多能干细胞的产生似乎至关重要。
细胞衰老的非细胞自主效应在不同的状态下有所不同。在体外,SASP因子IL-8,GROα,IL-6和IGFBP7以自分泌方式促进衰老表型。此外,可溶性或细胞外囊泡中的特定SASP成分可以旁分泌途径诱导衰老(包括DDR的激活)。这些因子可能会促进组织衰老,并导致组织和机体功能障碍。在体内,SASP所导致的结果更为复杂。例如,SASP成分干扰素-γ会在端粒缩短的小鼠中诱导细胞衰老和组织衰老,而抑制干扰素-γ信号传导可以改善衰老相关表型并延长寿命。同样,转化生长因子-β可通过阻断邻近未损伤肝细胞的旁分泌衰老,来促进损伤时的肝脏再生。相反地,体内短期的SASP处理可促进干细胞标志物的表达,并增强小鼠角质形成细胞和骨骼肌的再生能力。然而,长时间的SASP处理会导致旁分泌诱导的衰老。这表明至少在体内,SASP的作用取决于其组成和处理时间。从进化的角度看,SASP短期内可能通过增强干细胞功能,促进伤口愈合和组织损伤修复;而衰老细胞的长期存在则可能会导致SASP的有害作用。
细胞衰老是个体衰老的驱动力
BubR1小鼠早衰模型在证明细胞衰老细胞引发个体衰老和疾病方面发挥了重要的作用。BubR1是有丝分裂检验点机制的一部分,可确保在有丝分裂过程中将重复的染色体正确分配在两个相同的子细胞中。BubR1表达水平正常的小鼠中,约10%的小鼠会在生命早期表现出寿命缩短、白内障、驼背、脂肪代谢障碍和不育等各种早衰特征。BubR1小鼠的脂肪组织,骨骼肌和眼睛中出现了高水平的p16表达及其他衰老相关特征。为了减少高表达p16细胞的积累,研究人员将BubR1突变小鼠与Cdkn2ap16基因敲除小鼠进行了繁殖。在p16缺失的情况下,与衰老相关的脂肪组织、骨骼肌和眼睛的功能障碍有所缓解。重要的是,从基因水平上抑制了p19ARF的积累(一种通过影响MDM2介导的降解来调节p53稳定性的肿瘤抑制因子)并未导致类似的结果。这表明p16对这些疾病至关重要。
根据上述结果,研究人员已经构建了INK-ATTAC11和p16-3MR12两种不同的转基因小鼠模型,以严格检测清除衰老细胞是否会影响衰老以及衰老细胞积累相关的疾病。重要的是,从断奶开始对具有INK-ATTAC转基因的BubR1小鼠进行清除表达p16的细胞,能够减少骨骼肌、眼睛和脂肪组织中衰老细胞的积累并缓解早衰。在自然衰老小鼠中使用INK-ATTAC系统进行的第二项研究验证了上述发现,并且延长了不同的遗传背景雄性和雌性小鼠的中位寿命及健康寿命,还改善了肾脏瘢痕形成、心肌细胞肥大、心脏压力不耐受、白内障和脂肪代谢障碍。
p16-3MR小鼠模型表达了一种三态性报告基因融合蛋白。该蛋白由合成的海肾荧光素酶,单体红色荧光蛋白和截短的单纯疱疹病毒胸苷激酶组成,并由p16人工启动子控制。更昔洛韦是一种核苷类似物,会被单纯疱疹病毒胸苷激酶转化为有毒的DNA链终止剂,导致细胞死亡。在p16-3MR小鼠模型中,表达p16的细胞对更昔洛韦的清除敏感。这两种小鼠模型极大地促进了我们对衰老细胞是否会导致衰老与衰老相关关的疾病的理解,包括帕金森症、阿尔茨海默症、动脉粥样硬化、特发性肺纤维化、慢性阻塞性肺疾病和骨关节炎。然而,清除衰老细胞或其分泌的SASP是否是改善衰老及衰老相关疾病的关键因素,目前还不清楚。
探寻延缓衰老的新疗法
文献中充斥着衰老细胞在各种与年龄有关的疾病中积累的证据。因观察到消除衰老细胞在很大程度上是有益的,并且似乎缺乏长期的负面影响,学术界和工业界的研究人员旨在寻找在没有基因工程的情况下适用于人的消除衰老细胞新药物和策略。这些“治疗疗法”的策略可大致分为两类:第一个是通过选择性杀死衰老细胞(使用称为“senolytics”的药理学化合物)。第二个是通过使用senomorphics药物来消除细胞间通讯的有害影响,包括SASP。
Senolytics
最近,结合衰老的体外模型和体内的动物模型,开发了各种senolysis 策略(图4;表1)。衰老细胞经常上调凋亡的负调节因子,包括BCL-2 家族成员(包括BCL-2,BCL-W和BCL-XL),从而赋予对凋亡诱导信号的抗性。抗衰老药物ABT-737和ABT-263(也称为navitoclax)抑制BCL-2 家族成员的活性,从而使衰老细胞开始凋亡。另外,据认为抑制BCL-XL 的A-1331852 和A-1155463 也表现出抗衰老活性。最近,胭脂树苷哇巴因至少部分通过诱导NOXA(一种促凋亡的BCL-2 家族蛋白)表现出一定的抗衰老活性。通过EF24处理促进BCL-2的蛋白酶体降解也导致选择性杀死衰老细胞。服用proxofim(一种肽)会干扰p53与FOXO4的结合,从而促进抗衰老。在衰老细胞中,FOXO4 与p53结合,使其定位于细胞核。如果这种相互作用被反向肽破坏,则p53被排除在细胞核之外,从而启动细胞色素c 从线粒体的释放和细胞凋亡。单独或与泛酪氨酸激酶抑制剂达沙替尼组合使用各种天然类黄酮,包括槲皮素和非瑟酮,可在体外和体内的各种情况下刺激对抗衰老。与对INK-ATTAC 自然衰老小鼠进行的研究相一致,用达沙替尼和槲皮素的组合治疗高龄小鼠可改善身体机能并延长寿命。重要的是,在糖尿病肾病和特发性肺病患者的I期临床试验中,达沙替尼和槲皮素的给药已显示有效的降低p16和SA-β-gal 表达。其他senolytics,包括HSP90抑制剂和pipelongongine,也已被证明对衰老细胞具有选择性。最近,据报道,临床批准的抗生素通过代谢变化对DNA损伤诱导的衰老细胞具有抗衰老活性。这些策略针对广泛的细胞途径,表明可以通过多种途径去除衰老细胞。
研究人员利用一种监测SA-β-gal活性水平的升高新的方法来对抗衰老。发现含有被半乳寡糖包被的荧光团或细胞毒性剂的纳米颗粒优先将细胞毒性药物递送至衰老细胞,因为这些细胞中较高的SA-β-gal 水平。另外,最近的研究进一步显示了通过修饰半乳糖的前药或细胞毒性剂将细胞毒性因子传递给衰老细胞的溶酶体的潜力。
Senolytic 疗法最大的有优势在于利用生物体固有的生物分解系统——针对衰老细胞的免疫监视。实际上,衰老细胞通过先天性和适应性免疫的多种成分进行免疫监视,包括NK细胞,T细胞和巨噬细胞。因此,可以利用衰老细胞的免疫监视机制以及免疫系统来消除衰老细胞。由于免疫监视的下降,衰老细胞有可能在衰老和患病的组织中积累。因此,恢复或增强免疫系统特异性消除衰老细胞的能力可导致其成功地从组织中清除。这种方法是基于我们对衰老细胞免疫监视机制的理解,特别是NK和衰老细胞之间的相互作用。NK细胞通过穿孔素介导的颗粒胞吐作用,而不是死亡受体配体,通过与这些受体结合而诱导细胞死亡,从而靶向衰老细胞。该机制之所以受到青睐,是因为诱饵受体2(DCR2)在衰老细胞中高表达。DCR2 阻止表达其配体TRAIL151的多种细胞毒性细胞通过死亡受体(DR4和DR5)进行靶向。因此,阻断这种抑制机制可以导致抑制作用的消除,并通过天然存在的机制来增加衰老细胞的靶向性。
增强衰老细胞免疫清除能力的另一种方法是增强免疫细胞的活性并增加负责衰老细胞监视的免疫细胞的积累。用聚肌苷酸胞苷酸(可以模拟病毒感染)刺激先天免疫应答,可改善衰老细胞清除率。尽管由于可能的副作用,在医疗过程中几乎不考虑使用类似药物进行治疗,但是用能增强NK细胞的特定细胞因子对免疫系统刺激是一种可行的方法。细胞因子IL-21和IL-15已经用来增强NK细胞介导的针对癌细胞的免疫力。但是,尚未阐明这些细胞因子对衰老细胞免疫监视的作用,因此需要测试它们在疾病模型中作为senolytics的功效。
尽管增强衰老细胞监视的天然免疫机制可能在未来几年具有治疗潜力,但免疫肿瘤学领域中可应用的方法还是值得探讨的。定向细胞方法,例如使用嵌合抗原受体T(CAR T)细胞和NK细胞,以及通过阻断与PD1,细胞毒性T淋巴细胞相关蛋白4(CTLA4)和其他抑制分子的相互作用来阻断免疫抑制相互作用提供增加免疫监视的有力策略。此类方法取决于衰老细胞的细胞表面上特定标记的识别。最新令人激动的研究成果表明,senolytic CAR T细胞疗法可减轻衰老相关疾病。几项研究使用无差别法来识别此类标记。但是,当每个实验都鉴定出不同的标记物时,通过上述策略鉴定出的细胞外标记物之间的重叠率很低,这表明这些标记物可能对起源细胞和/或衰老诱导机制具有特异性。解决此问题的一种可能方法是使用衰老细胞上向NK细胞发出信号以消除它们的表面分子,即NKG2D受体配体。但是,这类配体的丰富成分,包括MICA,MICB和ULBP1-ULBP6,并且它们在不同来源的衰老细胞上表达水平的不同,导致使用它们作为标记识别衰老细胞后免疫清除带来挑战。当不同标记物存在于不同来源的细胞上时,最合理的方法是针对不同病理状况设计多种方法。
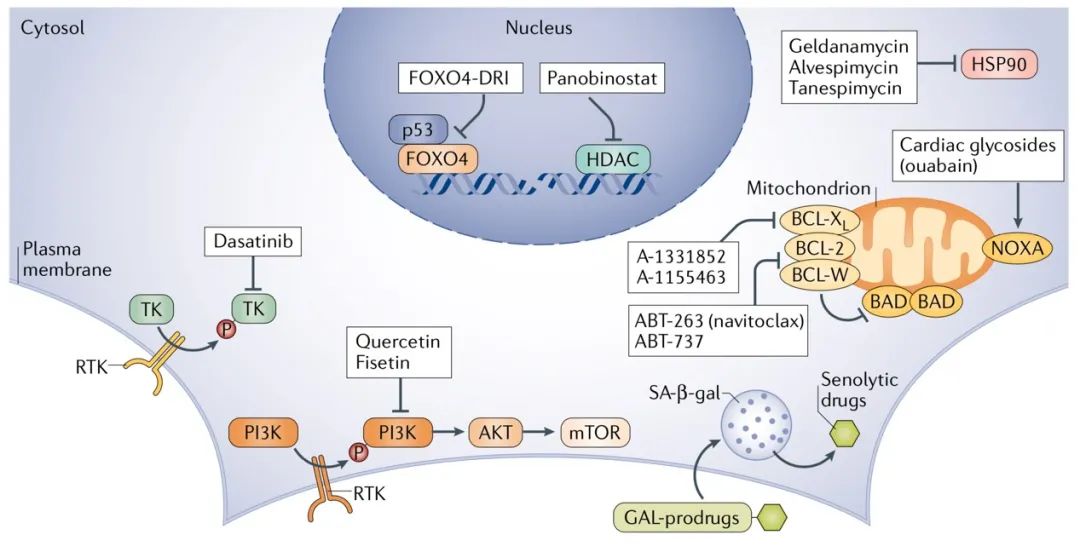
图4 | Senolytic干预疗法。促进衰老细胞死亡药物的治疗机制是多种多样的。图中展示了许多已知的衰老作用机理和用于关键节点的各种特定化合物。通过使用达沙替尼(单独使用或与类黄酮槲皮素组合使用)影响酪氨酸激酶(TK)能够引发某些衰老细胞类型的死亡。槲皮素和非瑟汀是影响哺乳动物雷帕霉素(mTOR)信号传递靶标的天然类黄酮。 BCL-2家族抗凋亡成员的抑制剂能够通过线粒体介导的机制诱导死亡,同时也可通过强心苷如哇巴因的作用引起。还已经提出了HSP90或组蛋白去乙酰基酶(HDAC)的抑制剂可促进衰老细胞的选择性凋亡。另外,通过使用小肽破坏在衰老细胞中发生的FOXO4与p53的结合,可以释放p53来激活细胞凋亡。通过与衰老相关的β-半乳糖苷酶(SA-β-gal)对半乳糖缀合的衰老药物(GAL-前药)进行处理,以发挥选择性衰老靶向作用。RTK,受体酪氨酸激酶。
Senomorphics
清除衰老细胞的另外一种方法是通过senomorphic 药物的使用。senomorphics的原理是破坏衰老的主要属性,主要是SASP的产生和分泌,同时保持细胞存活,或改变其维持稳定生长停滞的能力(图5)。这种方法可能会干扰衰老细胞的促炎性质,并成为延缓衰老和衰老相关疾病关键因素。
除了利用SASP因子表达的转录调控外,还通过假设驱动的策略和药物筛选等方式发现调控SASP的新机制。这些包括,例如,雷帕霉素(mTOR)途径的哺乳动物靶标,该靶标协调细胞对营养的生长和代谢,并通过增加mRNA 子集的翻译来促进SASP的产生,包括上游调控因子如NF-κB,IL-1α和丝氨酸/苏氨酸激酶MK2,它们间接稳定了许多编码细胞因子的转录物。这些机制的解析为在体内与衰老相关的病理环境中使用mTOR抑制剂雷帕霉素提供了分子基础。雷帕霉素(及其类似物RAD001)的处理可减弱自然衰老小鼠中的原癌基因SASP,防止衰老,改善的肝功能障碍。值得注意的是,雷帕霉素也可以通过不依赖衰老的机制起作用。此外,雷帕霉素治疗可延长小鼠的寿命并延缓某些与衰老相关的功能障碍。
现今已证明可调节NF-κB 信号的化合物包括二甲双胍,芹菜素,kaempferol和BAY 11-7082,同时可降低SASP的产生。 NAD+/ NADH 代谢被确定为与致癌基因激活相关的促炎性SASP量的关键调节通路,该调节可独立于衰老诱导的生长停滞。许多直接针对IL-6,IL-1α,IL-1β和TNF等SASP关键成分或其受体的中和抗体也表现出相似的特性(表 1)。此外,可以通过抑制HSP90来调节SASP因子产生与分泌。
最后,由于在许多情况下,细胞衰老是功能失调的端粒激活DDR途径的应激后果,端粒DDR 的抑制可能阻止或减少衰老的产生和维持。最近,反义寡核苷酸(ASOs)对DDR激活的序列特异性抑制及其在培养细胞和小鼠模型中特异性抑制端粒DDR的用途为这种方法提供了支持。在Hutchinson-Gilford早衰综合症(一种加速衰老综合症)的小鼠模型中使用端粒ASO可有效降低DDR激活,衰老标记和SASP诱导水平,改善组织稳态和延长寿命。这种或类似的方法不会使干细胞或祖细胞的储藏退化,反而会促进细胞增殖,这可能为Senolysis提供了另一种方法。
与senomorphics相比,senolytics具有潜在的优势。首先,尽管可能需要重复治疗,但去除衰老细胞方法的优点是可以间歇性地靶向其并且不需要连续施用SASP抑制剂。此外,去除衰老细胞消除了衰老可降低促受损细胞中肿瘤发生的可能性。此外,尽管SASP与衰老相关的组织和器官功能障碍之间有很强的相关性,但尚无直接证据表明SASP 会导致这些衰老相关的缺陷,因为无法通过转基因小鼠模型将SASP与senolysis 分离开。然而,尽管去除敏感细胞的INK-ATTAC转基因小鼠模型没有表现出明显的有害副作用,但是长时间或重复的衰老溶解对生物体最终是否有毒仍有待确定。此外,当器官衰老的细胞负荷高和高龄诱导的衰老情况下,尚不清楚衰老是有害的还是有益的。有人提出p16/SA-β-gal-阳性巨噬细胞亚群的药理学清除作用有助于senolysis的有益作用,但这种细胞类型导致与年龄相关的功能障碍的程度有待进一步研究。最后,新出现的证据表明,在小鼠中senolysis疗法对肝脏和血管周围组织可能具有很深的毒性,这是因为清除了衰老器官中具有结构功能且高表达p16的内皮细胞、脂肪细胞以及巨噬细胞
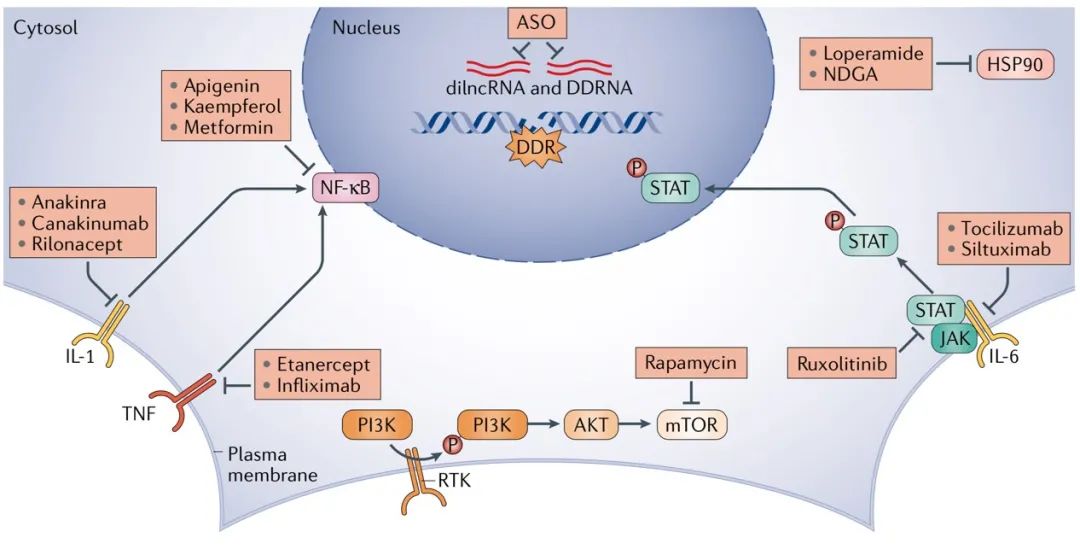
图5 | Senomorphic 干预疗法。作为主动杀死衰老细胞的一种替代方法,senomorphic方法试图主要通过调节衰老相关的分泌表型(SASP)来限制这些细胞的有害影响。至于senolytics,已经确定了不同的方式的共同的节点,这也是干预的最佳靶点。著名的靶标哺乳动物mTOR通路的雷帕霉素被证明可以延长实验小鼠的寿命。雷帕霉素会降低SASP的产生,这可解释了对生命的有益影响。核因子-κB(NF-κB)是产生SASP的关键成分,抑制NF-κB活性会降低细胞促炎的能力。另外,抑制HSP90能够调节SASP的产生。同样,Janus激酶(JAK)/信号转导子和转录激活子(STAT)抑制剂和白介素6(IL-6),IL-1和肿瘤坏死因子(TNF)的阻断。综上所述,这些分子开始阐明可以减弱衰老细胞的促炎信号传导的方式,以期希望减少衰老细胞在组织中积累的后果。 ASO,反义寡核苷酸; DDR,DNA损伤反应; DDRNA,DNA损伤反应RNA; dilncRNA,损伤诱导的长非编码RNA; NDGA,去氢二氢愈创木酸; RTK,受体酪氨酸激酶。
表格1 | Senolytic 和 senomorphic 化合物
化合物
靶点
临床实验进程
Senolytic
达沙替尼
泛受体酪氨酸激酶(包括ephrin B1)
II期(NCT02848131)用于慢性肾脏疾病,II期(NCT04313634)用于骨骼健康,III期(NCT04063124)用于阿尔茨海默病
槲皮素
多种(包括PI3K)
II期(NCT02848131)用于慢性肾脏疾病,II期(NCT04313634)用于骨骼健康,III期(NCT04063124)用于老年痴呆症
非瑟酮
PI3K/AKT/mTOR
III期(NCT04210986)用于膝关节骨关节炎,II期(NCT04313634)用于骨骼健康
ABT-737
BCL-2, BCL-XL和BCL-W(促生存蛋白)
临床前动物模型
ABT-263 (navitoclax)
BCL-2, BCL-XL和BCL-W(促生存蛋白)
III期(NCT00445198),II期(NCT02591095), I期(NCT02520778), II期(NCT02079740)用于各种癌症
A-1331852
BCL-XL(促生存蛋白)
衰老的体外临床前模型
A-1155463
BCL-XL (促生存蛋白)
衰老的体外临床前模型
EF24
BCL-2家族蛋白(促生存蛋白)的蛋白体降解
衰老的体外临床前模型
强心苷(包括oubain and digoxin)
BCL-2, BCL-XL和BCL-W(促生存蛋白)
临床前动物模型
阿奇霉素
自噬,代谢变化
衰老的体外临床前模型
罗红霉素
自噬,代谢变化
衰老的体外临床前模型
Proxifim
p53
临床前动物模型
UBX0101
MDM2和p32
II期(NCT04129944)治疗膝关节骨关节炎
帕比司他
HDAC
Approved for multiple myeloma
格尔德霉素
HSP90
衰老的体外临床前模型
坦螺旋霉素
HSP90
衰老的体外临床前模型
Alevspimycin
(17-DMAG)
HSP90
衰老的体外临床前模型
荜茇酰胺
OXR1
衰老的体外临床前模型
半乳糖偶联纳米颗粒
衰老细胞的溶酶体活性
临床前动物模型
半乳糖改良细胞毒性药物
衰老细胞的溶酶体活性
临床前动物模型
BET蛋白降解器
Bromo结构域
临床前动物模型
Senomorphic
二甲双胍
IKK和NF-κB
被批准用于治疗2型糖尿病
芹黄素
NF-κB p65亚基和IκB
天然黄酮类
山柰酚
NF-κB p65亚基和IκB
天然黄酮类
BAY 11-7082
NF-κB p65亚基和IκB
衰老的体外临床前模型
雷帕霉素
mTOR
被批准用于免疫抑制
RAD001
mTOR
被批准用于结节性硬化症复杂相关疾病
SB203580
p38 MAPK
衰老的体外临床前模型
(5Z)-7-Oxozeaenol
TAK1
衰老的体外临床前模型
Ruxolitinib
JAK
被批准用于移植物抗宿主病
KU-60019
ATM
临床前动物模型
NDGA
HSP90
天然抗氧化剂
洛派丁胺
HSP90
被批准用于治疗腹泻
辛伐他汀
IL-6, IL-8, MCP1
衰老的体外临床前模型
皮质醇
IL-6 分泌
甾类激素
阿那白滞素
IL-1R
被批准用于类风湿性关节炎
康纳单抗
IL-1β
冷吡啉相关周期性综合征
利纳西普
IL-1α and IL-1β
冷吡啉相关周期性综合征
依那西普
TNF
被批准用于治疗自身免疫性疾病
英夫利昔
TNF
被批准用于治疗自身免疫性疾病
托珠单抗
IL-6R
被批准用于治疗自身免疫性疾病
司妥昔单抗
IL-6
被批准用于多中心Castleman病
端粒的反义寡核苷酸
端粒非编码RNA促进DDR
临床前动物模型
“返老还童”疗法细胞衰老之间的相互作用
热量限制已被证明是延长健康期和寿命的最有效策略,并且对从酵母到灵长类动物的各种物种都有效。对于它是否能影响衰老细胞的数量或活性已经有了令人欣喜的研究。据报道,热量限制降低了小鼠和健康人结肠中p16水平以及与细胞衰老相关的基因(包括SASP基因)的转录表达。小鼠的热量限制减少了DDR,并改善了端粒的长度维持。还发现热量限制可降低有丝分裂后神经元中DDR标记和SASP调节剂的水平。热量限制与降低的DDR 信号传导和降低的衰老负担之间的联系可能与培养物中血清增强衰老细胞中的DDR信号传导有关。
在衰老和所谓的端粒综合症的背景下,抑制端粒缩短作为预防和减少细胞衰老被认为是一种可靠的治疗方法。除了使用端粒ASO调节DDR之外,科学家们还在探索其他选择,例如使用天然化合物重新激活内源性端粒酶基因,但功效有限,并且通过性激素进行激活(具有一些明显的临床弊端)。端粒酶编码基因(Tert)的病毒传递已在多种环境下得到了成功的测试。Tert 的全身递送减少了一些衰老标记和与衰老相关的条件,并延长了野生型小鼠的寿命,因此证明端粒DDR 激活在自然衰老中起作用。特发性肺纤维化与端粒缩短和人类细胞衰老的标志物有关。在概括了这些特征的小鼠模型中,已证明传递Tert的腺相关病毒颗粒可降低DDR和衰老标记的水平并改善肺功能。重要的是,最近的研究表明,表达癌基因的小鼠在这种治疗中没有显示出加速的肿瘤发生,从而减轻了人们对端粒酶在受损组织中强迫表达的安全性的担忧,特别对癌症影响。
在过去的几十年中,异体共生(通过外科手术将年幼的小动物和年老的小动物连接起来,确定了年轻血液中存在的系统性因素)可以改善包括肝脏、肌肉、心脏、大脑在内的多个老年器官的功能。在大脑中,通过更简单的将年轻人血浆转移到老年动物中的过程,可以观察到相似的恢复活力。研究还表明,暴露于较年轻的系统环境中可以缓解衰老的端粒酶缺乏症小鼠的年龄相关组织功能障碍。最近,有报道说,年轻小鼠和老年小鼠之间的血液交换导致许多衰老组织中的细胞衰老和SASP 标记物表达显着降低,而同时暴露在年老血液中的年轻小鼠的衰老标记物明显增加。这些观察表明,系统性因素逆转了与衰老相关的一些特征,包括干细胞和祖细胞功能缺陷,慢性炎症和衰老负担,并支持人间断性血液交换可以用作与年龄有关的疾病的治疗方式的假说。与此相符,目前正在对患有急性败血症或肝损伤的患者进行治疗性血浆置换的测试。
挑战与未来方向
虽然我们对体外和体内衰老细胞特征的了解不断增加,但目前仍然存在许多挑战。例如,目前尚不清楚有多少种“衰老表型”。在衰老状态下,在单细胞水平上以及在细胞类型之间,并且在不同的衰老诱导的刺激下,衰老表型可能存在非常高的异质性。细胞衰老是一个随着时间而发展的动态过程,这一新兴观念进一步增加了它的复杂性。一个真正通用的衰老标记物的确立将有助于衰老细胞的分离和表征。此外,鉴定更加详细的标志物以区分不同类型的衰老细胞,将对衰老细胞的表征和起源追溯十分有利。目前,单细胞转录组学方法(包括空间转录组学)是唯一可以充分了解衰老细胞复杂性的方法,它不仅可以检测衰老细胞在细胞分化、衰老等受调控过程中的异同点还可以检测衰老细胞对已经分化的,非增殖细胞的影响的异同点。区分衰老细胞亚型并确定引发每种衰老亚型的原因有助于我们识别对组织功能最有害的衰老细胞的特定子集,并且它们的靶向作用将进一步扩大senolytic和senomorphic方法的益处,同时将有害影响最小化。
另外,我们对体内衰老的生理触发机制了解甚少。端粒功能障碍可能是其中重要的一个,而特定的端粒DDR抑制剂则可以改善各种生理疾病状况。端粒ASO既可以作为研究工具,又可以作为潜在的治疗手段,对端粒损伤引起的细胞衰老具有选择性。
在似乎与DDR无关的条件下,衰老的诱因仍然是难以捉摸的。SASP是所有衰老细胞表型中最重要的一种。然而,随着更多的SASP成分在不同的细胞类型和不同的环境中被鉴定出来,对SASP调控机制的复杂性的认识也在逐渐加深。目前同样需要考虑的是细胞衰老对非衰老细胞的强大旁分泌影响,对其调控机制的了解将阐明其在机体衰老和与细胞衰老诱导的疾病中预期但未证实的作用。
模拟人类疾病的动物模型有助于我们理解衰老细胞对疾病的影响。然而,在senolysis用于患者治疗之前,其安全有效性仍有待证明。研究senolysis对啮齿动物的长期影响的最长年限仅有2-3年,这比它们对人类的长期影响要短得多。因此,使用我们目前的模型和工具无法简单地评估衰老细胞清除的长期毒性作用或负面影响。很明显,在某些情况下,免疫系统具有清除衰老细胞的能力,然而,随着年龄的增长和疾病的发生,清除过程似乎变得不正常,这可能解释了衰老细胞随着年龄的增长而积累的原因。由于免疫系统的细胞成分也会发生衰老,因此确定senotherapies是否会消除这些细胞以及消除衰老的免疫细胞是否有助于或介导senotherapies的效果将是非常重要的。此外,利用免疫系统的内在能力来靶向这些细胞,可能为新的治疗带来希望,如利用工程化的T细胞。总之,细胞衰老显然比最初认为的更为复杂和微妙,对生理学和衰老做出了不同的贡献。重要的是,多年来在这一领域的基础研究已经为现在爆炸性的生物技术以及将知识转化为治疗手段的工业研究奠定了基础。

来源:老顽童说
原标题:《【重磅综述】从机制到治疗——深度解析细胞衰老与个体衰老》
本文为澎湃号作者或机构在澎湃新闻上传并发布,仅代表该作者或机构观点,不代表澎湃新闻的观点或立场,澎湃新闻仅提供信息发布平台。申请澎湃号请用电脑访问http://renzheng.thepaper.cn。



- 报料热线: 021-962866
- 报料邮箱: news@thepaper.cn
互联网新闻信息服务许可证:31120170006
增值电信业务经营许可证:沪B2-2017116
© 2014-2024 上海东方报业有限公司

