铜蓝蛋白和血清铜都偏低,是肝豆状核病变吗?
4 个回答

单纯铜蓝蛋白和血清铜偏低是不能确诊肝豆状核变性的
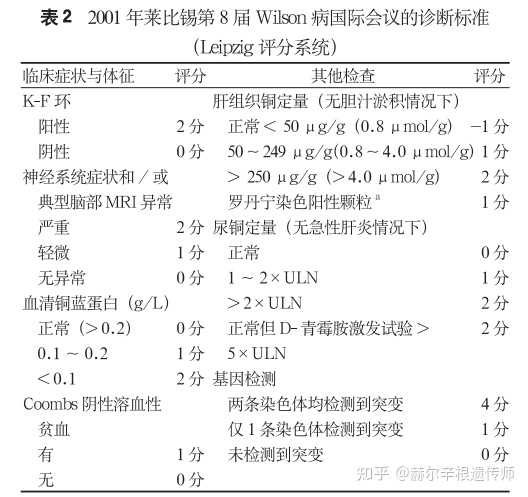
要确诊肝豆状核变性,需要结合各方面的临床表现和实验室检查结果,目前主要通过2001年莱比锡第八届Wilson病国际会议制定的Leipzig评分系统进行诊断。根据各方面的表现进行评分,分值总合≥4可确诊,3分为疑似诊断需进一步检查,≤2基本可排除。

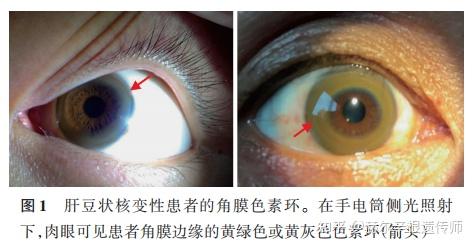
角膜K‐F环:K‐F环为角膜边缘的黄绿色或黄灰色色素环,一般在手电筒侧光照射下肉眼可见,如未见到,需采用眼科裂隙灯检查明确角膜K‐F环。7岁以下患者一般无法检出角膜K‐F环。

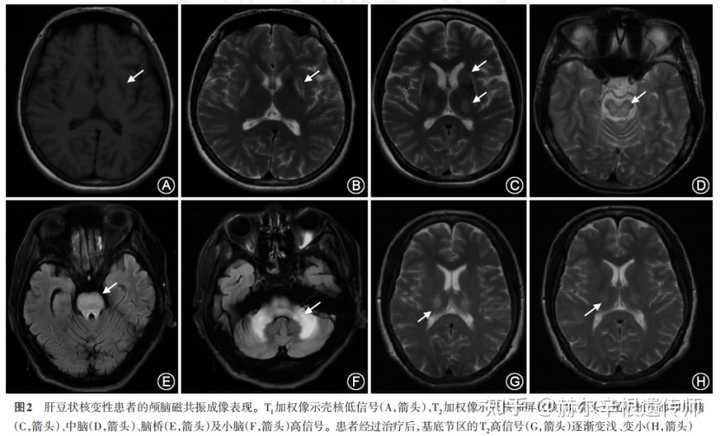
颅脑MRI检查:肝豆状核变性患者的颅脑MRI病灶主要表现为壳核、尾状核头部、丘脑、中脑、脑桥及小脑T1低信号、T2高信号,少数情况下可出现 T1高信号或 T1、T2均低信号。T2加权像时,壳核和丘脑容易出现混杂信号,苍白球容易出现低信号,尾状核等其他部位多为高信号。此外,可有不同程度的脑沟增宽、脑室扩大及额叶皮质软化灶等。T2加权成像上的高信号和低信号可反应肝豆状核变性患者脑部的病理改变过程。MRI病灶可随着治疗逐渐变浅、变小。

血清铜蓝蛋白:检测方法主要为免疫学和酶学方法,因酶学方法复杂且费用昂贵,目前大多数医疗机构采用免疫比浊法。铜蓝蛋白正常为200~500 mg/L,患者一般小于 200 mg/L。Wilson 病患者在妊娠期和接受雌激素治疗时,铜蓝蛋白可能大于200 mg/L。出生后至2岁的婴幼儿,20%以上的 ATP7B基因杂合致病变异携带者,以及慢性肝病、重症肝炎、慢性严重消耗性疾病患者的铜蓝蛋白亦可低于200 mg/L,在临床上需进行鉴别。铜蓝蛋白<80 mg/L是诊断Wilson 病的强烈证据,若铜蓝蛋白<120 mg/L应引起高度重视,需进行ATP7B基因检测明确诊断。
肝铜量:正常值<40~55 μg/g(肝脏干重),Wilson病患者>250 μg/g(肝脏干重)。
24 h尿铜:目前多采用电感耦合等离子体发射光谱法或石墨炉原子吸收光谱法检测尿铜含量。在规范的 24 h 尿液收集及正常肌酐清除率的前提下,正常人 24 h 尿铜<100 μg,Wilson 病患者24 h 尿铜≥100 μg。不明原因肝酶增高的儿童 24 h尿铜≥40 μg应引起高度重视。对于经24h尿铜检测仍不能确诊的儿童,如基础尿铜排泄量铜<100 μg/24h,可进行D-青霉胺激发试验。在24h尿液收集期间,开始时和12h后分别口服500mg,当尿铜排泄量>1600ug/24h,对诊断具有重要意义,此方法对成人无效。
ATP7B基因检测:检测ATP7B基因致病变异对诊断具有指导意义。尽管目前已报道的 ATP7B 基因致病变异多达900余种,但我国肝豆状核变性病患者主要有3个高频致病变异,即 p.R778L 、p.P992L 和p. T935M,占所有致病变异的50%~60%,而10种常见致病变异包括R778L、p.P992L、p.T935M、p.A874V、p.I1148T、p.Q511X、p.N1270S、p.G943D、p.R919G和p.R778Q,可占所有致病变异的67%。因此,临床上高度怀疑WD的患者可先进行ATP7B基因测序。
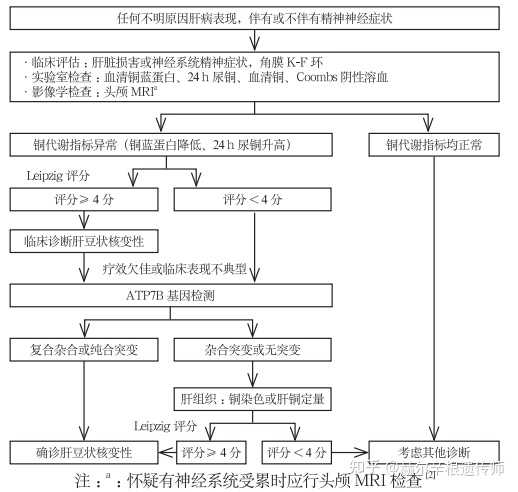
肝豆状核变性诊断流程如下:


铜蓝蛋白和血清铜都偏低,不能简单的断定为肝豆状核病变。
肝豆状核变性(或称Wilson病),是一种常染色体隐性遗传的铜代谢障碍疾病,其致病基因ATP7B编码一种铜转运P型ATP酶,该基因的致病变异导致ATP酶的功能缺陷或丧失,造成胆道排铜障碍,大量铜蓄积于肝、脑、肾、骨关节、角膜等组织和脏器,从而出现铜在各个器官的病理性沉积,最终导致大脑及肝脏等器官损害的病理过程。
Wilson病患者多见于5-35岁起病,但发病年龄不能作为诊断或排除Wilson病的依据,该病的临床表现为肝脏损害、神经精神表现、肾脏损害、骨关节病及角膜色素环(K-F环)等。
Wilson病的诊断标准包括:
1.神经和(或)精神症状。
2.原因不明的肝脏损害。
3.血清铜蓝蛋白降低和(或)24 h尿铜升高。
4.角膜K‐F环阳性。
5.经家系共分离及基因变异致病性分析确定患者的2条染色体均携带ATP7B基因致病变异。
符合(1或2)+(3和4)或(1或2)+5时均可确诊Wilson病。为诊断肝豆状核变性,原先医疗机构会要求进行肝穿刺活组织检查,但随着国内 ATP7B基因检测技术的普及,此项肝铜量检测的重要性已降低,且肝穿刺是有创检查,故国内专家一般不再推荐该项检查。
Wilson病采用早期治疗、终身治疗、终身检测的治疗原则,由于肝豆状核变性可能引起的临床症状多变,故医疗机构往往需根据患者的临床表现采取不同的治疗策略。值得注意的是,一旦怀疑患者罹患Wilson病,应立即开始低铜饮食,有效控制铜蓄积对靶器官的损害。
虽然Wilson病未经治疗通常是致残或致命的,患者病死率在5.0%~6.1%左右,但Wilson病作为少数可治的神经遗传病之一,经过长期规范的治疗,可以大幅延长患者的寿命。在疾病早期,神经症状出现之前进行干预,大部分患者可回归正常的工作和生活。经过治疗症状稳定后可正常结婚和生育,但需注意配偶也应进行ATP7B基因检测,以免再次生育Wilson病患儿。
具体内容可参见我之前发布的案例
成功案例 | 简析重疾险合同约定的疾病标准与医学标准不一致情形下的法律适用——以“肝豆状核变性”判断为例 - 陈禹彦商事律师的文章 - 知乎