
案例分享 | 基因检测辅助诊断遗传病——遗传性淀粉样变性

案例分享:遗传性淀粉样变性
临床信息:女,50岁,全身肌营养不良;视力:R0.12,L0.15;左眼外转受限,不过中线;双眼上睑下垂,双眼角膜营养不良;面部:无表情,面瘫;舌萎缩;喝水呛、吞咽困难;患者父母为近亲结婚。
检测项目:送检基信源科全外显子组测序
检出1个匹配受检者临床表型的可能致病性的基因变异:GSN基因c.G487A:p.D163N纯合变异。


基因关联疾病表型简介:


ACMG致病性评级:
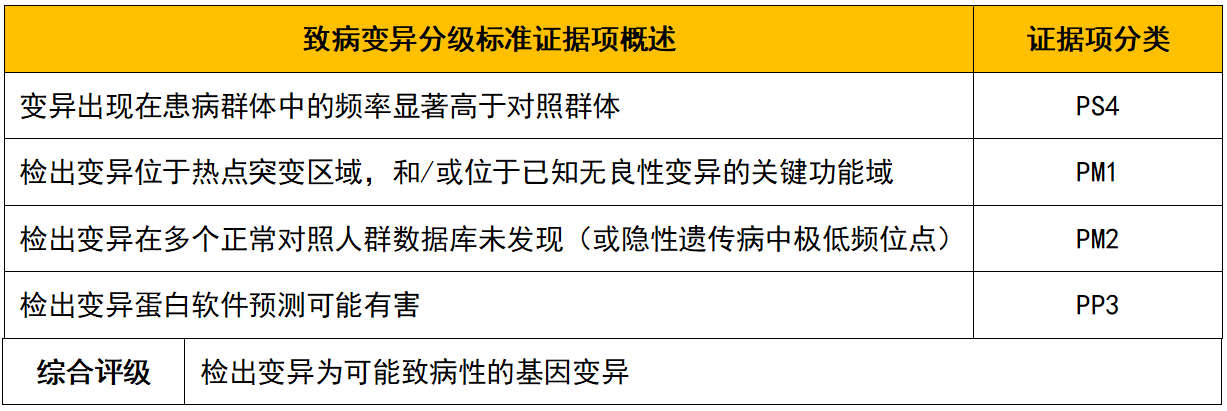

变异位点在数据库收录或文献报道情况:
检出变异已被ClinVar数据库收录,临床意义注释为致病性,信息参见:https://www.ncbi.nlm.nih.gov/clinvar/variation/16180/,已有文献报道在淀粉样变性芬兰型表型患者样本中检出[1]。
遗传咨询建议:
①建议结合实际,对受检者亲属如父母、儿子进行该变异位点的一代测序验证,并随诊临床表型,必要时行肾功能、皮肤等相关检查,进一步明确检出变异临床意义。
②受检者检出该纯合变异,有100%概率将上述两个变异之一遗传给子代,为杂合子携带者,结合随诊行遗传咨询。
一、淀粉样变性概述
淀粉样变性是由于蛋白的错误折叠形成不溶性纤维沉积(淀粉样纤维)于细胞外基质,最终导致受累器官功能障碍的一组互不相同的疾病。这些蛋白可以在机体局部聚集,引起相对较少的症状,也可以在机体多个器官沉积而产生严重的多器官功能衰竭。
淀粉样蛋白沉积物可以被苏木精和伊红染成粉红色,其碳水化合物组分可以被高碘酸-希夫或阿尔辛染成蓝色,但更具有特征性的是刚果红染色后在偏振光显微镜下可以观察到苹果绿双折射。肉眼观察,受累器官呈现蜡状改变。
二、病因及分类
淀粉样变性的病因与分型非常复杂多样,主要原因是突变的蛋白更易发生错误折叠与聚集(淀粉样蛋白)。一方面是变性的淀粉样蛋白产生过多。另一方面,变性蛋白的清除障碍也是淀粉样变性病情进展的重要机制。淀粉样沉积物无代谢活性,但是会干扰器官组织的结构和功能。然而,淀粉样蛋白的一些前纤维低聚物具有直接的细胞毒性,这也是发病机制的重要组成部分。
淀粉样蛋白的类型决定了淀粉样变的亚型及疾病的临床表现,尽管不同亚型的临床表现可能会重叠。
1. 系统性淀粉样变性时, 循环淀粉样蛋白可以在全身多个脏器沉积。主要分类:
① AL(原发性淀粉样变性):最常见,系获得性免疫球蛋白轻链过表达所致,主要见于单克隆浆细胞病或其他B淋巴细胞增殖疾病。有些原发性淀粉样变性病患者同时患有多发性骨髓瘤。淀粉沉积部位通常包括皮肤、神经、心脏、胃肠道(包括舌)、肾脏、肝脏、脾脏和血管。
② AF(家族性淀粉样变性):因为遗传致某基因突变,在绝大多数病例中,它与转甲状腺素(TTR)基因突变有关,在特殊情况下与凝溶胶蛋白基因(GSN)、β2微球蛋白或APOA1有关。
③ SSA(老年性系统性淀粉样变性):由野生型的转甲状腺素(TTR)错误折叠和聚集引起(因此也称为ATTRwt),主要是在心脏。
④ AA(继发性淀粉样变性):可以继发于感染性(如结核病、支气管扩张症、骨髓炎和麻风病)、炎症性疾病(如类风湿性关节炎、克罗恩病、遗传性周期性发热综合征如家族性地中海热)和恶性病变,其发生是由于急性期反应物-血清淀粉样蛋白A(SAA)的降解。AA好发于脾脏、肝脏、肾脏、肾上腺和淋巴结。心脏及周围或自主神经很少受累。
⑤ β2-微球蛋白淀粉样变性:β2微球蛋白(Aβ2)堆积亦可引起淀粉样变。常发生于长期血液透析患者,随着先进的高流量透析膜逐步推广,发生率已趋于降低。
2. 局部的淀粉样变性:脑外的局限性淀粉样变性一般是源于免疫球蛋白轻链的堆积,而脑部局限性淀粉样变性由Aβ蛋白沉积导致。常受累部位包括中枢神经系统(如阿尔茨海默病或脑血管淀粉样病),皮肤,上或下呼吸道,膀胱和其他部位。
三、诊断
① 活检:
发现受累器官内是否有纤维状沉积物。腹部皮下脂肪活检(AL或ATTR筛查阳性率约80%,但SSA的筛查阳性率仅50%)。或对受累器官进行活检如唾液腺、消化道、或肾脏活检。病理解剖学实验室采用特定的颜色和技术来确定组织中淀粉样变性的存在(刚果红染色)并帮助鉴定类型(免疫组织化学)。
② 淀粉蛋白分型:
如疑诊AL,应测定血清游离的免疫球蛋白轻链,免疫固定电泳法定量测定血、尿中的单克隆轻链。同时应行骨髓活检,应用流式细胞术或免疫组织化学法测定浆细胞克隆数。针对AF可行基因测序,通过质谱法进行生化鉴定等。
③ 器官受累的判定:
如有症状提示器官受累,应首先实施针对肾、肝、胃肠道、神经系统、心功能的无创检查。
四、家族性淀粉样变性
家族性淀粉样变性(AF)是一种罕见的常染色体显性遗传性疾病。症状是由淀粉样沉积引起的,主要发生在周围和营养神经系统,但也可能发生在所有器官(心脏、眼睛、肾脏)。在绝大多数病例中,它与转甲状腺素蛋白、载脂蛋白A-1、溶菌酶、纤维蛋白原、凝溶蛋白、A β蛋白和半胱氨酸蛋白酶抑制蛋白C等有关(下表为已鉴定致病基因的AF亚型)。近期研究发现,血清蛋白白细胞趋化因子2(LECT2)与某种家族性淀粉样变性有关。然而,这一亚型的遗传学机制尚不清楚。


芬兰型(FAF)家族性淀粉样变性,又称遗传性凝溶胶蛋白淀粉样变性、AGel淀粉样变性,它是由Meretoja在芬兰于1969年首次描述,是一种常染色体显性遗传的缓慢进行性的系统性淀粉样变性,其特征是由于凝溶胶蛋白淀粉样纤维在眼睛,神经系统和皮肤中的沉积。临床表现包括晶格角膜营养不良,颅神经病变,特别是影响面神经,延髓体征,皮肤松弛,以及较少见的周围神经病变和肾衰竭。
1. 症状
症状在同一个家庭内以及不同家庭之间可能有所不同。最初的症状通常在20岁以后出现,但可能会在生活中更晚出现。该疾病通常发展缓慢,并且随着年龄的增长,症状变得更加麻烦。
首发表现是眼睛,如干燥和刺激。检查角膜时,会看到II型晶格角膜营养不良,这会损害视力,并最终导致严重的视力障碍或失明。青光眼和白内障最终可能也会发生。
由于颅神经损伤,面部瘫痪发生在40岁左右。麻痹症是从顶部开始,并逐渐扩散到面部。吞咽困难和构音障碍也可能发生。
同时(40岁时),皮肤增厚,下垂,变薄变脆,形成大的皱纹,尤其是头皮,前额,眼睑和下唇。汗液的分泌能力下降,使皮肤干燥并导致瘙痒。
发生由感觉神经损伤(内耳或听神经的损伤)引起的听力障碍。
感觉周围神经病变主要表现为远端麻木和感觉异常,震颤感和肌腱反射丧失,一些人晚期由于脊髓后部的变性和在最靠近脊髓的神经部分周围区域中强烈的淀粉样蛋白积累而导致步态共济失调和手笨拙。轻微的自主神经功能障碍和腕管综合征也会发生。
在疾病的后期,通常纯合突变的人会有更严重表型,如淀粉样变性肾小球病、肾病综合症、蛋白质尿,还有一些心脏受累。
2. 原因
是9号染色体(9q33.2)上GSN基因发生突变。凝溶胶蛋白是一种主要的肌动蛋白调节蛋白,涉及多种生物过程,也涉及神经系统,例如轴突运输,髓鞘形成,神经突起生长和神经保护作用。
形成凝溶胶蛋白时,会产生两种蛋白质变体:一种在细胞内部,另一种在血浆中分泌并发现(血浆凝溶胶蛋白)。突变影响血浆凝胶维持其构象的能力。这导致血浆凝溶胶蛋白分解成较小的碎片,在血管壁和细胞膜中,尤其是在眼睛和颅神经中,形成淀粉样蛋白的积累。在皮肤和血管壁中,堆积物也位于弹性蛋白纤维周围,然后分解。
3. 治疗/支持
目前尚无凝溶胶蛋白淀粉样变性的治疗方法,重点是缓解症状并补偿功能障碍。
对于干眼症,用人工泪液。检查眼压,因为可能会发生青光眼。用眼药水或手术治疗青光眼可以降低眼内压力,并防止进一步损害视神经。对于角膜营养不良,穿透性角膜移植术可能是有益的。
皮肤局部治疗,以减轻瘙痒和干燥。
用尿液和血液样本检查肾功能。重患有严重肾衰竭的人需要透析和/或肾脏移植。
心电图和超声监测心脏功能。
参考文献:
[1] Steiner R D, Evans J P, Paunio T, et al. Asp187Asn mutation of gelsolin in an American kindred with familial amyloidosis, Finnish type (FAP IV)[J]. Human genetics, 1995, 95(3): 327-330.[2]《默克家庭诊疗手册》—淀粉样变性https://www.socialstyrelsen.se/stod-i-arbetet/ovanliga-diagnoser/gelsolinamyloidos/
