
颅缝早闭专家共识(3)--综合征型颅缝早闭的分类、临床表现

以下内容节选自中华小儿外科杂志2021年9月第42卷第9期《儿童颅缝早闭症诊治专家共识》
综合征型颅缝早闭患儿除颅缝早闭所致颅骨畸形外,常伴有严重的面部发育畸形和四肢末端畸形,多有颅内压增高和脑发育落后,基因检查常可找到突变基因。综合征型颅缝早闭临床表现复杂多样,文献报道的综合征型颅缝早闭有一百余种,常见的有以下几种。
Crouzon综合征 最常见的综合征型颅缝早闭,发病率约为1/25000,典型临床表现为双侧冠状缝早闭,有时也表现为额缝或者矢状缝早闭,根据闭合的颅缝不同,其头形既可以呈短头畸形,亦可为舟状头畸形;由于眼眶周围骨骼过早闭合,眼眶较浅,表现为眼球突出,眼距过宽。眼球突出严重者眼睛无法闭合。同时由于上颌骨发育不全,下颌骨前突,中面部内陷,压迫阻塞呼吸道可导致阻塞性睡眠呼吸暂停,表现为睡眠时鼾声大作并伴有憋气[13-14]。黑棘皮症型的Crouzon综合征皮肤改变主要位于颈部和关节屈曲部位的皮肤。临床表现为皮肤增厚和色素加深。另外,9%-26%的患儿有脑积水,部分患儿还有主动脉狭窄和动脉导管未闭;本综合征具有家族性,亦为染色体显性遗传,发病率约为1/25000,绝大多数患儿是由于10号染色体FGF2基因突变所致[15],另外FGFR3基因突变与Crouzon综合征发生也有关,FGFR3蛋白391位丙氨酸被谷氨酸取代可导致Crouzon综合征伴黑棘皮病[16-17],根据临床表现、辅助检查结合基因检查结果,诊断并不困难。
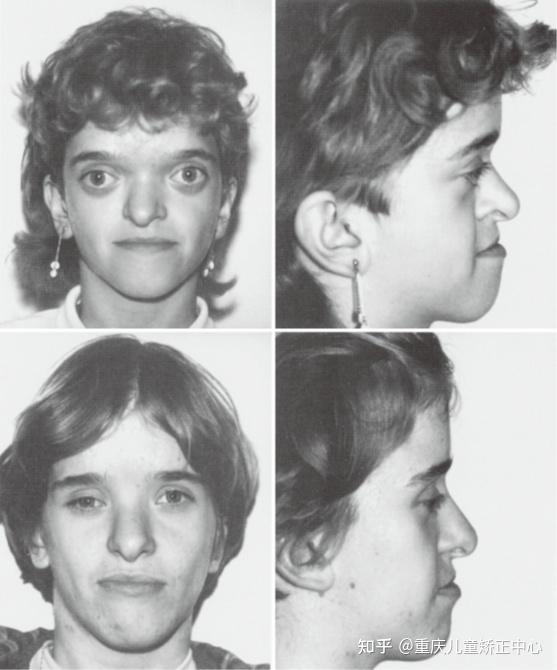

Apert综合征,又称尖头并指畸形,临床表现为双侧冠状缝早闭,身材矮小,面中部发育不全,眼距过宽,突眼以及并指(趾)畸形。由于双侧冠状缝早闭,额缝代偿性增宽,前囟也异常增大,前额部明显扁平和后倾,头颅畸形多为尖头和短头,颜面部畸形主要为严重上颌骨发育不良。临床表现为反颌、鹰嘴鼻、颜面宽阔、眼距增宽和严重突眼,部分患儿可有严重腭裂。由于上颌骨发育不良,可引起患儿呼吸道狭窄,导致代偿性张口呼吸;骨性融合的并指(趾)畸形可以表现为第二、三、四指合并成为一个形似团块的巨大中指,拇指(趾)关节呈骨性融合[14,18];椎体融合多见于第五颈椎和第六颈椎。Apert综合征可有不同程度的智力发育迟缓,45%左右患儿有颅内压升高,40%-90% 的患儿有心室扩大;Apert尖头并指综合征为常染色体显性遗传性疾病,最常见的两个致病突变位点位于10号染色体的FGFR2基因上[19]。
Pfeiffer综合征,临床常有冠状缝早闭和矢状缝早闭,严重者呈三叶草状头颅畸形,表现为额顶与颞骨交界处狭窄,双颞部突出,与额顶突出形成三叶草形状[20];同时可伴有上颌骨发育不良,面中部塌陷,眼距增宽,眼球突出,上呼吸道狭窄,气道梗阻,常伴有脑积水,小脑扁桃体下疝,突变基因常为FGFR1和FGFR2。
Muenke综合征,临床表现为单侧冠状缝早闭,有的表现为双侧冠状缝早闭,有研究表明,约12.5%的患儿可无颅缝早闭,而表现为巨脑症,患儿有不同程度的面中部发育不全、眼距增宽、耳下垂,并常见中枢性听力障碍、斜视,无并指(趾)、多指(趾)现象,但可有腕骨和跗骨融合,约1/3的患儿出现生长发育迟缓和智力障碍[21];该综合征为常染色体显性遗传性疾病,发病率约为1/10000,所有患儿都有FGFR3基因突变[22],结合临床表现及基因检查结果可确诊。
Carpenter综合征,患儿一般首先出现人字缝和矢状缝早闭,这种早闭逐渐累积冠状缝进而造成综合征型颅缝早闭症,畸形严重者可出现三叶草状头颅;眼距可增大或者缩小,合并突眼、眼眶变浅、内眦赘皮折叠[23],其他眼部畸形有视盘水肿、角膜浑浊、小角膜和眼睑下垂,耳部发育不全,鼻背低平,上颌骨弓背抬高,手指短胖弯曲,并指(趾)为指(趾)间软组织粘连成蹼,非骨性融合,常累及第3、4指(趾),同时伴有多指(趾)畸形,智力发育受限。该综合征的致病基因为6号染色体上的RAB23基因,是一种常染色体隐形遗传病[24]。
Saethre-Chotzen综合征,临床表现多样、诊断困难,这类综合征可有一条或多条骨缝的早闭,因早闭的骨缝不同,其颅面部畸形也各不相同,多数为双侧的冠状缝早闭,表现为额部平坦、发际线低、鹰嘴鼻、眼距增宽、斜视、上睑下垂和耳朵畸形,单侧冠状缝早闭时可导致面部不对称[25],该综合征的耳朵畸形表现为小而圆的耳廓,上颌骨畸形特点为上腭裂或者悬雍垂裂;鼻背部扁平,额鼻角呈直线样畸形;该综合征四肢短小而拇指(趾)巨大,巨大的拇趾呈外翻畸形,并指(趾)呈软组织蹼,非骨性融合,并指常发生于第二、三或第二、三、四指间,并趾常发生于第二、三或第四、五趾间,部分患儿有贯通掌,隐睾。
有研究表明42%的患儿伴有顽固性颅内高压;Saethre-Chotzen综合征是一种罕见的以颅缝早闭为特点的先天性颅面部畸形,是一种常染色体显性遗传病,其发病率在1/50000至1/25000之间,基因研究表明位于7号染色体的TWIST1基因的碱基突变、缺失或者移位会导致Saethre-Chotzen综合征[26]。
参考文献:
13. 许震宇,鲍南,张臻,等. Crouzon综合征的临床和遗传学研究[J]. 中华神经外科杂志,2014,30(6):592-595. DOI:10.3760/cma.j.issn.1001-2346.2014.06.016
14. Derderian C, Seaward J. Syndromic craniosynostosis.[J] Semin Plast Surg. 2012 May;26(2):64-75. doi: 10.1055/s-0032-1320064.
15, W Reardon, R M Winter, P Rutland, et al. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome[J]. Nat Genet. 1994 Sep;8(1):98-103. doi: 10.1038/ng0994-98.
16. D N Schweitzer, J M Graham Jr, R S Lachman, et al. Subtle radiographic findings of achondroplasia in patients with Crouzon syndrome with acanthosis nigricans due to an Ala391Glu substitution in FGFR3[J]. Am J Med Genet. 2001 Jan 1;98(1):75-91. DOI:10.1002/1096-8628(20010101)98:1<75::AID-AJMG1010>3.0.CO;2-6.
17. Arnaud-López L, Fragoso R, Mantilla-Capacho J, et al. Crouzon with acanthosis nigricans. Further delineation of the syndrome[J]. Clin Genet. 2007 Nov;72(5):405-410. doi: 10.1111/j.1399-0004.2007.00884.x.
18. Cohen MM Jr, Kreiborg S.. A clinical study of the craniofacial features in Apert syndrome[J]. Int J Oral Maxillofac Surg. 1996 Feb;25(1):45-53. doi: 10.1016/s0901-5027(96)80011-7.
19. A O Wilkie , S F Slaney, M Oldridge, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome[J]. Nat Genet. 1995 Feb;9(2):165-172. doi: 21.1038/ng0295-165.
20. M M Cohen Jr. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet[J]. 1993 Feb 1;45(3):300-307. doi: 10.1002/ajmg.1320450305.
21. M Muenke , K W Gripp, D M McDonald-McGinn, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome[J]. Am J Hum Genet. 1997 Mar;60(3):555-564.
22. Paul Kruszka , Yonit A Addissie , Colin M P Yarnell, et al. Muenke syndrome: An international multicenter natural history study[J]. Am J Med Genet A. 2016 Apr;170A(4):918-929. doi: 10.1002/ajmg.a.37528.
23. D M Cohen , J G Green, J Miller, et al. Acrocephalopolysyndactyly type II--Carpenter syndrome: clinical spectrum and an attempt at unification with Goodman and Summit syndromes[J]. Am J Med Genet. 1987 Oct;28(2):311-324. doi: 10.1002/ajmg.1320280208.
24. Jenkins D,Seelow D,Jehee FS,et a1.RAB23 mutations in carpenter syndrome imply an unexpected role for hedgehog signaling in cranial—suture development and obesity[J]. Am J Hum Genet. 2007 Jun;80(6):1162-1170. doi: 10.1086/518047.
25. O A Pantke, M M Cohen Jr, C J Witkop Jr, et al. The Saethre-Chotzen syndrome[J]. Birth Defects Orig Artic Ser. 1975;11(2):190-225.
26. Emily R Gallagher, Chootima Ratisoontorn, Michael L Cunningham, et al. Saethre-Chotzen Syndrome[J]. Initial Posting: May 16, 2003.
