
创造奇迹的神药——GLP-1/GIP双受体激动剂替尔泊肽结构-活性浅析(二)

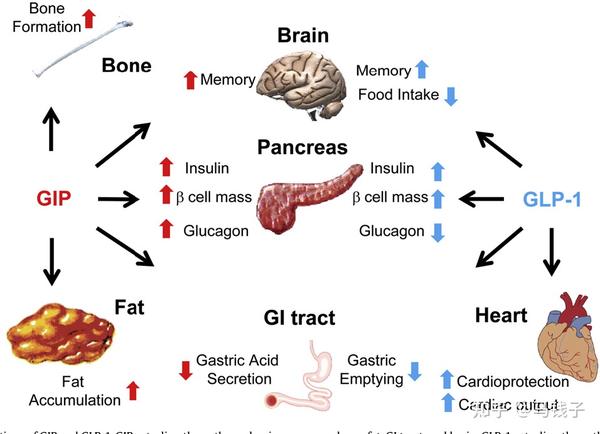
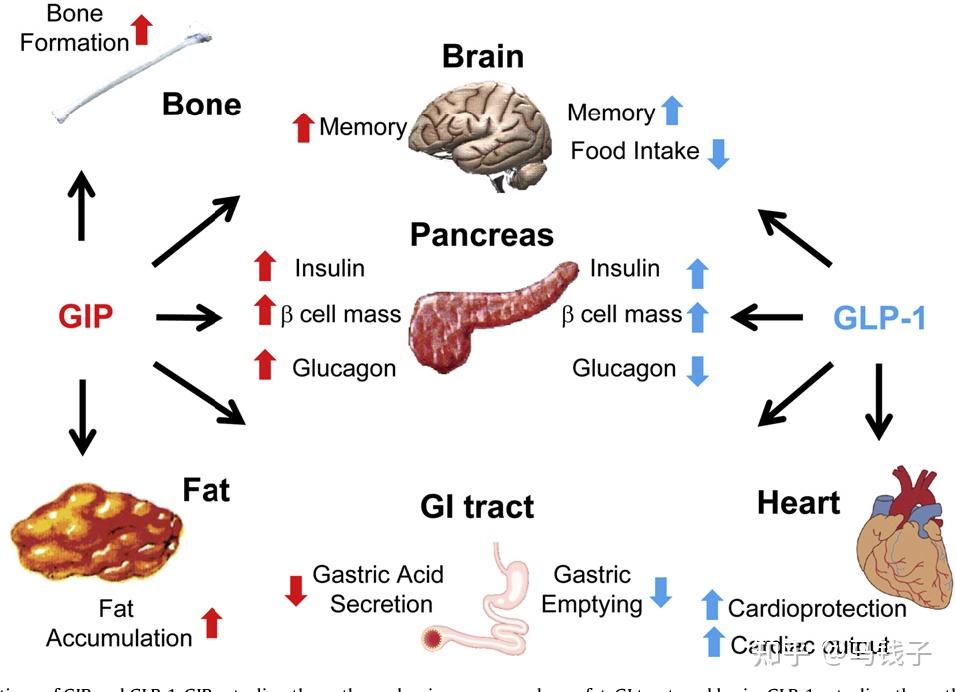
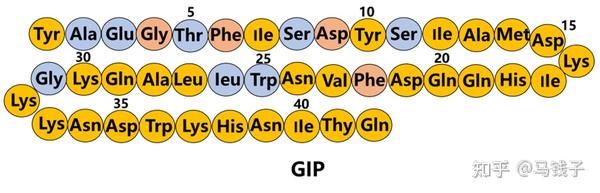
GIP是第一个被发现的肠促胰素,目前已知的肠促胰素也就是GIP和GLP-1。在正常的人体中肠促胰素效应(进餐以后)刺激的胰岛素分泌,约占整体的胰岛素分泌总量70%, GIP的在整个肠促胰素效应中占到了2/3,比重远高于GLP-1。
GIP是一个单链的肽类激素,它由42个氨基酸残基组成的一个多肽分子,它是由小肠中的十二指肠、空肠的肠黏膜的K细胞分泌的。主要的调节机制就是在我们摄食以后,营养物质对小肠刺激,小肠中的内分泌细胞(K细胞)分泌GIP,GIP分泌释放入血以后,几分钟内GIP就快速地升高,达到高峰,但是它降解速度也很快,最主要的是它会被我们体内无处不在的降解酶——二肽基肽酶Ⅳ(DPP-4)降解。DPP-4降解GIP跟GLP-1一样,都从N端的第二位和第三位之间的肽链剪切掉N端的两个氨基酸,就成为没有活性/不具备刺激胰岛素分泌活性的一类降解产物。在正常人,GIP的半衰期大约7分钟,2型糖尿病(T2DM)患者甚至更短,因为T2DM患者的DPP-4酶的表达水平增加,活性也增加,所以GIP的半衰期就缩短到5分钟。而被DPP-4酶降解以后的GIP变成3-42片段,没有肠促胰素效应,不再刺激胰岛素分泌。尽管它没有刺激胰岛素分泌的生物活性,但它保留跟GIP受体(GIPR)结合的作用,因此它等同于内源性GIPR的拮抗剂。
GLP-1受体主要表达在β细胞,α细胞并没有表达,而跟它不一样的GIPR在α和β细胞都有表达。一些基础的研究已经证实肠促胰素在肠道摄入营养物质以后分泌的肠促胰素包括GIP。GIP能够把我们营养素摄入的这种信号分别传递给β细胞、α细胞。所以GIP能够直接调控机体的代谢[19]。且GIP对于α细胞跟β细胞调节作用的比例,跟摄入的营养物质的成分有关系:如果我们摄入的营养物质是以碳水化合物为主,GIP会起到直接的作用,即被β细胞所感知*。在血糖浓度升高与GIP共同的协同下(GIP作用于GIPR),提高β细胞内环磷酸腺苷(cAMP)的水平,可以呈葡萄糖依赖性地促进β细胞的胰岛素分泌。此外GIP还可以上调胰岛素的生物合成,并促进β细胞的存活。其中包括促进β细胞的增殖。
GIP跟GLP-1不太一样,除了对β细胞的调控以外,对α细胞也有直接调控作用。而GLP-1对α细胞的调控,绝大多数学者认为是间接,GLP-1可能通过β细胞分泌的胰岛素,或者通过δ细胞分泌的生长抑素来间接调控α细胞的功能。而GIP不同,它可以直接对α细胞进行生物学调控,原因是α细胞有明确的GIPR的表达*。
GIP其实它在分泌胰高糖素的α细胞中,GIPR是有明确的表达的,跟GLP-1不一样,GIP对胰高糖素的调节是非常精细化的调节,取决于机体不同的健康状态,也取决于血糖的水平
研究显示,在健康受试者中GIP可以通过葡萄糖依赖的方式来调节胰高糖素分泌。具体表现在血糖特别高的状态下,GIP就不再明显地刺激胰高糖素的分泌,而GIP在高血糖的时候,对β细胞能够促进胰岛素的分泌和C肽释放。相反,在空腹或者低血糖状态下,GIP就能促进α细胞的胰高糖素的分泌。
在正常人中的肠促胰素,尽管GIP跟GLP-1它们都是进餐以后分泌,这两者有一个相互协调和相互作用,。GIP和GLP-1两者联合,或能让肠促胰素效应达到最大化。
不幸的是,GIP的可成药性很低,在T2DM患者中,达到超生理GIP浓度的输注未能引发显著的胰岛素分泌反应;因此,GIP输注不能快速使T2DM患者的血糖水平正常化。这种迟钝的反应可能是由高水平的循环葡萄糖下调GIP受体(GIPR)引起的。然而,大量数据表明,降低循环葡萄糖水平的药物可以在很大程度上克服GIP耐药性,为考虑将GIP作为降血糖疗法(如GLP-1.6)的补充铺平了道路。此外,GIPR信号传导阻断呕吐并减轻GLP-1受体(GLP-1R)激活的其他负面副作用。
故基于上述研究基础,现多家制药企业开发研究GIP/ GLP-1双受体激动剂,如替尔泊肽。
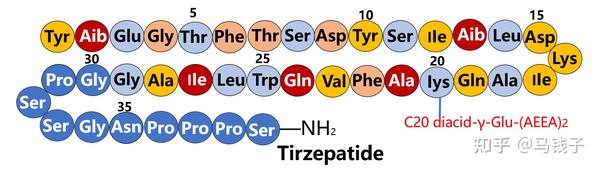
替尔泊肽tirzepatide是礼来公司研发并成功上市的一种单分子双功能肽,可同时激活GLP-1R和GIPR。替尔泊肽由39个氨基酸组成,在C端被酰胺化,通过连接到Lys20的间隔区结合C20脂肪二酸部分,半衰期为116.7小时。替尔泊肽与GLP-1R的亲和力高于GLP-1,其cAMP激活活性显著较低。替尔泊肽与GIPR的亲和力明显低于GIP,其cAMP活性与GIP的活性大致相等。
为了获得双重活性,替尔泊肽不仅融合了主要来自GLP-1和GIP的氨基酸残基,还使用了一些独特的氨基酸残基。在下文中,根据现有的报告来分析替尔泊肽是如何构建的。
通过对替尔泊肽氨基酸结构分析,其多肽构建主要来自GLP-1、GIP、艾塞那肽和司美格鲁肽,少数残留是独特的。
替尔泊肽与GLP-1受体
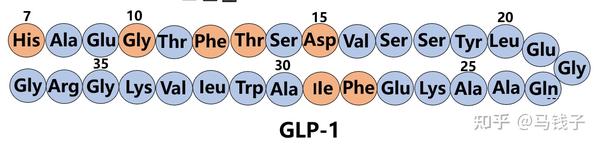


内源性GLP-1的早期结构-活性研究揭示了与效力和酶降解相关的关键序列结构域,研究表明,N-末端残基His7、Gly10、Phe12、 Thr13和Asp15对与GLP-1受体的相互作用至关重要,Phe28和Ile29可能对肽的二级结构以及因此对受体识别的构象更重要,而不是参与受体相互作用。故替尔泊肽构建时,保留了Gly4(Gly10)、Phe6(Phe12)、 Thr7(Thr13)和Asp9(Asp15)、Phe22(Phe28),以保证对GLP-1受体活性。
替尔泊肽与GIP受体活性

Tyr1:
替尔泊肽采用GIP中Tyr1可能牺牲其GLP-1活性,同时支持其GIP活性。
Aib2:
GLP-1(7-37)Ala8和GIP-Ala2是DPP-4切割位点,故许多研究的方向就是通过取代或修饰前三个残基来减少这种降解。,使用Aib2可能仅对GLP-1和GIP活性产生最小影响。尽管Gly2在一定程度上影响GLP-1活性,但总体而言,比如艾塞那肽显示出比天然GLP-1更好的活性。因此,如果生物合成双激动剂,Gly2可能是可行的。此外,由于Ala2的不稳定性是由DPP-4的降解引起的,Ala2可能不需要改变,这会产生空间位阻以阻止酶接近。
Thr7:
GLP-1和GIP是部分同源的,将GLP-1中的Thr7结合到替尔帕肽中,取代GIP中IIe7保留了GLP-1活性,可能会略微降低其GIP活性。
Tyr10、Ile12、Asp 15、Lys16、IIe17、Gln19、Val23、Ala28:
替尔泊肽保留GIP的高活性氨基酸残基。艾塞那肽(全GLP-1R激动剂)的位置10、12、13和14与GLP-1的位置不同,表明这些残基对GLP-1不重要。GLP-1第16、17、18和20位突变为Ala显示出较小的影响。GIP第10和11位的Ala取代对促胰岛素活性几乎没有负面影响,而第12和14位的突变具有更大的负面影响。Ile12在GIPR 15的激活中起关键作用。Tyr10和Ile12也用于替尔泊肽中。在GLP-1(7-37)NH2中,当用Tyr16取代Val16时,亲和力和活性都略有提高。因此,在替尔帕肽可能略微降低GLP1活性,但增强了GIP活性。
Aib13
GLP-1、GIP、GCG(胰高血糖素)和GLP-2具有高度同源性,因此应认为所设计的GLP-1/GIP双重激动剂应避开GCGR和GLP-2R的激活。在实验中,用Aib取代13位使激动剂对GCGR无活性,并轻微改变GLP-1和GIP活性。用Ala取代GLP-1(7-37)Tyr19显著降低了亲和力和活性。
在GIP中,用Aib取代Ala13对活性产生影响的可能性很小。替尔帕肽的Aib13似乎降低了其GLP-1活性而不影响其GIP活性,并破坏了其GCG活性,,但这一假设缺乏支持性证据。然而,Aib13是不必要的,因为其他替代物可以发挥同样的作用。此外,考虑到艾塞那肽中的Gln13,GLP-1可能通过氢键与GLP-1R相互作用。用Tyr、Gln、Thr、Ser和Lys也可以在这个位置上值得尝试。
Ala18:
GIP His18突变为Ala增强了其促胰岛素作用。通过用丙氨酸取代GCG中的Arg18同时提高了GLP-1和GIP的活性。使用来自GLP-1的Ala18可能增加GIP活性机率。
Lys20-X:
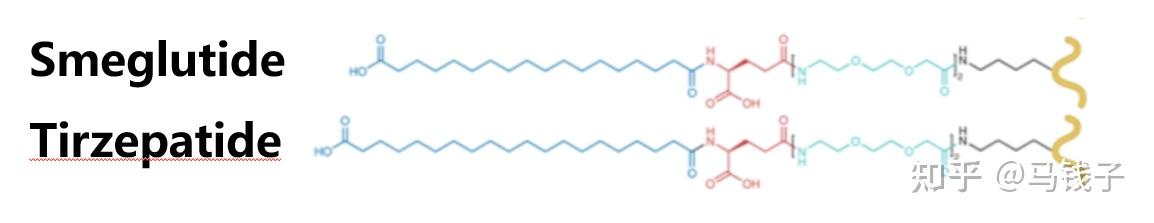

用Ala取代GLP-1 Lys26会导致亲和力和活性略有下降。在semaglutide中,脂肪酸链连接在该位点,亲和力和活性增强。GLP-1 Lys26可能与受体形成极性相互作用,这可能被艾塞那肽中的Arg20增强,但不会产生空间位阻。GIP类似,因为GIP-Gln20似乎与GIPR-Asn120形成氢键,并不受阻碍地暴露在细胞外基质中。与替尔泊肽中的Lys20连接的脂肪酸链,与司美格鲁肽结构相近,只是中间多了两个碳链,连接处与末端一致,研究结果表明可以提高GLP-1活性,而GIP活性不受影响,更为重要的时候延长替尔泊肽吸收和代谢。
Ala21:
GLP-1 Glu27突变为Ala不改变亲和力,但略微提高活性。在实验中,GIP的Asp21被Ala取代;GLP-1活性变化很小,但在GIP活性中观察到轻微下降。因此,GLP-1的Glu27不是与GLP-1R相互作用的关键点。没有GIP(31-42)尾,选择Ala21可能会增加GLP-1活性,尽管它会略微降低GIP活性。
Val23, Ile27:
GLP-1(7-37)Phe28、Trp31和Leu32与GIP的22、25和26位对称。GLP-1、GIP和艾塞那肽在这里似乎具有相同的模式:Phe22居中与受体特异性相互作用,位置23、25和26的相邻疏水残基用于形成疏水区。更远处的位置24的残基可以与受体极性相互作用,并且可能有利于定位疏水区域。GLP-1 Ile29和Val33具有与GIP Val23和Leu27类似的性质。GLP-1 Phe28突变为Ala导致其失去几乎所有的亲和力,并且Ile29突变显著降低亲和力,而Trp31、Leu32和Val33突变几乎没有影响,表明它们可能只是次要的。替尔泊肽中使用的Val23和Ile27可能对GLP-1或GIP活性几乎没有影响。因为它们是同一种残基,并且在这些位点中未检测到特异性相互作用。
Gln24:
用Gln取代GLP-1 Ala30降低了亲和力,但增加了活性。GIP Asn24与Gln相似;因此,替尔泊肽中采用的Gln24可以提高GLP-1活性并支持GIP活性。
Ile27:
从GLP-1的33–35位,每次用Ala取代,IC50增加约五倍,但EC50略有降低。尽管这些取代降低了亲和力,但配体是用来“纠正”结构的。将位置30和34与内酰胺桥连接同时提高亲和力和活性。结构“修正”可能意味着形成更稳定的α-螺旋。用Ala取代GLP-1 Arg36明显阻碍了亲和力和活性。如果GLP-1的Arg36被切除,亲和力和活性都会降低10倍,这表明它对相互作用至关重要。此外,Gly35、Arg36和Gly37可能有助于GLP-1的选择性。
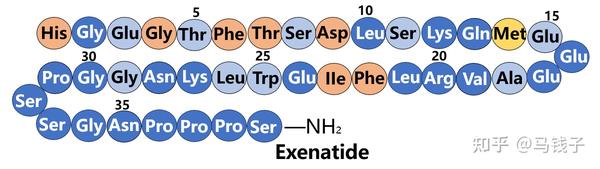

与GLP-1相比,艾塞那肽在C末端有9个额外的残基,它们构成了所谓的“Trp笼”结构,折叠并保护Trp免受暴露。随着C末端的截短,艾塞那肽暴露于NEP 24.11降解。此外,将GLP-1 C-末端连接到艾塞那肽(31-39)上可以防止DPP-4降解。随着C末端的减少,艾塞那肽的促胰岛素活性逐渐下降。有趣的是,当减少到艾塞那肽(1-28)时,其促胰岛素活性几乎恢复了所有的完整性,但当达到艾塞那苷(1-26)时,它的促胰岛素活性突然消失。显然,艾塞那肽的Lys27和Asn28在与受体结合中起着重要作用。之前的大多数实验都是艾塞那肽(1-30)和GLP-1之间的比较,从而探索了艾塞那肽C末端额外残基(31-39)的功能。然而,在上述实验中,应在艾塞那肽(1-28)和GLP-1之间进行比较,因为艾塞那肽(29-39)确实是额外的片段。同时对肽的二级结构以及因此对受体识别的构象更重要。
与第一代GLP-1R激动剂相比,替尔泊肽作为GLP-1/GIP双受体激动剂,有三个关键的改进:首先,肽骨架中的许多残基被改变以获得GIPR激活活性;第二,艾塞那肽的C末端序列延长了C末端;第三,一个脂肪酸侧链是共轭的类似于semaglutide延长半衰期。通过研究每个位置采用的不同残基,可以定制双激动剂的活性。最初,主要关注的是受体激活活性,而最近,GLP-1R和GIPR的下游效应越来越多地表明,受体激活不仅促进cAMP和刺激胰岛素释放,而且其他因素(β-抑制蛋白募集、停留时间等)也会影响受体内化,从而进一步在体内引发长期效应。
现有研究证明:与司美格鲁肽降糖效果的对比中,替尔泊肽使受试者糖化血红蛋白平均降低2.0-2.3%,而司美格鲁肽平均降低1.9%;减脂方面,替尔泊肽使试验者平均减脂17-25磅,而司美格鲁肽为13磅。替尔泊肽展现出了比司美格鲁肽更加出众的降糖、减脂水平。替尔泊肽的安全性同其他肠促胰素类药物类似。替尔泊肽的胃肠道反应(如反胃、腹泻和便秘)较安慰剂有明显提升,但大部分为轻中度至中度。其余不良反应特别是低血糖仍未发现两组之间有突出差别。
随着,GLP-1类似物被广泛用于治疗T2DM及减肥人群中,肠促胰岛素GIP却显得比较神秘,而且在临床上应用有限,但科学家们从未停止探索,替尔泊肽其成功上市突出的临床效果,是这些努力的回报。
参考文献:
1.M. Yu, et al., Battle of GLP-1 delivery technologies, Adv. Drug Deliv. Rev. (2018),
2.Lijing Wang, Designing a Dual GLP-1R/GIPR Agonist from Tirzepatide: Comparing Residues Between Tirzepatide, GLP-1, and GIP Drug Design, Drug Design, Development and Therapy 2022:16 1547–1559
资料来源互联网,欢迎交流!