
人体氨基酸-有机酸代谢途径:蛋白质代谢障碍类疾病尿素循环障碍和有机酸血症

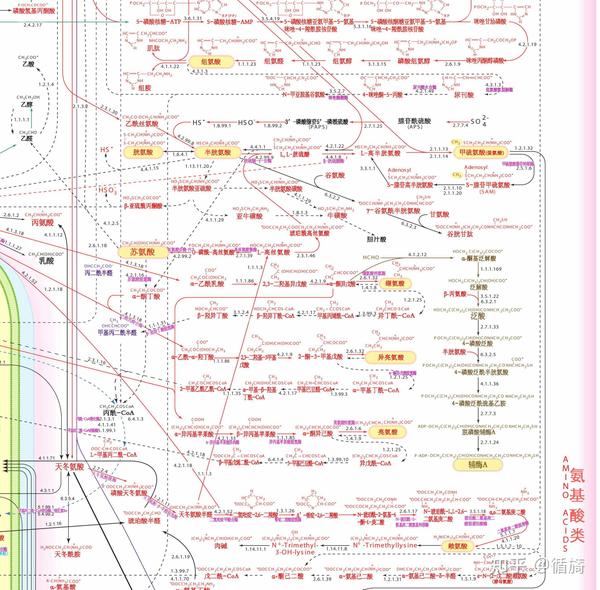


人体代谢障碍类疾病是指正常新陈代谢过程出现紊乱或者障碍,身体将食物转化为能量的过程因为疾病受阻。通常参与众多相互依赖的代谢途径的数千种酶执行这一过程,代谢疾病影响细胞执行关键功能的能力涉及蛋白质(氨基酸)、碳水化合物(糖和淀粉)或脂质(脂肪酸)加工或运输的生化反应。
代谢性疾病通常是遗传性的,但大多数受代谢性疾病影响的人可能会在数天、数月甚至数年内表现出健康状态。症状通常发生在身体的新陈代谢受到压力时,例如,长时间工作后禁食或发烧期间。对于某些代谢紊乱,可以获得产前诊断筛查,此类分析通常提供给曾经有过患有代谢疾病的孩子或属于特定种族群体的家庭。例如,泰-萨克斯病检测在德系犹太人中相对常见。在出生时进行足跟血代谢性疾病筛查通常会测试多达 十几 种不同的病症。串联质谱是一项新技术,几乎可以同时检测多种异常代谢物,从而可以在新生儿可检测的疾病清单中添加大约 30 种疾病。如果已知婴儿出生后不久患有代谢紊乱,则可以尽早开始适当的治疗,这可能会带来更好的预后。

代谢途径
食物通过细胞的一系列步骤分解酶(催化化合物转化的蛋白质底物)转化为具有不同生化结构的产品。这些产物然后成为代谢途径中下一种酶的底物。如果酶缺失或活性减弱,途径就会被阻断,最终产物的形成就会有缺陷,从而导致疾病。酶的低活性可能会导致酶底物的积累,高浓度时可能会产生毒性。此外,当底物积累时,通常处于休眠状态的次要代谢途径可能会被激活,可能形成非典型的、具有潜在毒性的产物。体内的每个细胞都包含数千条代谢途径,所有这些代谢途径在某种程度上都是相互关联的,因此一次堵塞可能会影响众多的生化过程。
代谢失衡的后果可能很严重;可能会出现智力障碍、癫痫发作、肌张力下降、器官衰竭、失明和耳聋,具体取决于哪种酶功能失调。近年来,人们已经发现,甚至一些与多种先天性异常相关的疾病(例如史密斯-莱姆利-奥皮茨综合征)也有潜在的代谢原因。
遗传突变
几乎所有酶、结构蛋白、细胞运输的分子蓝图蛋白质和其他负责执行新陈代谢中复杂反应的成分以脱氧核糖核酸 ( DNA ) 的形式储存在细胞核中。线粒体中的细胞器中还含有少量对新陈代谢至关重要的 DNA 。DNA 被组织成更小的单位,称为基因,它们指导特定蛋白质或酶的产生。1945年美国遗传学家乔治·比德尔和爱德华·塔图姆提出了分子生物学的一个中心原则,““一个基因一个酶”的原理,指出单个基因指导单个酶的合成。这一原则已被细化,以考虑到并非所有的事实基因产物是酶,有些酶是由不同基因编码的多个结构单元组成的。然而,当应用于加罗德关于先天性代谢缺陷的最初理论时,单基因一酶理论具有直接的意义。据推测,当基因突变产生功能减弱或缺失的有缺陷的酶时,就会发生遗传性代谢疾病。1948年高铁血红蛋白尿症成为第一个被确定为由酶缺陷引起的人类遗传病。1949年 美国化学家Linus Pauling及其同事证明,突变会导致蛋白质结构发生改变;从正常人红细胞中提取的血红蛋白(红细胞中将氧气输送到身体组织的蛋白质)被证明与从患有遗传性疾病镰状细胞贫血症的人中提取的血红蛋白的行为 不同。因此,确定引导功能改变的异常蛋白质形成的突变基因会导致先天性代谢错误。
遗传
先天性代谢缺陷的遗传最常见常染色体隐性遗传,意味着需要两个突变基因才能产生疾病的体征和症状。受影响儿童的父母通常是无症状携带者,因为 50% 的正常酶活性足以维持足够的健康。当有害物质的两个携带者然而,有 25% 的机会生出受影响的孩子,25% 的机会生出没有突变等位基因的孩子,50% 的机会生出同时也是携带者的孩子。从遗传学角度来说,常染色体隐性遗传病的携带者只有一个突变基因(杂合子),而受影响的个体则有两个突变基因(纯合子)。所有人类的基因组中都有大约六个隐性突变等位基因,但个体与携带同一基因突变的人交配的情况相对较少。然而,在父母近亲结婚的情况下,由于有共同的遗传背景,生出患有常染色体隐性遗传疾病的孩子的风险会增加。
与常染色体隐性遗传病不同,当仅存在一个突变基因时,就会表达常染色体显性遗传疾病。这些疾病有很强的家族史,除非该疾病是由个体新的自发突变引起的。杂合子个体有 50% 的机会将这种疾病遗传给他的后代。患有常染色体显性遗传疾病的个体表现出广泛的疾病严重程度,显性特征的携带者甚至可能表现为无症状。
细胞核中的遗传物质被发现被包装成 DNA-蛋白质复合物,称为染色体. 女性有两个X染色体,而男性有一个X和一个Y染色体。如果突变基因是 X 染色体的一部分,则由此产生的疾病称为 X 连锁疾病。所有继承了X连锁突变受到影响,因为XY对的Y染色体没有补偿性正常基因。由于突变发生在 X 染色体上,而男性在受精过程中仅将 Y 染色体传给儿子,因此父亲不会将疾病传给儿子。然而,她们可以将携带者状态(即突变的 X 染色体)传递给她们的女儿。与此同时,杂合女性携带者有 50% 的机会生出携带者女儿或受影响的儿子。
X连锁遗传因女性X染色体失活(lyonization)过程而变得复杂。尽管雌性携带两条 X 染色体,但在胚胎发育早期,每个细胞中的一条 X 染色体失活。X染色体失活的过程通常是随机的,导致携带X连锁疾病突变的特定女性体内形成两种细胞系;一种细胞系具有失活的正常 X 染色体,另一种细胞系具有失活的异常 X 染色体。然而,特定个体中较高比例的正常 X 染色体可能会失活,从而出现不同程度的疾病症状。这样的女性被称为表现出杂合子。X连锁疾病的例子包括鸟氨酸转氨甲酰酶缺乏症(一种酶缺乏症,导致血液中氨含量升高和尿素形成受损),X连锁肾上腺脑白质营养不良(一种以进行性精神和身体恶化以及肾上腺功能不全为特征的疾病),以及Lesch-Nyhan 综合征(一种嘌呤代谢紊乱,其特征是尿液中排泄大量尿酸、神经系统紊乱和自残)。
位于线粒体(即不包含在细胞核中)的基因的传递被称为母系(线粒体)遗传。线粒体DNA(mtDNA)虽然比核DNA小得多,但在细胞代谢中至关重要。细胞驱动其新陈代谢所需的大部分能量是由蛋白质在线粒体中通过一系列电子供体-受体反应产生的,这些反应构成了电子传输或呼吸链。线粒体位于卵子的细胞质中,遗传自母亲。精子也有线粒体,但它们不会融入发育中的胚胎。当细胞分裂时,线粒体随机分配给子细胞。每个线粒体含有2至10个线粒体DNA拷贝,每个细胞含有无数线粒体。在患有线粒体疾病的人的特定细胞中,正常线粒体的数量可能大于异常线粒体的数量,细胞可能功能良好。另一方面,如果细胞含有显着比例的异常线粒体,则该细胞和含有许多此类细胞的任何组织将表现出功能受损。受影响的儿童可能表现出一系列异常,从表现正常或轻度受影响到严重受损,具体取决于线粒体功能障碍的程度和组织受累的程度。
疾病氨基酸代谢
二十种氨基酸,其中九种是人体无法合成、必须通过食物获得的,参与新陈代谢。氨基酸是蛋白质的组成部分;有些还充当或合成为体内重要分子,例如神经递质、激素、色素和携氧分子。每种氨基酸进一步分解为氨、二氧化碳和水。影响氨基酸代谢的疾病包括苯丙酮尿症、酪氨酸血症、高胱氨酸尿症、非酮症高甘氨酸血症和枫糖浆尿病。这些疾病是常染色体隐性遗传,所有疾病都可以通过分析体液中的氨基酸浓度来诊断。(枫糖浆尿病还具有有机酸的产生的特点,并在有机酸血症部分进行讨论。)
食物中如果含有所有的必需氨基酸,称为完全蛋白质。肉类中的蛋白质是完全蛋白质,可以提供人体所需的全部氨基酸种类,属于优质蛋白质,素食里面蛋白质低点,且氨基酸不完整。
蛋白质有两种主要类型:
1、动物蛋白:优质蛋白,完整的蛋白质来源、含有人体所需的所有必需氨基酸, 优质蛋白第一阶梯:鱼、虾、蛋、牛肉、羊肉、牛奶等,第二阶梯:猪肉、鸡肉等
2、植物蛋白:蛋白质来源不完整,缺一种或者多种人体所需的必需氨基酸,常见于豆类、坚果蔬菜等食物
人体无法合成的9种氨基酸(必需氨基酸)包括:
- 苯丙氨酸 (Phenylalanine)(Phe)
- 缬氨酸 (Valine)(Val)
- 苏氨酸(羟丁氨酸)(Threonine)(Thr)
- 色氨酸 (Tryptophan)(Trp)
- 异亮氨酸 (Isoleucine)(Ile)
- 亮氨酸 (Leucine)(Leu)
- 甲硫氨酸 (蛋氨酸)(Methionine)(Met)
- 赖氨酸 (Lysine)(Lys)
- 组氨酸 (Histidine)(His)
过去人们曾认为组氨酸只针对婴幼儿是必需的,然而后续较长期的研究表明,它也是成年人必不可少的必需氨基酸。
人体可以自身合成的11种氨基酸(非必需氨基酸):甘氨酸、丙氨酸、脯氨酸、丝氨酸、酪氨酸、半胱氨酸、谷氨酰胺、天冬氨酸、天冬氨酰、谷氨酸、精氨酸。
非必需氨基酸合成途径各异,有些由代谢中间物合成、少数由必需氨基酸生成。
苯丙酮尿症(PKU) 是由于苯丙酮尿症活性降低引起的苯丙氨酸羟化酶(PAH),一种将氨基酸转化为氨基酸的酶苯丙氨酸酪氨酸,几种重要激素以及皮肤、头发和眼睛色素的前体。PAH 活性降低会导致苯丙氨酸的积累以及酪氨酸和其他代谢物含量的减少。血液中持续高水平的苯丙氨酸反过来会导致进行性发育迟缓、头围变小、行为障碍和癫痫发作。由于色素量减少黑色素,与其他未患有该疾病的家庭成员相比,患有 PKU 的人往往具有较浅的特征,例如金发和蓝眼睛。使用特殊配方以及苯丙氨酸和蛋白质含量低的食物进行治疗可以将苯丙氨酸水平降至正常并维持正常智力。然而,极少数 PKU 病例是由代谢受损引起的生物蝶呤是苯丙氨酸羟化酶反应中的重要辅助因子,可能不会始终对治疗产生反应。
经典型(肝肾型或 I 型)酪氨酸血症是由延胡索酰乙酰乙酸水解酶 (FAH) 缺乏引起的,FAH 是酪氨酸分解代谢中的最后一种酶。典型的酪氨酸血症的特征包括严重的肝脏疾病、体重增加不理想、周围神经疾病和肾脏缺陷。大约 40% 的人患有这种疾病如果未经治疗,5 岁时就会罹患肝癌。使用酪氨酸分解代谢途径的有效抑制剂 2-(2-硝基-4-三氟甲基苯甲酰基)-1,3-环己烷二酮 (NTBC) 进行治疗,可以防止有毒代谢物的产生。虽然这会改善肝脏、肾脏和神经系统症状,但可能无法预防肝癌的发生。严重肝病或癌症发展可能需要肝移植。还存在一种良性的、短暂的新生儿酪氨酸血症,对蛋白质限制和维生素 C治疗有反应。
高胱氨酸尿症是由参与蛋氨酸代谢的胱硫醚β-合酶(或β-合酶)缺陷引起的,导致同型半胱氨酸积聚。症状包括脸颊明显潮红、身材高瘦、晶状体脱位、血管疾病和骨骼变薄(骨质疏松症)。还可能存在智力障碍和精神障碍。大约 50% 的高胱氨酸尿症患者对以下药物治疗有反应维生素 B 6(吡哆醇),这些人往往有更好的智力预后。叶酸治疗,甜菜碱(一种从体内去除多余同型半胱氨酸的药物)、阿司匹林以及限制蛋白质和蛋氨酸的饮食也可能有益。
非酮症高甘氨酸血症的特征是癫痫发作、低肌张力、打嗝、屏气和严重发育障碍。它是由中枢神经系统中神经递质甘氨酸水平升高引起的,而甘氨酸水平又是由负责裂解氨基酸的酶系统缺陷引起的。甘氨酸。阻断甘氨酸作用的药物(例如右美沙芬)、低蛋白饮食和甘氨酸清除药物(例如苯甲酸钠)可能会缓解症状,但无法治愈这种严重的病症。
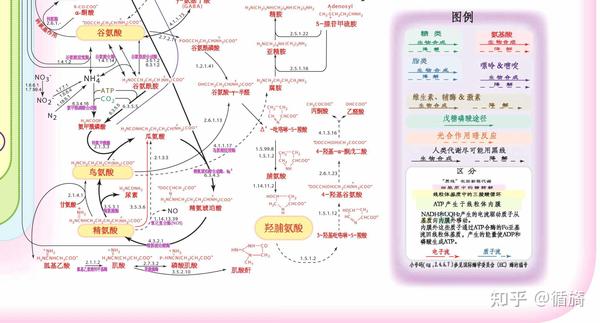
尿素循环障碍
肝细胞通过形成化合物在处理含氮废物方面发挥着关键作用 通过尿素循环的作用产生尿素(尿液的主要固体成分)。当氨基酸被降解时,分子一端的氨氮被分解,与尿素结合,并从尿液中排出。尿素循环中任何酶的缺陷都会导致血液中有毒的氨积累。反过来,这会导致出生后两三天内进食不良、呕吐、嗜睡,甚至可能昏迷(精氨酸酶缺乏症除外,这种情况会在儿童期后期出现)。
尿素循环障碍专题:
氨基酸转运障碍
将许多氨基酸从肠道转移到血液中或通过肾脏中的特殊细胞从尿液中回收它们需要能量。这种氨基酸的转运不涉及代谢途径中的酶,而是转运嵌入细胞或细胞内细胞器膜中的蛋白质。转运活性降低的突变蛋白可能会阻止膳食氨基酸的吸收或导致其从尿液中流失。例如,在胱氨酸尿症中,尿液中胱氨酸、鸟氨酸、精氨酸和赖氨酸的排泄增加,从而导致肾结石。胱氨酸病的特征是由于胱氨酸转运体的缺陷,胱氨酸无法从称为溶酶体的细胞器中排出。患有这种疾病的人会出现角膜沉积物和肾脏疾病,并且可能需要进行肾移植。肠道内赖氨酸、精氨酸和鸟氨酸的膜运输缺陷会导致赖氨酸尿蛋白不耐受(LPI),一种以蛋白质不耐受、腹泻、体重增加不理想、骨质疏松和皮疹为特征的疾病;LPI 的晚期并发症包括肾脏和肺部疾病。Hartnup 病是一种肠道和肾脏氨基酸转运障碍;共济失调、光敏性皮疹和精神异常是主要症状。
有机酸血症
有机酸是碳基化合物,当涉及特定酶的代谢途径被阻断时,有机酸的水平会异常升高。有机酸血症是以有机酸在身体组织和体液(尤其是尿液)中积累为特征的病症。这些疾病中最常见的是常染色体隐性遗传病,涉及支链氨基酸的代谢亮氨酸,异亮氨酸,和缬氨酸。有机酸血症有许多共同特征,包括血液中酸含量增加(酸血症)、低血糖(低血糖)、白细胞计数低(中性粒细胞减少症)、生长发育不良以及不同程度的精神障碍。这些疾病可能会在婴儿期或儿童后期出现。
丙酸PA血症是由于丙酰辅酶A羧化酶缺乏引起的,导致丙酸积聚。患有这种疾病的人通常在婴儿期早期就出现危及生命的疾病。酸血症、脱水、白细胞计数低、肌张力低和昏睡进展为昏迷是典型特征。血液中的氨水平也可能很高,因为异常代谢物会抑制尿素循环的正常运作。丙酸血症的主要治疗方法是限制支链氨基酸的饮食、补充肉碱以及用静脉输液、葡萄糖和碳酸氢盐大力治疗代谢危机。
具有以下经典形式的人甲基丙二酸血症 (MMA) 是由甲基丙二酰辅酶 A 变位酶缺陷引起的,其症状与丙酸血症患者相似,但也可能出现肾衰竭的长期并发症。肝肾联合移植可能对一些患有严重肾脏疾病的患者有益。经典无效型MMA 的一种形式对维生素 B 12治疗有反应(部分有效型)。较罕见的形式是由维生素 B 12加工缺陷引起的,通常在儿童后期出现,并伴有进行性神经功能障碍,还有种甲基丙二酸合并同型半胱氨酸型的可以查看往期文章:
枫糖浆尿病(MSUD) 是一种支链氨基酸代谢紊乱,导致亮氨酸、异亮氨酸、缬氨酸及其相应的含氧酸在体液中积聚,导致某些患者的尿液中出现特有的枫糖浆气味。这种疾病在宾夕法尼亚州的门诺派教徒中很常见。MSUD 的典型形式在婴儿期表现为嗜睡和进行性神经功能恶化以癫痫发作和昏迷为特征。与大多数有机酸血症不同,显着的酸血症很少见。治疗包括限制蛋白质和喂养缺乏支链氨基酸的配方奶粉。尽管接受治疗,患有 MSUD 的人仍可能出现智力障碍,但早期和仔细的治疗可以导致正常的智力发育。较轻的 MSUD 可以通过简单的蛋白质限制或服用硫胺素(维生素 B 1)来治疗。往期文章:
新生儿足跟血筛查需要检测哪些有机酸代谢紊乱?
1、3-甲基巴豆酰辅酶A羧化酶缺乏症 (3MCC)
是由于身体无法分解氨基酸亮氨酸。所有含蛋白质的食物都含有必需氨基酸亮氨酸。对于患有 3MCC 的人来说,吃含蛋白质的食物会导致健康问题。所以需要低蛋白高碳水饮食低蛋白高碳水+适量高脂肪之饮食管理
2、β-酮硫解酶缺乏症 (BKT)
如果您的宝宝患有这种疾病,她的身体就无法使用氨基酸异亮氨酸,也无法使用酮。在美国,每年出生的婴儿中只有不到十万之一患有 BKT。
3、1 型戊二酸血症 (GA1)
由于身体无法代谢赖氨酸、羟赖氨酸和色氨酸这三种氨基酸,所有含蛋白质的食物都含有这些氨基酸。
4、羟甲基戊二酸尿症 (HMG)
患有 HMG 的患者难以分解氨基酸亮氨酸。他的身体也停止产生酮,他可能会出现低血糖。患有 HMG 的人可能需要吃富含碳水化合物的食物,如面包、面食、水果和蔬菜。他们可能还需要限制富含蛋白质和脂肪的食物。在美国,每年只有不到十万分之一的婴儿出生时患有 HMG。
5、异戊酸血症(IVA)
与 3MCC 一样,在这种情况下,您的宝宝无法分解亮氨酸。在美国,每年只有不到十分之一的婴儿出生时患有 IVA。
6、甲基丙二酸血症,CBl A 和 CBl B 形式
这种疾病会导致患者的身体难以分解食物中的脂肪和四种氨基酸(异亮氨酸、蛋氨酸、苏氨酸和缬氨酸),靶向损伤器官是肾脏。
7、甲基丙二酸血症,变位酶缺乏症 (MUT)
这种疾病与 Cbl A、B 类似。
8、多种羧化酶缺乏症 (MCD)
在 MCD 中,患者的身体无法使用一种称为生物素的维生素。生物素是一种 B 族维生素,存在于鸡蛋和牛奶等食物中。身体在制造和分解蛋白质、脂肪和碳水化合物时会使用生物素。
9、丙酸血症 (PA)
与甲基丙二酸血症 Cbl A、B 和 MUT 一样,患者的身体无法使用异亮氨酸、蛋氨酸、苏氨酸和缬氨酸这四种氨基酸。吃高蛋白质的食物会导致危险物质在血液中积聚。靶向损伤器官是心脏
这些疾病的体征和症状是什么?
有些患有此类疾病的婴儿从未出现过严重的体征或症状。有的晚发型、中间型患者,体征和症状可能要到童年、青少年、晚年才会出现,此外,体征和症状可能会出现,然后随着时间的推移而消失。每种疾病都有不同的体征和症状,大部分患者发病基本是由于感染、手术、剧烈运动后没补充电解质、添加辅食期间、母乳过渡天然食物期间、大量饮用高蛋白质食物诱发急性感染期等,且临床发现大部分患者,在未确诊疾病期间最先都是就诊于消化科、肝病科、神经内科(精神异常)。
患者急性期体征包括:
- 电解质紊乱、脱水。脱水的迹象包括感到头晕或头晕、心跳加快以及口唇干燥。
- 感觉疲倦或昏昏欲睡
- 挑食、偏食、食欲不好、消化不好
- 低血糖
- 体温低
- 代谢性酸中毒(体液中酸性过多)
- 恶心、腹泻和呕吐
- 饮食不健康且体重增加困难
- 皮疹或感染
- 肌肉无力或肌肉痉挛
- 疾病或感染、吃错种类的食物或长时间不进食都可能导致这些疾病的体征和症状
这些疾病会导致哪些健康问题?
如果不治疗,其中一些疾病可能会导致健康问题,包括:
- 脑损伤
- 昏迷
- 眼睛问题和视力丧失
- 智力和发育障碍这些是大脑工作方式的问题,可能导致一个人在身体发育、学习、沟通、照顾自己或与他人相处方面出现问题或延迟
- 骨质疏松症——导致骨骼变薄变脆
- 心脏、肝脏、肾脏或胰腺出现问题
- 癫痫发作、精神异常
- 中风
- 如果不治疗,某些疾病可能导致死亡
有机酸紊乱的治疗方法是什么?
需要主治医生将与代谢医生和营养师合作。需要及时治疗以防止精神发育迟滞和严重的医疗问题。大多数患者需要遵循特定的饮食习惯并饮用特殊的医疗配方奶粉。
以下是通常推荐用于患有有机酸疾病的儿童的治疗方法:
1、维生素 B12、生物素、甜菜碱、左旋肉碱等药物
2、饮食计划如低蛋白饮食、低亮氨酸饮食、低缬氨酸饮食。需要联系营养师制定一个饮食计划,其中含有适量的蛋白质、营养素和能量。大部分患者一生都需要遵循特殊的饮食计划。
3、医疗配方食品和食品 - 特殊医疗配方食品含有正常生长和发育所需的适量蛋白质和营养素。特殊低蛋白面粉、面食和大米等医疗食品是专门为有机酸紊乱患者制作的。
4、避免长时间不进食:婴儿和幼儿需要经常进食以避免代谢危机和内源性分解。大多数儿童禁食时间不应超过 4 至 6 小时。有些孩子可能需要吃得比这更多。夜间喂养婴儿很重要。如果他们不自己醒来,可能需要被叫醒才能吃饭。
5、定期进行血液和尿液检查——您的孩子的饮食和药物可能需要根据这些检查的结果进行调整。
许多患有有机酸紊乱的儿童在患病期间可能需要在医院接受治疗,以避免出现严重的健康问题。患者应该携带一份的该患者的急救卡给当地主治医师,其中包含患者的护理医疗说明。
因为特殊的饮食限制,所以需要保证患者热量,以免引起内源性分解(一种因为蛋白质或者脂肪营养,不能满足身体需求,身体在需求的情况下自身分解身体存储的营养,分解的同时释放了很多不可代谢“毒素”),饮食管理对此类患者的疾病治疗非常关键与重要,相关饮食管理可以查看往期文章:
成人以精神异常起病的代谢类疾病
| 紊乱 | 临床体征 | 诱发 | 眼科检查 | 生物标志物 |
|---|---|---|---|---|
| 威尔逊病 (WD) | 震颤 | – | 凯泽-弗莱舍环 | 铜蓝蛋白 |
| 肌张力障碍 | ||||
| 构音障碍 | ||||
| 尿素循环障碍 (UCD) | 困惑 | 蛋白质饮食 | – | 高氨血症 |
| 腹痛 | 术后 | |||
| 恶心,呕吐 | 药物* | |||
| 同型半胱氨酸血症(MTHFR) | 共济失调 | – | – | 同型半胱氨酸血症 |
| 精神退化 | 蛋氨酸血症 | |||
| 同型半胱氨酸血症 (CbS) | 血栓栓塞 | 蛋白质饮食 | 严重近视 | 同型半胱氨酸血症 |
| 脊柱侧弯 | 术后 | 异位晶状体 | 蛋氨酸血症 | |
| 马方样小脑体征 | ||||
| C 型尼曼-匹克病 (NP-C) | 肌张力障碍+共济失调 | 新生儿黄疸 | 核上垂直 | 皮肤活检 |
| 脾肿大 | 进展缓慢 | 凝视麻痹 | 菲律宾测试 | |
| NPC1和NPC2基因检测 | ||||
| 脑腱黄瘤病 (CTX) | 慢性腹泻 | – | 青少年白内障 | 高胆固醇 |
| 痉挛性瘫痪 | ||||
| 卟啉症 (POR) | 黑色或红色尿液 | – | 胆色素原(尿液) | |
| 便秘 | ||||
| 困惑 | ||||
| 腹痛 | ||||
| 恶心,呕吐 |
- *示例药物:丙戊酸/皮质激素。
文献参考:
Lauterbach MD、Stanislawski-Zygaj AL、Benjamin S:儿童和青年发病疾病(包括精神病)的鉴别诊断。J 神经精神病学临床神经科学。2008, 20(4): 409-418。


