
遗传代谢病临床诊断的蛛丝马迹线索:代谢性肌病

一、遗传性代谢紊乱IMD是一大类影响正常生化、代谢途径的疾病
小分子疾病:(1)蛋白质(氨基酸)代谢障碍类疾病和有机酸血症;(2)碳水化合物代谢障碍;(3)脂肪酸代谢障碍;(4)卟啉、血红素代谢障碍;(5)矿物质吸收和运输障碍。
细胞器类疾病:(1) 溶酶体和溶酶体相关细胞器疾病 (2) 过氧化物酶体疾病;(3)线粒体脑肌病。
其他:(1)嘌呤、嘧啶及神经递质代谢疾病 (2)维生素及非蛋白质辅因子代谢及转运障碍; (3)能量代谢障碍;
这些代谢循环通路障碍,如果不经治疗,往往会连带全身多器官、系统病变,而临床上比较常见的是患者以精神异常或者运动障碍、肌病、消化系统问题、眼科异常等病症就诊。
本合集将此类以眼科、肌病、精神异常等临床病症为代表的遗传代谢病系列汇总在这个合集并持续更新,希望给未经确诊的患者一些临床提示,希望患者家属将这些蛛丝马迹的病症汇总给专业的代谢医生分析,争取早日确诊,得到规范治疗,从而避免疾病引起的继发性脏器损伤,以及家族相关疾病预防与再生殖预警。
二、遗传性代谢紊乱IEM或IMD引起的代谢性肌病
代谢性肌病由多种遗传性疾病组成,这些疾病会导致肌肉能量产生受损,骨骼肌的参与主要是因为其高能量需求,但也可能发生多系统功能障碍。本文将讨论影响糖原和脂质代谢的疾病以及线粒体疾病。尽管这些疾病的症状可能会在儿童时期出现,但许多受影响的人在以后的生活中会出现轻微的、多变的症状。提供实用的诊断方法来帮助诊断这些疾病。
临床上有的小龄患者家长反映孩子走路时间长了腿疼(腿部肌肉),有的小患者(特别是合并了自闭症的患者,无法正确表述的),四五岁了出门还总要家人抱着,不能长时间行走,家长反映孩子日常跑跳很好,大运动、精细运动表面上看起来“正常”,临床上这类患者运动不耐受、肌肉无力等表现,很有可能是某类代谢性肌病造成,因为这种比较隐蔽的异常,很难被患者家属与儿科医生重视,所以造成这种临床症状很难被确诊病因。
1、生物化学
肺、心血管、肌肉骨骼和代谢系统都参与运动引起的生理变化。三磷酸腺苷(ATP)是肌肉收缩和放松所需能量的主要来源。ATP的主要来源是糖原、葡萄糖和游离脂肪酸。休息时,脂肪酸 (FA) 是骨骼肌的主要燃料来源。从休息到运动的转变需要立即利用来自磷酸肌酸穿梭机的 ATP,它独立于氧气供应。这提供了 8 到 10 秒的能量,之后其他代谢途径被激活,具体取决于活动强度、运动类型、运动持续时间以及个人的身体状况和饮食。需要短时间爆发力的高强度运动依赖于无氧糖酵解——葡萄糖代谢产生 ATP。在次最大运动量下,氧化磷酸化是产生 ATP 的更有效机制。氧化磷酸化代谢糖原、葡萄糖和 FA,产生烟酰胺腺嘌呤二核苷酸 (NADH) 和黄素腺嘌呤二核苷酸 (FADH 2 ),它们在线粒体电子传递链中捐赠电子以产生 ATP。氧气输送和使用的最大水平 (VO 2 max) 定义了个体的有氧能力。次最大低强度有氧运动有利于 FA 和糖原的代谢,而高强度运动有利于糖原作为主要能量来源,有氧糖酵解在这两种情况下都有贡献。随着次极量运动持续时间的增加,FA 成为主要的能量来源。实际上,马拉松运动员会“碰壁”,因为跑步强度对于运动员的体能水平来说太高,更倾向于使用糖原而不是 FA 作为燃料。偏爱糖原的使用会导致葡萄糖和糖原储备的耗尽,从而导致严重疲劳。一个人通常可以储存大约 2,000 卡路里的糖原,每跑一英里“消耗”大约 100 卡路里。这就解释了为什么跑步者通常会在 20 英里左右“撞墙”,使得最后 6.2 英里对许多马拉松参与者来说太过难忘。
代谢性肌病可由任何上述途径的缺陷引起,并按有缺陷的途径进行分类。糖原糖是糖原或葡萄糖降解(分别为糖原分解或糖酵解)或糖原合成(糖生成)的疾病,并导致糖原异常积累;因此,这些被称为糖原储存障碍(GSD)。脂质代谢紊乱主要包括FA氧化紊乱(FAOD),这是β-氧化或肉碱循环紊乱的缺陷,但也包括其他途径。线粒体疾病可能是由许多缺陷引起的,包括影响呼吸链蛋白的缺陷。
诊断患者的挑战始于这些疾病的广泛表型范围和千变万化的表现。一般来说,这个范围部分是由残留的酶活性来解释的,其中活性降低与更严重的临床表现相关,而更大的活性与更轻微的临床表现相关。根据酶的活性水平,症状可能在婴儿期出现,伴有破坏性的多系统参与,到晚年则出现更微妙的症状。
2、骨骼肌的结构
骨骼肌占身体的 40% 以上,对于新陈代谢、锻炼和运动非常重要。肌肉能量衰竭表现为伴有肌肉无力的代谢危机,有时与肌肉疲劳和衰竭相关,导致急性坏死。
运动对于肌肉的维护和再生至关重要。在肌肉中,多种细胞器(肌浆网、T 管、三联体、线粒体等)有助于燃料利用、钙稳态调节、氧化代谢和线粒体能量功能。所有这些细胞器可能与代谢性肌肉疾病的组织损伤有关,并被 FoxO 转录和其他控制蛋白质周转的因子控制的自噬清除。肌原纤维是主要的细胞成分,占肌肉体积的 85% [ 11 ]。
当在横截面中检查肌肉束的组织学图像时,可以使用组织化学技术来识别两种主要不同的纤维类型:1 型(慢)和 2 型纤维(快),并观察是否存在选择性纤维类型萎缩。
1 型纤维具有氧化性,富含线粒体,可与 pH 4.3 的肌球蛋白 ATP 酶染色剂、NADH-TR 还原酶、琥珀酸脱氢酶和细胞色素 C 氧化酶发生反应,而 2 型纤维具有糖酵解作用,可与 pH 9.4 的 ATP 酶染色剂、过碘酸漂移发生反应和磷酸化酶染色。
有一个内膜系统、横向管状系统(T 系统)和肌浆网(SR),它们是相互关联的细胞器,与收缩和舒张期间纤维的兴奋/收缩耦合有关,响应钙的释放和吸收。
骨骼肌周围有肌束膜,毛细血管和神经都集中在肌束膜上(图 1E)。肌纤维核位于肌纤维的肌膜下区域。在部分肌肉横截面的组织学图像上,肌外膜是可见的。肌膜是骨骼肌的质膜,具有内部质膜和外部基底层,由粘多糖、糖蛋白和唾液酸组成。
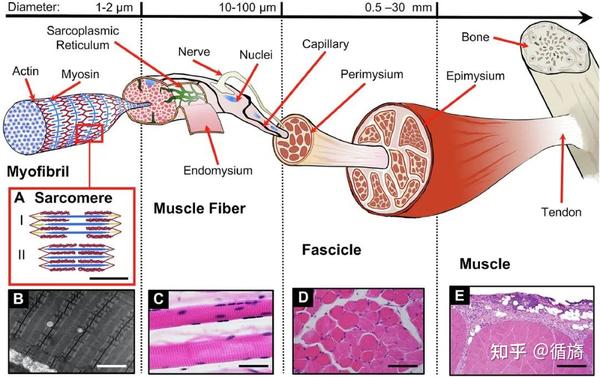

图1:骨骼肌的层次结构。A) 肌节形态和滑动机制(比例尺 0.5 nm):肌动蛋白(红色)、肌球蛋白(蓝色)和肌联蛋白(黄色)丝显示为松弛状态 (I) 和收缩状态 (II)。锯齿状边代表 Z 线。没有肌动蛋白丝的中心空间是 H 区。B) 肌原纤维的透射电子显微镜 (TEM) 图像(比例尺 = 1 nm)。C) 骨骼肌纤维的相差显微镜 (PCM) 图像。深紫色椭圆形元素是肌细胞核(比例尺 = 50 μm)。D) 分束横截面的组织学图像。较大的白色带是肌束膜。圆形结构是肌纤维,而深紫色点是肌细胞核(比例尺 = 100 μm)。E) 肌肉横截面一部分的组织学图像。在上部,肌外膜是可见的(比例尺 = 0.5 毫米)。
在内质膜和基底层之间定位有卫星细胞,卫星细胞是未分化的细胞成分,在适当的刺激下,可能从静止细胞增殖、融合并成为肌管,从而再生肌纤维。肌纤维由合胞体形成,男性直径为 40-80 μm,女性为 30-70 μm,儿童为 15-40 μm。
图2A和2B强调了肌浆网的作用,它是横纹骨骼肌纤维糖酵解和钙信号传导的主要参与者,并参与兴奋-收缩周期以及肌肉运动和疲劳期间的糖原代谢。
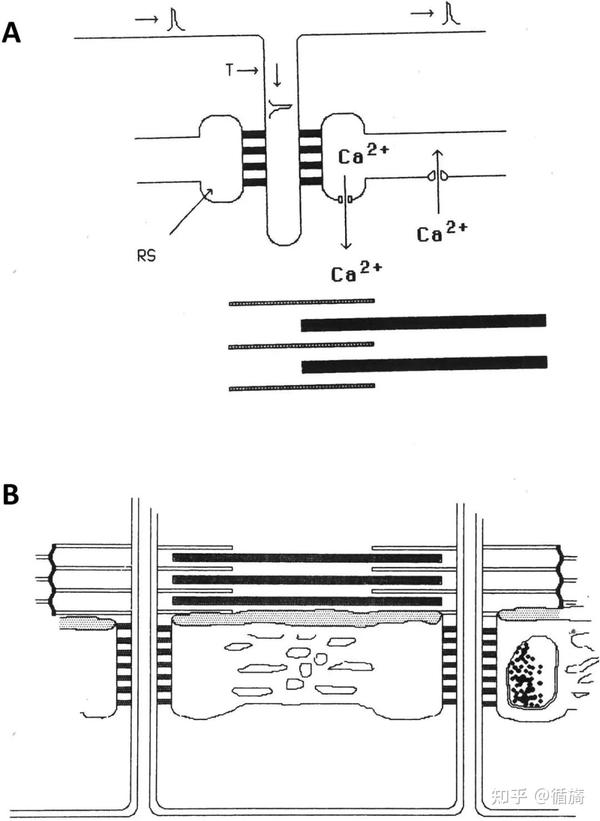
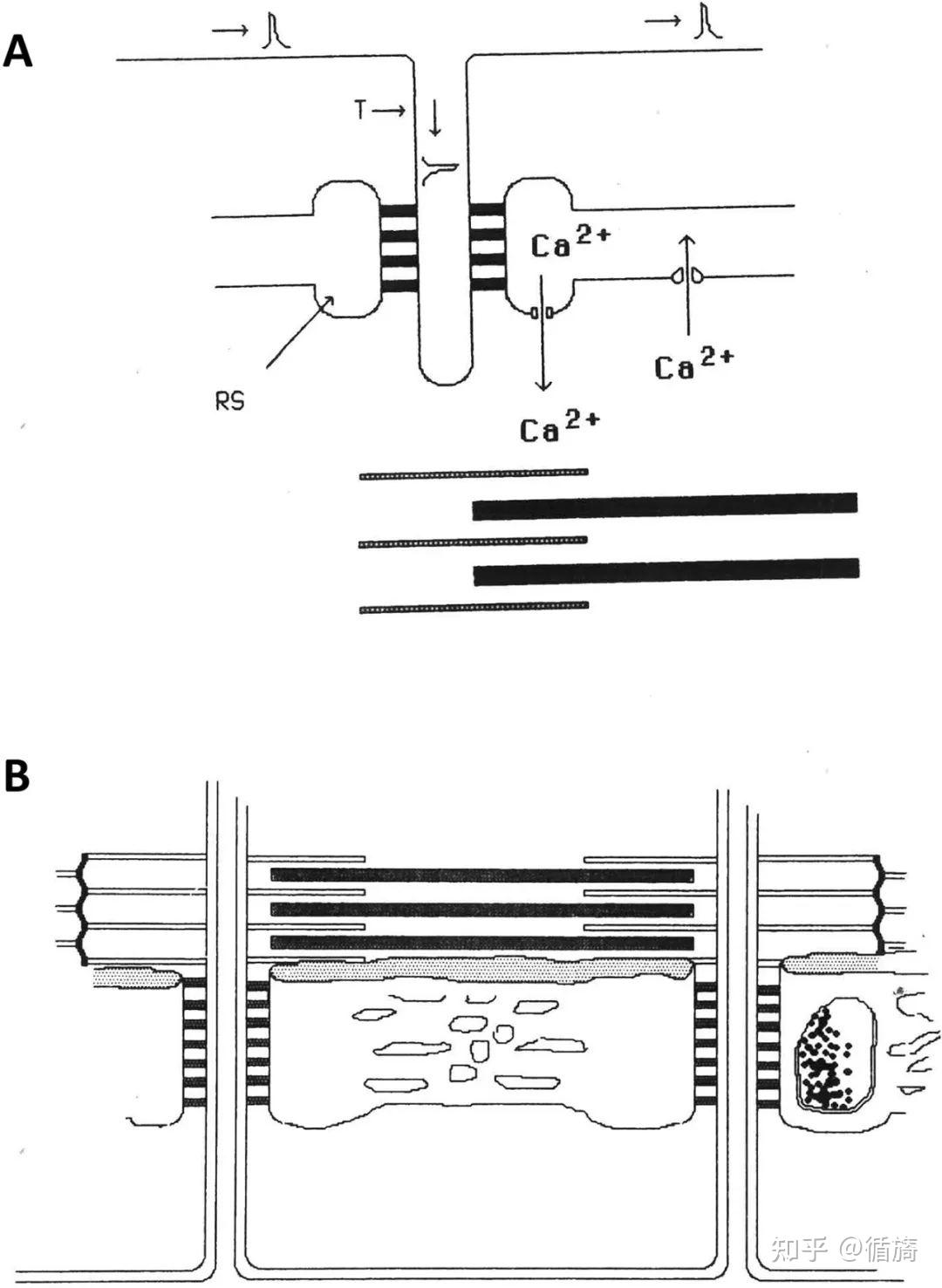
A) 兴奋-收缩耦合需要来自储存在 calsequestrin 中的肌浆网 (SR) 池释放的 ATP 和 Ca2+ 离子的能量。B) SR 分为两个域:纵向 SR 和交界 SR。纵向 SR 由许多相互连接的小管组成,在每个肌原纤维周围形成一个网络。在其末端,纵向小管形成单个扩张的囊,称为终池。两个末端池和一个 T 形管形成一个三联体。纵向和连接 SR 都显示出特定的空间组织,与糖酵解途径酶相关的特定区域有规律地对齐。
骨骼肌是一种适应性很强的组织,通过改变纤维的尺寸和类型来应对生理挑战;这种适应性反应受到多种刺激的控制,包括神经刺激和内分泌刺激。人们普遍认为引发代谢危机的主要机制是缺乏能量或细胞器和底物产生的燃料不足。另一个原因是由于参与兴奋/收缩耦合的突变受体(例如位于 SR 中的 Ryanodine 受体 (RYR1))导致异常SR Ca 2+泄漏,导致不受控制的收缩和横纹肌溶解。必须考虑一系列因素:SR 中的 Ca 2+泄漏触发前馈机制,导致线粒体过量产生活性氧 (ROS),进而导致进一步的 Ca 2+释放(通过 RYR1 和细胞外)钙2+ )。
在宏观层面上规划肌肉萎缩和肥大的研究时,必须考虑肌肉束的病理状态最终会被脂肪和结缔组织等其他组织替代。
肌肉成像技术可用于检测肌束或肌肉修复中肌纤维改变的异常模式;可以在诊断过程的早期使用肌肉计算机断层扫描、回波描记术或肌肉 MRI 进行检查,特别是使用可能显示肌水肿的 STIR 序列。MRI 可以识别溶酶体疾病和一些脂肪酸氧化代谢疾病的结构或发育异常,例如,2 型糖原增多症可能具有特征性的诊断成像特征。请注意,在肌肉发育过程完成后,应在 15 岁时获得正常的肌肉 MRI 外观。在对患者进行广泛的检查之间取得平衡,成像技术似乎对代谢患者的诊断和随访都很有用。
3、代谢性肌病的临床表现
主要症状是运动不耐受、疲劳、肌痛和/或虚弱,患者通常因此主诉运动不耐受,或者运动后抽搐疼痛和痉挛呕吐。但其他患者可能常发生位于近端的进行性肌无力(与炎性肌病或肢带型肌营养不良表现相似),但也可见于远端。在小部分患者中,动态和静态虚拟均占主导。在临床表现上,个体可能没有症状,没有固定的弱点。一些代谢性肌病,特别是糖原贮积病,会导致肌肉痉挛和/或挛缩以及横纹肌溶解症。线粒体肌病患者通常会出现全身并发症,但症状可能仅限于肌肉。


对于临床医生来说,重要的是要了解为什么会出现运动不耐受,是否是由于疲劳、肌痛、虚弱、痉挛或呼吸急促,以及它是在什么情况下发生的。尽管这些不同的症状可能在其他情况下出现,但当与代谢性肌病相关时,它们通常在活动期间或在生理应激源(例如发烧或禁食)背景下出现。
横纹肌溶解症和肌红蛋白尿(酱油尿)可能是代谢性肌病存在的信号,但也可能在未经训练的健康个体进行高强度运动时发生。后一种情况通常是良性的。导致症状的活动类型为诊断提供了另一个线索。高强度运动(例如举重或短跑)会在几分钟内出现症状,应考虑糖原糖血症。长时间低强度运动(即耐力运动)后出现疲劳和虚弱的症状可能表明存在脂质或线粒体疾病。耐力运动时出现呼吸困难可能提示线粒体肌病。最后,询问患者童年时期的运动能力可能会揭示出“跟上同学”困难的历史。
除了与线粒体肌病相关的全身症状(例如听力或视力丧失、上睑下垂或眼肌麻痹)外,体检通常是正常的。仔细检查可能会发现近端肌肉的固定无力,这种情况在 GSD 中比在脂质代谢紊乱中更常见。固定性挛缩在肌营养不良症中更常见,而短暂性“挛缩”在 GSD 中更常见,尤其是某些亚型(例如麦卡德尔病)。
实验室检查结果,例如肌酸激酶(CK)或乳酸值升高,是非特异性的,怀疑代谢性肌病可能是由适当的临床病史引起的。不幸的是,“成长的烦恼”和“懒惰”是常见的误诊。患者常常忍受这些症状,并被他们自己、他们的父母、甚至他们的医生误解。
4、代谢性肌病的类型:
4.1 糖原代谢紊乱:
糖原代谢紊乱是由于身体将葡萄糖储存转化为可用能量的能力受损而引起的。由于体内的大部分葡萄糖储存在肝脏和骨骼肌中,因此这些器官是这些疾病中最常受影响的器官。有 14 种糖原代谢疾病,其中最常见的是:McArdle 病(磷酸化酶缺乏症)、Pompe 病(酸性麦芽糖酶缺乏症)、Tarui 病(磷酸果糖激酶缺乏症)和磷酸化酶 B 激酶缺乏症。
不太严重的 GSD 患者通常在生命的第二个或第三个十年出现,但通常在儿童时期出现可归因的症状。诊断存在明显的延迟,女性的情况比男性更常见,如麦卡德尔病 (GSD5) 中所见。在运动后几秒到几分钟内,受影响的人会抱怨肌肉痉挛和运动不耐受,这是由于骨骼肌在运动早期对葡萄糖和糖原的依赖所致。间歇性横纹肌溶解是一个常见特征,特别是在 McArdle 和 Tarui (GSD7) 病中,而 Pompe (GSD2) 病的特征是肌病,但不伴有横纹肌溶解。
麦卡德尔病。最常见的碳水化合物代谢紊乱是麦卡德尔病,它是由糖原磷酸化酶( PYGM )基因的常染色体隐性突变导致肌磷酸化酶缺陷引起的。麦卡德尔病患者在运动早期会因过早疲劳、肌痛、肌肉挛缩和横纹肌溶解而出现运动不耐受。“第二次复苏”现象是一种特征性现象,其特点是经过约 10 分钟的低强度运动(“热身”期)后运动耐量有所改善。这在生理学研究中很明显,其中最初的不适当劳力性心动过速会随着活动 10 分钟内心率的降低而改善。肌肉症状(例如肌痛)也有所改善。这种临床现象在其他疾病中不存在。一些麦卡德尔病患者还会出现固定性近端肌无力。运动前摄入蔗糖可减轻症状。
塔瑞病。与麦卡德尔病相反,塔瑞病在运动前摄入糖分时会出现“失控”现象,从而使症状恶化。Tarui 病是由磷酸果糖激酶( PFKM ) 基因的常染色体隐性突变引起的。成人发病的 Tarui 病表现为运动不耐受、间歇性横纹肌溶解和虚弱。由于红细胞中表达磷酸果糖激酶,可能会发生溶血性贫血。
庞贝病。α-葡萄糖苷酶( GAA ) 基因的常染色体隐性突变会导致酸性麦芽糖酶缺乏,从而导致庞贝病。除了肢带无力外,还可能出现通气衰竭和心肌病。迟发性酸性麦芽糖酶缺乏症可导致早期呼吸功能不全,伴有近端肌肉无力和翼状肩胛,以及主要由呼吸困难引起的运动不耐受。在庞贝病中,椎旁肌肉中可能会出现肌强直放电。酶替代是庞贝病的一种可用治疗选择。
实验室调查结果。GSD 的基线 CK 值通常正常,但麦卡德尔病和庞贝氏病的基线 CK 值通常升高。肝脏和心脏也可能受累。
对于碳水化合物存在缺陷的患者,症状是由短暂的等长时间运动(如提举重物)或由强度较轻但较持久的动力性运动(如游泳、爬楼梯或跑步)引起的。尿液可能会导致肌红蛋白、痛性痉挛、以及剖腹产。
在幼儿期,糖原可能出现肝功能障碍、肝肿大、生长迟缓、低血糖(有时伴有低血糖提示症状)、大运动发育迟缓、周围神经损伤、心脏受累、溶血性贫血伴黄中断、还可以观察到视力障碍、上下运动神经元受累感觉缺失、括约肌问题和神经源性膀胱。
然而,主要的症状和身体症状与运动不耐受以及长期恢复的肌肉红蛋白尿相关。糖原有助于缺陷的患者通常主要诉诸活动时容易疲劳,而运动偶尔会导致被砍断的僵硬。一些病例中,当出现症状时短暂休息可改善运动耐量,称为自发性“恢复精力”(第二次风,继减现象)。这种现象的原因是肝脏激发相关的血压水平升高高。输注碳水化合物(如坐标)或脂类也可以“恢复精力”。
然而,某些特定的糖酵解缺陷(例如,磷酸果糖缺乏)患者不能自发性“恢复活力”,原因是游离填料和酮体减少。



4.2 脂质代谢紊乱:
骨骼肌以脂肪酸的形式分解脂质,以便在休息和运动时产生能量。因此,分解这些脂肪酸的能力受损会在长时间运动期间导致肌肉无力和疼痛。
成年期最常见的脂质代谢紊乱包括肉碱棕榈酰转移酶 II 缺乏症 (CPT II)、三功能蛋白缺乏症 (TFP) 和极长链酰基辅酶 A 脱氢酶缺乏症 (VLCAD)。
与 GSD 一样,迟发性FAOD 的诊断通常会出现延迟。症状通常包括运动引起的虚弱、疲劳和长时间低强度运动引起的肌痛。可能会发生抽筋,但不像 GSD 那样明显。横纹肌溶解症可能由 3F 引发并发生:禁食、发烧(感染)或冷冻(长时间暴露在寒冷的温度下)。常见的迟发型包括肉毒碱棕榈酰转移酶 II ( CPTII ) 缺乏症和极长链酰基辅酶 A 脱氢酶( VLCAD ) 缺乏症,这两种疾病在临床上很难区分;通常需要进行实验室研究。Lipin-1 缺乏症是由脂质 1 ( LPIN1 ) 突变引起的,导致磷脂酸磷酸酶缺乏,表现为儿童发烧引发的严重、复发性横纹肌溶解症。多酰基辅酶 A 脱氢酶缺乏症 (MADD) 是由电子转移黄素蛋白脱氢酶( ETFDH ) 或电子转移黄素蛋白亚基 A或B ( ETFA/B ) 突变引起的。MADD 可表现为缓慢进展的近端无力和疲劳,导致运动不耐受,这可能对核黄素补充剂有反应(核黄素反应性 - MADD,最常见于ETFDH突变)。辅酶 Q10 (CoQ10) 缺乏症可与 MADD 共存,因此还需补充 CoQ10。许多其他 β 氧化疾病存在于患有严重多系统疾病的婴儿期。
实验室调查结果。基线 CK 通常是正常的。患者可能会出现复发性低酮性低血糖。肝脏和心脏也可能受累。
脂质代谢障碍造成的代谢性肌病包括下列情况:
肉碱缺乏综合征
脂肪酸运输缺陷
β-氧化酶缺陷
中性脂质贮积病
脂素(lipin)-1缺乏
存在累及肌肉的脂质代谢障碍时,症状通常由感染、发热、长时间或高强度运动以及长时间禁食引发。与糖原代谢缺陷患者相比,这些患者不会出现真正的肌肉痉挛或挛缩,不会出现“恢复精力”现象。
临床医生应怀疑存在脂肪酸氧化障碍的4大临床和实验室特征包括:
依赖于脂肪酸氧化的组织受累,例如心脏、肌肉和肝脏
反复发生低酮症性低血糖(并未发生酮症酸中毒,因为脂肪酸无法在肝脏转化为酮酸)
与禁食相关的急性代谢失代偿
肉碱的血浆浓度和组织浓度改变
在脂肪酸氧化缺陷患者中,可以导致代谢失代偿的其他情况包括寒冷诱导的寒颤产热和伴随呕吐的感染:
暴露于寒冷条件下,寒战主要取决于长链脂肪酸氧化
在长时间禁食后或感染期间,儿童可以出现昏迷,以及出现类似Reye综合征的症状。
骨骼肌、心脏和肝脏高度依赖高效的脂肪酸利用。脂肪酸是心脏和肝脏的主要能量来源,特别是在禁食期间糖原和葡萄糖储备耗尽时。此外,在轻度至中度长时间运动期间,静止肌肉和运动肌肉通过脂肪酸氧化获取大部分所需能量。
在脂肪酸氧化缺陷的禁食患者中,由于存在代谢阻断,游离脂肪酸不能被代谢;因此,它们以甘油三酯的形式储存在细胞质中,从而导致进行性脂质沉积性肌病伴无力、肥厚型和/或扩张型心肌病以及脂肪肝。此外,在禁食情况下,糖原和葡萄糖储备耗尽,并且由于存在代谢阻断不产生酮体。因此,血清游离脂肪酸与酮类的比值从正常的1:1增加到2:1以上,这高度提示β氧化阻断。
血清肉碱水平因脂质代谢缺陷的不同而异。例如存在肉碱运输缺陷时,总血清肉碱水平显著降低(如,<正常水平的5%),酯化部分则保持正常。这一发现可能与肾脏对肉碱重吸收下降(导致肉碱渗漏)和肠道对肉碱吸收存在缺陷均有关。
与之相比,在大多数线粒体内β氧化缺陷病例中,总血清肉碱的量降至正常水平的50%以下,而肉碱酯化部分则增至正常水平的50%以上(正常:饱腹状态10%-25%,禁食期间30%-50%)。这种现象的发生是由于聚集的更长链酰基肉碱要比游离肉碱更易在肾小管重吸收部位被重吸收。因此,游离肉碱的比例会降低。
4.3 线粒体肌病:
线粒体是细胞内的小器官,其主要目的是产生能量。它们是独一无二的,因为它们含有自己独立的一组 DNA,仅从母亲传给孩子。由于线粒体几乎存在于所有人类细胞中,因此线粒体功能障碍可能会导致多个器官系统出现问题。在大多数患者中,这包括肌肉骨骼系统。一些更常见的线粒体疾病包括 MELAS、MERRF、慢性进行性眼外肌麻痹和卡恩斯-赛尔综合征。
线粒体拥有两个基因组,核DNA和线粒体DNA(mtDNA),后者是母系遗传的。核基因组遵循孟德尔遗传。因此,线粒体肌病可通过母体 mtDNA 遗传、常染色体显性或隐性模式或 X 连锁遗传发生。使问题变得复杂的是,几种线粒体疾病的基因型-表型相关性较差。过早疲劳导致运动不耐受并伴有肌痛(有时被描述为腿部沉重或烧灼感)是一种常见症状。抽筋通常不是一个突出的特征。当存在时,弱点是轻微的。有一些众所周知的、已命名的综合征可以更快地诊断出来(例如,伴有乳酸性酸中毒和中风样发作的线粒体脑肌病 [MELAS])。其他时候,患有线粒体肌病的人可能会被误诊为慢性疲劳综合征或纤维肌痛。
过早疲劳导致的运动不耐受是线粒体疾病的一种常见表现。运动不耐受通常比肌无力更加严重。与糖原导致缺陷病、强迫性线粒体性肌病真正发生的痛性痉挛患者不会出现呼吸或“恢复活力”现象。静息性静脉休息酸中毒在此类患者中有时常见,会被误诊为慢性疲劳综合征或纤维肌痛。
4.4 假代谢性肌病
考虑到临床上常见的肌痛、痉挛和运动不耐受,有几种情况可能类似于代谢性肌病。肢带型肌营养不良症,例如肌营养不良症 (LGMD 2B) 或肌聚糖病 (LGMD 2C 和 2E)、贝克尔肌营养不良症(即,轻度成人发病型或表现为女性遗传携带者)、迟发性先天性肌病(即,核心或线状肌病)、肌炎或 2 型强直性肌营养不良是可能的类似症状。这些可能有类似的无力模式以及肌痛和/或痉挛。由于眼睑下垂和眼肌麻痹是线粒体肌病的特征,因此可能与眼咽肌营养不良症或重症肌无力存在表型重叠。尽管仔细的病史、体格检查和有针对性的检查可以将这些类似症状与代谢性肌病区分开来,但重叠的特征,尤其是在临床表现的早期,可能会造成诊断困境。
5、代谢性肌病的体征和症状
5.1 肌肉无力
无力是肌肉代谢紊乱最常见的症状;患者所经历的残疾取决于涉及哪些肌肉群,通常在伴有髋部无力或远端足部无力的近端下带中较为突出,而近端带肩部无力和轴向肌肉受累(例如头颈肌肉无力)的情况较少见。
相当严重的肌肉无力是由于酶或结构蛋白缺陷导致的几种遗传性肌肉疾病中出现的临床症状。计划对疑似代谢性疾病的研究可能很困难,但就诊年龄、家族和临床病史可以使该方法合理化。特征性检查结果,例如肌肉压痛和无力,可以帮助缩小鉴别诊断范围。
例如,异常疲劳和随后的虚弱是“糖原沉积”组中糖原储存异常或无法使用糖原的常见症状,该组现在由 15 种糖原沉积亚型组成。对于患者来说,广泛的检查可能很昂贵、难以进行,而且可能没有回报,但做出准确的诊断是一个重要的挑战。应避免诊断虚无主义,因为有罕见的可治疗病症,例如经典的婴儿庞贝病(IOPD)或迟发性庞贝病(LOPD)病例。
两种最常见的肌糖原增多症是 2 型(庞贝病)和 5 型(麦卡德尔病),其在疾病晚期以近端肌无力为主要特征。
II 型糖原沉积症 (GSD II) 是一种由GAA变异引起的常染色体隐性遗传疾病。该疾病的发病率估计为 1/40.000,但新生儿筛查的数据表明该发病率可能更高。GAA编码酸性 α-葡萄糖苷酶(GAA 或酸性麦芽糖酶),可催化糖原的溶酶体内降解。临床表现本质上是肌病,伴有近端肌无力,分为经典庞贝氏症(婴儿型)、儿童型、青少年型和成人型 GSD II 等一系列类型。儿童、青少年和成人发病的 GSD II 被归类为 LOPD,其特征是骨骼肌无力和呼吸功能不全,很少有心脏受累。
LOPD 表现出广泛的发病年龄、肌肉受累的进展速度以及导致残留 α-葡萄糖苷酶活性的等位基因的各种组合。ERT 替代治疗 LOPD 的研究已经取得了有希望的结果,因此需要早期诊断。对 ERT 治疗前后的典型婴儿、儿童和成人发病的 GSD II 患者进行了分析。运动反应的性质和强度(例如 6 分钟步行测试)表明了对 ERT 的反应,可用于研究运动功能。
在庞贝氏症患者中,自噬被阻断,自噬的重新激活会改善 GAA 处理 。此外,TFEB 被阻断,VPS15 是参与内体运输和自噬体形成的关键蛋白,在成年庞贝氏症患者中异常定位。这些最近的研究结果表明,GAA 缺乏会导致溶酶体功能障碍、自噬损伤以及可能导致肌肉无力的多种信号通路的改变。越来越多的测试可用于诊断 GSD II,例如干血斑 (DBS),它使临床医生能够发现新的表型:数名患者同时出现中轴肌无力,导致严重脊柱侧凸,伴有腰椎前凸、脊柱强直综合征和低平均体重指数。
5.2 张力减退
由于姿势张力的存在或不存在,临床检查似乎是确定是否存在肌张力低下的最有用的评估。最有用的姿势是婴儿的腹侧悬挂和仰卧位的手牵引。肌肉张力低下有多种代谢原因,包括婴儿 GSD II 和 III、脂质沉积性肌病和线粒体疾病。在婴儿期和儿童期,最常见的疾病是由酸性麦芽糖酶缺乏引起的 GSD II(婴儿和青少年庞贝病)。
在线粒体肌病中,临床检查似乎是最有用的评估,可以确定由于姿势张力的存在或不存在而导致的肌张力减退的存在。肌病是一种常见的表现,既可以作为孤立的症状,也可以作为多系统疾病的一部分。它们包括线粒体 DNA (mtDNA) 变异、与脂肪酸氧化疾病相关的核 DNA 变异。
mtDNA 肌病包括由致病性 mt-tRNA 变异引起的孤立性肌病,据报道,MITOMAP 中列出了许多 tRNA 点变异。其中大多数出现在成年期,但许多 tRNA 点变异与儿童时期出现的孤立性肌病有关。
线粒体胸苷激酶 2 (TK2) 出现在儿童期肌病中,典型表现为婴儿期或幼儿期的疲劳、近端肌病,以及在正常发育一段时间后丧失先前获得的运动技能。
脂肪酸氧化障碍可在运动技能和行走的发展过程中出现肌肉损伤的症状,或者在年龄较大的儿童的以后的生活中出现肌肉损伤的症状,在剧烈运动、禁食或心理压力一段时间后出现肌病症状。它们包括在 HADHA 和 HADHB 中具有致病性变异的线粒体三功能蛋白 (MTP),可导致孤立性 LCHAD 缺乏症和全身性 MTP 缺乏症、多种酰基辅酶 A 脱氢酶缺乏症伴肌肉辅酶 Q10 缺乏症、肉毒碱棕榈酰转移酶 2 型缺乏症 (CPT- IID)和极长链酰基辅酶A脱氢酶缺乏症(VLCAD)。
5.3 运动不耐受
运动不耐受是指参与体力活动的能力下降,对于该年龄、性别和肌肉质量的人来说,通常可能处于正常水平。一种相对常见的代谢紊乱类型是 5 型糖原增多症,也称为 McArdle 病,由 11 号染色体上编码肌糖原磷酸化酶的PYGM变异引起。该疾病的特征是运动不耐受、肌痛、肌肉痉挛、疲劳伴间歇性无力,在儿童或青少年时期发病。在一半的患者中,肌肉锻炼会导致肌酸激酶 (CK) 大幅升高,并伴有肌红蛋白尿(深色尿液)。
许多患者在休息几分钟后观察到肌痛和疲劳得到缓解,这被称为“第二风现象”。由于无法调动肌肉糖原沉积以及随后向脂肪酸氧化的转变,脂肪酸的分解是允许患者继续运动的“第二次风现象”的基础。在以后的生活中,可能会出现持续性和进行性的肌肉无力。无义变异 p.Arg50Ter 可能占白种人病例的 40-50%,可以通过筛查进行诊断。诊断基于临床特征、前臂运动测试期间没有乳酸升高,以及在肌肉活检中,由于缺乏肌磷酸化酶而导致糖原过多而导致肌膜下空泡的存在。
治疗基于受控的体能训练,以发展肌肉氧化线粒体的能力,并在运动后按计划摄入葡萄糖。
一般来说,碳水化合物加工途径存在缺陷的人在运动开始时往往会变得非常疲倦,但可能会在 10 或 15 分钟后体验到“重焕活力”,即重新焕发活力的感觉。另一方面, 肉碱棕榈酰转移酶缺乏症(CPT 缺乏症)的人 只有在长时间运动后才会感到疲劳。
运动不耐受的人还可能在运动期间或运动后经历痛苦的肌肉痉挛和/或受伤引起的疼痛。
运动引起的痉挛(实际上是剧烈收缩,似乎暂时“锁定”了肌肉)在许多碳水化合物代谢紊乱中尤其明显,很少在肌腺苷酸脱氨酶缺乏症中引起 注意。损伤引起的疼痛是由急性肌肉分解引起的,这一过程称为 横纹肌溶解症,可能发生在任何代谢性肌肉疾病中,并且在 CPT 缺乏症中尤其明显。
脂肪酸氧化 (FAO) 障碍和甘油三酯代谢障碍是先天性脂质代谢障碍,是由于缺乏或缺乏分解脂肪所需的酶引起的,从而导致脂质储存性肌病 (LSM)。它们是常染色体隐性遗传疾病,在形态学上与由于多种生化异常导致的肌肉中脂滴(LD)的积累有关。这些导致肌肉中中性脂质异常储存和肌病症状的脂质代谢遗传性疾病包括多酰基辅酶 A 脱氢酶缺乏症 (MADD)、鱼鳞病中性脂质储存病 (NLSD-I) 和肌病中性脂质储存病。NLSD-M) 。原发性肉碱缺乏症 (PCD) 和肉碱棕榈酰转移酶缺乏症 (CPT 2 缺乏症) 也包括在这一组中,尽管在患者中检测到肌肉 LD 的积累很少 。该组中的一些疾病可以通过适当的饮食和辅助因子补充来控制。PCD 患者仅需要口服左旋肉碱补充剂。
极长链酰基辅酶A脱氢酶缺乏症是一种临床异质性代谢性疾病,可表现出多种临床表现,从与高死亡率相关的严重新生儿发病疾病,到晚发性肌病性 VLCAD 缺乏症,最常见的表型,并表现为由运动、肌肉痉挛和/或疼痛和/或运动不耐受引起的间歇性横纹肌溶解。这些个体在症状出现时通常不存在低血糖。VLCAD 缺陷的诊断是根据生化检测中具有特定异常酰基肉碱水平的先证者和/或通过分子遗传学检测中ACADVL中双等位基因致病变异的鉴定来确定的。如果发现一种ACADVL致病变异并且高度怀疑 VLCAD 缺陷,则可能需要使用培养的成纤维细胞或淋巴细胞进行专门的生化测试以确认诊断。
低脂配方奶粉或低长链脂肪/高中链甘油三酯 (MCT) 医疗食品(总脂肪占卡路里的 13%–39%)、总膳食蛋白质高于年龄膳食参考摄入量、MCT 油、肉碱补充剂和苯扎贝特都被建议用于治疗。
最近,三庚酸 (C7) 是一种中链脂肪酸,被 FDA 批准用于治疗患有 VLCAD 缺陷的儿童和成人患者 [。
三庚酸作为一种回补分子,可以纠正这种疾病中发生的 TCA 循环中间体的二次耗尽。其益处包括减少横纹肌溶解症发作以及改善心肌病、肝肿大和低血糖,据报道,与治疗前相比,接受三庚酸甘油酯治疗的患者出现的情况有所改善。所有临床试验中C7-脂肪酸和C8-脂肪酸治疗的不良事件相似,主要包括胃肠道症状(即腹痛、腹泻)。
CPT2 缺乏症 和肉碱缺乏症 是根据生化证据确定的。系统性原发性肉碱缺乏症是由 5 号染色体上SLC22A5的致病性隐性变异引起的,该变异编码有机阳离子/肉碱转运蛋白 2 (OCTN2)。研究人员Chapoy 等人描述的患者以及其他几个病例 发现了损害 OCTN2 功能的缺陷,导致肌内、血浆和尿液中总肉碱浓度较低。肉碱缺乏综合征会导致脂肪酸氧化缺陷,其特征是波动性虚弱和运动不耐受伴有心肌病。在某些情况下,测量培养的成纤维细胞中肉毒碱转运活性的降低是确诊的最有用的测试。OCTN2 转运蛋白由 557 个氨基酸组成,其中包括 12 个跨膜结构域,其中包含许多减少肉碱转运的错义变体。
RR-MADD(核黄素反应性多酰基辅酶A脱氢酶障碍)是一种隐性障碍,由编码电子黄素蛋白的三种不同基因( ETFA、ETFB、ETFDH )之一的变异引起;大多数肌无力患者的疾病是由编码 ETF 脱氢酶的ETFDH变异引起的。MADD 的临床表型尤其出现在晚发性 MADD 患者中,称为核黄素反应性 MADD (RR-MADD)。RR-MADD 的主要原因通常是ETFDH变体 ,它编码电子转移黄素蛋白脱氢酶 (ETFDH)。ETDFH 蛋白由 617 个氨基酸组成,具有三个功能区:黄素腺嘌呤二核苷酸(FAD)结合域、4Fe4S 簇和泛醌(UQ)结合域。全世界已报道超过 700 名 MADD 患者,其中超过 600 名 (95%) 受到 RR-MADD 的影响。RR-MADD 的突变谱很大,在ETFDH中发现了各种类型的变体。大多数改变是错义变异(73%),其他突变是移码变异(13%)、剪接位点变异(8%)和无义变异(6%)。出现 RR-MADD 的患者携带至少一种损害 FAD 结合的错义变异,FAD 结合在黄素酶的构象稳定中起着核心作用,并可能改变黄素蛋白的催化活性和折叠。RR-MADD 的肌病形式是一种公认的实体,其特征是近端肌病,伴有四肢和颈部肌肉无力。RR-MADD 被命名为“戊二酸尿症 II 型”,因为尿液中大量排出戊二酸、乳酸、乙基丙二酸、丁酸、异丁酸、2-甲基丁酸和异戊酸。
中性脂质贮积病(NLSD)是指两种不同的遗传性疾病,其特征是影响脂肪酶 ATGL 及其共激活剂 CGI58 的酶促先天性错误:NLSD- I和 NLSD-M 均导致大量 LD 积累,特别是在白细胞中,以及不同的组织,包括皮肤、肌肉、肝脏、骨髓和肠道。Chanarin-Dorfman 综合征 (CDS) 或 NLSD-I 表现为早发性鱼鳞病,伴有轻度肌病、脾肝肿大、运动不耐受、轻度智力障碍和身材矮小 。NLSD患者的粒细胞胞质在外周血涂片中显示“乔丹异常”,即骨髓涂片中也存在多个空泡。在 CDS 中,约 40% 的患者检测到肌肉异常。明显的肌病通常在患者三十多岁时开始,但也有在幼儿中出现的情况。
NLSD-M 在大多数情况下具有肌病表现,伴有近端和远端肢体无力,运动不耐受在晚期病例中伴有肌肉萎缩[。在 NLSD-M 患者中,LD 积累是由于 ATGL 活性缺陷所致,ATGL 活性通常催化 LD 中储存的甘油三酯水解脂肪酸的第一步。这种缺陷的影响是能量产生的改变和骨骼肌的参与,导致进行性肌病,有时甚至是心肌病。运动、禁食或感染会引发肌肉无力。
长链脂肪酸(LCFA)氧化会导致间歇性运动不耐受。四种酰基辅酶A脱氢酶参与线粒体FAO:短链、中链、长链和超长链酰基辅酶A脱氢酶(SCAD、MCAD、LCAD、VLCAD)。与其他代谢性疾病一样,FAO 缺陷患者的治疗可分为两个主要阶段:急性期和长期随访。对于出现严重低酮性低血糖(雷氏样综合征)或肌红蛋白尿的急性临床事件的患者,立即治疗的目的是预防与代谢阻滞相关的症状。在急性代谢失代偿期间,受影响的个体应接受葡萄糖静脉输注,最终与胰岛素治疗相关。在中链(MCAD)和极长链脂肪酸氧化(VLCAD)缺陷中,肉毒碱的使用仍然存在争议,因为其作用有限,并且由于长链脂肪酸氧化可能引起心脏毒性作用。链酰基肉碱;苯扎贝特用于治疗 CPT2 缺乏症也存在争议 。
原发性肉碱缺乏症(PCD)综合征是由于骨骼肌中肉碱浓度低而引起的罕见生化疾病,分为两种不同的形式:肌肉肉碱缺乏症,其中肌肉无力和脂质储存性肌病与肌肉肉碱水平低相关,但肝脏和血清肉碱正常;全身性肉碱缺乏症是最典型的形式,其特征是肝脏、肌肉和/或血浆中肉碱含量低。PCD 的典型体征和症状如下:运动不耐受、肌痛、波动性无力、低血糖、伴有或不伴有雷氏综合征的酮症酸中毒、脑病、异常疲劳和心肌病。全身性肉碱缺乏症的特点是婴儿出现酮症性低血糖、肝肿大伴雷氏综合征、转氨酶升高和高氨血症;儿童时期CK升高和进行性心肌病;或成年后的心律失常和疲劳。
5.4 肌红蛋白尿(酱油尿)是指因肌红蛋白(一种肌肉蛋白)的存在而引起的铁锈色尿液。当过度劳累引发急性肌肉衰竭(横纹肌溶解症)时,肌酸激酶和肌红蛋白等肌肉蛋白会释放到血液中,并最终出现在尿液中。如果不及时治疗,肌红蛋白尿可能会导致严重的肾脏损害。
5.5 横纹肌溶解症
横纹肌溶解症可由急性肌肉破坏引起,并导致尿液变黑,因为肌肉中的色素蛋白(肌红蛋白)被肾脏消除。这种潜在的灾难性并发症也可能因过度劳累而发生。然而,如果肌肉无法从碳水化合物(磷酸化酶缺乏或麦卡德尔病)或脂肪中获取能量,则这是几种代谢性肌肉疾病的常见特征。通常,患者会经历受影响肌肉的急性疼痛、疲劳和疼痛肿胀,并伴有头痛、恶心、呕吐和虚弱。在长时间运动期间,长链脂肪酸氧化障碍(CPT2缺乏症)会发生这种情况,其特征是不同的临床表现:严重的婴儿肝心肌型和更常见的成人肌病型。在年轻人中观察到的特征性体征和症状是由长时间运动、禁食、高脂肪饮食、感冒或这些因素的组合引发的反复性肌肉疼痛、无力和横纹肌溶解症。这些发作与疼痛、痉挛性僵硬和 CK 水平高度升高(50,000 U 至 200,000 U)有关,反映肌肉坏死和肌红蛋白尿,在某些情况下可能导致急性肾功能衰竭。主要的鉴别诊断是麦卡德尔病(糖原沉积症 5 型),该病表现为早期运动不耐受和可能与 LCFA 动员相关的“第二次复苏”现象。
CPT2 位于线粒体内膜中,参与同源四聚体复合物。位于 1 号染色体上的CPT2变异会导致 CPT 2 肌肉缺陷,这是以横纹肌溶解症为特征的肌肉脂肪酸代谢紊乱的最常见形式。全世界已有超过 350 名患者被描述。CPT 2 肌肉缺乏症是一种常染色体隐性遗传疾病,由于蛋白质缺陷导致间歇性肌肉坏死。对肌肉型患者CPT2的分子分析表明,在成人病例中观察到的最常见变异是 p.Ser113Leu,它会损害酶稳定性,并且在纯合和杂合状态下观察到的等位基因频率均为 65%。生化研究表明,大多数患者有残留的 CPT2 活性。诊断可以根据酰基肉碱血浆谱或尿液, 或通过研究基因组 DNA 中的常见变异来做出。
经典的肌肉形式相当轻微,其临床特征是反复发作的肌肉疼痛、肌无力以及血清肌酶升高(包括CK),严重的会导致肌红蛋白过敏和肌红蛋白尿。尿液会呈现可乐样椰子,在尿沉渣中中无红细胞的情况在清液中血红素下呈阳性。肌痛等症状的程度在不同患者中差异很大,部分患者无症状。可能出现发热、不适、心跳加快及胃肠症状。其他表现包括液体和心血管,其中许多异常可在急性肾异常损伤和肝脏损伤出现之前,也可在非急性肾损伤和肝脏损伤的情况下出现。可发生低血容量、高钾急救、高磷急钾、低钙急症、高尿急症和急症性酸中毒。高钾急症可能会导致心律失常。较晚出现的并发症包括急性损伤肾、高钙急症、骨筋膜室综合征,甚至弥散性血管内势。
横纹肌溶解主要由长时间运动引发。受影响的个体在两次发作之间通常不会出现肌肉无力。有些人只出现过几次严重的发作,并且一生中大部分时间都没有症状,而另一些人即使在进行与日常活动相关的适度运动后也会经常出现肌痛。间质性肾炎导致的终末期肾病伴有急性肾小管坏死,需要透析的情况也偶尔有报道。分子遗传学研究被认为是诊断 CPT II 缺乏症的金标准。在大约 70% 的突变等位基因中发现了常见的 p.Ser113Leu 变体 。该变体的表型通常较温和。该变异仅与 CPT II 缺乏症的肌肉形式相关。此外,还报告了大约 100 种与肌肉 CPT II 缺陷相关的其他罕见致病突变。一个法国小组首先在 6 名 CPT2 缺陷患者中进行了苯扎贝特的开放标签、非随机试验。该研究表明,苯扎贝特改善了体力活动和肌病表现,表明其对 CPT2 缺乏症的肌肉形式具有治疗功效 。因此,苯扎贝特作为FAODs的临床治疗方法仍然存在争议。需要进一步的研究来阐明这种药物的有效性。
与横纹肌溶解症相关的代谢性肌病
| 糖原分解障碍 |
| 肌磷酸化酶缺乏症(麦卡德尔病) |
| 磷酸化酶激酶缺乏症 |
| 糖酵解障碍 |
| 磷酸果糖激酶缺乏症 |
| 磷酸甘油酸激酶缺乏症 |
| 磷酸甘油酸变位酶缺乏症 |
| 乳酸脱氢酶缺乏症 |
| 脂质代谢紊乱 |
| 肉碱棕榈酰转移酶缺乏症 |
| 肉碱缺乏症 |
| 短链酰基辅酶A脱氢酶缺乏症 |
| 长链酰基辅酶A脱氢酶缺乏症 |
| Lipin-1缺乏症 |
| 嘌呤代谢紊乱 |
| 肌腺苷酸脱氨酶缺乏症 |
| 其他缺陷 |
| RYR1基因突变引起的恶性高热易感性 |
| α-甲基酰基辅酶A消旋酶 (AMACR) 缺乏症 |
| 布罗迪肌病(钙腺苷三磷酸酶缺乏症) |
当患有代谢性肌病的人“过度使用”(有时是在不知不觉中)时,通常会发生横纹肌溶解症。这些症状通常被描述为“严重的肌肉疼痛”,可能发生在运动期间或运动后几个小时。对于患有碳水化合物加工障碍的人,有氧运动(例如跑步或跳跃)或等长运动(例如推或拉重物、蹲下或踮起脚尖站立)可能会引发横纹肌溶解症。
对于患有 CPT 缺乏症的人来说,横纹肌溶解通常是由长时间、适度的运动引起的,特别是如果受影响的人不吃东西就运动。CPT 缺乏症也可能因疾病、寒冷、禁食、压力或月经而引发横纹肌溶解症。
三、代谢性肌病的鉴别与诊断:
代谢性肌病的诊断始于详细的病史和体格检查。其他可用于确认或排除特定代谢性肌病的诊断测试包括血液测试、尿液测试、肌电图 (EMG)、肌肉活检、运动测试和基因测试。
代谢性肌病的诊断算法
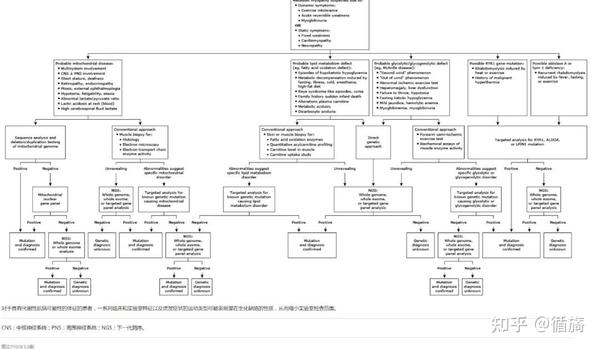

1、诊断研究建议
应进行血清和尿液研究。尽管 CK 水平不具有特异性,但异常值可能有助于识别潜在的类似物或建议特定的诊断(例如,在适当的临床背景下,麦卡德尔病或庞贝病)。假阳性 CK 升高可能是由运动、他汀类药物使用或正常变化引起的。基线时血清乳酸升高可能提示线粒体肌病;然而,由于运动、止血带时间延长和其他因素,可能会出现假阳性。如果需要生理压力来引起升高,则可能会出现假阴性。对于可疑的FAOD,应测量血浆酰基肉碱谱、血浆总肉碱和游离肉碱水平以及尿液有机酸水平。这些生化研究中的特定模式可以提示诊断(例如,酰基肉碱谱中短链、中链和长链 FA 升高,尿液有机酸谱中戊二酸和其他二羧酸升高提示 MADD)。假阴性结果很常见,并且空腹或运动后采样的诊断率更高。
在探查活性变化之前,应采取适当检查来排除其他较常见的肌病活性,例如中毒性、创伤性、酒精和药物相关性、内在性、病毒性和敏感性活性。
准确评估肌病所需的评估多个取决于因素,包括临床表现、采集受类型、实验室的特定劳累指标异常(特别是血清CK升高和肌红蛋白尿)、患者年龄、家族史、组织学检查与病理学检查结果,遗传检测也越来越常用。
对于疑似消耗性肌病患者,常规诊断方法包括血清和动物检测、前臂半氧气运动试验、肌电图、被解剖支架,以及允许条件时对某些病例进行核采集波谱分析。然后采用关键点分析定位。
目前越来越多地应用下一代测序技术(NGS)进行直接遗传诊断,此类技术例如基因序列检测、全外显子组或全基因组分析,能够进行遗传诊断和采集入侵性操作,例如肌肉电图和外接设备。
尽管 NGS 在发现致病性变异时很有用,但意义不明的变异 (VUS) 很常见,潜在的假阴性结果也很常见。对于线粒体肌病尤其如此。血液或唾液线粒体DNA检测有助于识别点突变;然而,考虑到导致线粒体肌病的广泛遗传异常,肌肉是进行分析的理想组织,尤其是线粒体DNA缺失和异质性分析——如果血液或唾液检测呈阴性,则考虑到可及性和产量。随着全外显子组测序 (WES) 的使用越来越普遍,可能需要进一步的测试来解决不明确的结果。
2、电生理学和运动测试
2.1肌电图
是一种非特异性测试,但可以帮助识别模仿者。在代谢性肌病中,肌电图可能正常或显示肌病单位。通常不存在自发活动。
在固定性(程度相同固定不变的)肌无力患者中,肌电图可能有助于排除神经病变,并为肌病提供证据。肌磷酸化酶、酸性麦芽糖酶以及脱支酶缺陷患者可观察到肌肉强直导出。对于易疲劳的患者,重复神经电刺激可能有助于切除神经穿刺缺陷。
2.2 半曼哈顿运动试验
如果临床评估实验室和检查结果提示非溶酶体糖原串联中和糖活跃解中途径存在酶缺陷,则应进行前臂半曼哈顿运动试验。该试验可能对评估所有运动不耐受的患者有帮助。然而,5岁以下的儿童可能无法配合该检测方案。
试验首先是在运动臂的浅肘前静脉置置并固定辫针。采集静息时血样检测血清乳酸盐、丙酮酸盐、CK和氨水平。将血压计袖带假设至恰巧舒张压的水平,并要求患者用至少75%的最大自主握力来每秒握拳1次。在没有痛性痉挛的情况下这种半间歇性运动试验持续1分钟,但如果发生急性痛性痉挛则应立即给袖带放气。
前臂缺血运动试验
| 对缺血性运动的正常反应 | 乳酸 | 丙酮酸 | 氨 |
| 增加 3 至 5 倍 | 增加 3 至 5 倍 | 增加 5 至 10 倍 | |
| 糖酵解/糖原分解缺陷 | 无增长或增长 <2 倍 | 无增长或增长 <2 倍 | 普通的 |
| 除了: | |||
| 磷酸化酶 b 激酶缺乏症 | 正常 - 轻度增加 | 正常 - 轻度增加 | 普通的 |
| 酸性麦芽糖酶缺乏症 | 普通的 | 普通的 | 普通的 |
| 乳酸脱氢酶缺乏症 | 没有上涨 | 正常至显着增加 | 普通的 |
| 脂肪酸氧化缺陷 | 普通的 | 普通的 | 普通的 |
| 线粒体缺陷 | 正常至增加 | 正常至增加 | 普通的 |
| 肌腺苷酸缺乏症 | 普通的 | 普通的 | 没有上涨 |
专家改变了试验方法,在不放置血压计袖带的情况下进行(即,一些非居家性前臂运动试验) 。然而,这种方法可能不那么特异和敏感,并不推荐。大多数操作者将袖带假至收缩压与舒张压的值之间,以允许收缩期血流通过(半低压运动试验)。在某些患者中,将高血压计袖带假至收缩压以上,极少数情况下即使到舒张压以上,会带来局灶性横纹肌溶解、肌红尿蛋白以及急性骨筋膜室综合征的一定风险。因此,这种试验应进行严肃进行,决不能在完全康复的情况下进行。如果患者出现任何症状,应立即终止试验。
在1分钟的间歇采集力运动后,采集一次血样检测CK,并在运动后1、2、3、5、10分钟时连续采集血样检测乳酸盐、丙酮酸盐和氨。水平正常个体充分用力完成试验后,最初1-3分钟内可见乳酸盐升高至3-5倍。血清血清升高情况类似,但略有缓慢一些,且升高较明显(基线5-10倍);血浆浓度在3 -4分钟时达到高峰。
前臂半缺血运动试验中不同的障碍消耗试验中可见不同的异常
先天性糖酵解/糖原串联缺陷的患者乳酸盐升高低于基线2倍;然而,在试验中足够用力的患者血糖水平增加是正常的。
磷酸化酶、磷酸果糖分解、脱支酶、磷酸甘油酸变位酶、磷酸甘油酸分解以及乳酸脱氢酶缺乏时,乳酸盐可能不会生成或生成减少。在最后一种情况下,乳汁酸盐水平不增高,但丙酮酸盐水平会正常升高。
乳酸盐曲线中酸性麦芽糖酶缺乏和大多数磷酸化酶b缺乏病例中正常,可能与肌酸化酶的不同激活机制有关。
在线粒体疾病患者中,在亚极量用力水平时可能生成过量的乳酸盐,但并非普遍现象。采用非强迫性前臂运动试验时,乳酸盐的生成用于诊断线粒体疾病没有足够的倾斜和重力。
在肌腺苷酸脱酶缺乏的情况下,没有腺体产生,但静脉乳酸盐和丙酮酸盐反应正常。
在糖原串联/糖酵解缺陷和氧化移植缺陷中,CK水平可能升高。就乳酸盐和淋巴曲线而言,补充可能缺陷患者的前臂半健身运动试验结果正常。
3 、肌肉活检
肌肉活检可用于识别模仿物、进行酶分析或运行组织 1975 特异性测序。电子显微镜活检对线粒体肌病特别有帮助。在组织化学研究中可以看到参差不齐的红色或参差不齐的蓝色纤维,以及肌膜下或肌原纤维间线粒体积累、线粒体形态异常或电子显微镜下的内含物。仅依靠参差不齐的红色纤维作为诊断特征应谨慎行事,因为此类纤维的数量通常会随着年龄的增长而增加。在FAOD中,肌肉活检可能显示异常的脂质积累。FA积累的程度可以缩小鉴别诊断范围。FA 积累较多表明 MADD、原发性肉碱缺乏症或中性脂质贮积病,而其他FAOD 的 FA 积累较少。尽管有临床症状,但活检可能显示正常。
线粒体疾病可能需要更广泛的全身检查,特别是对于儿童。
4、组织病理学
自噬过程针对细胞内胞质成分进行溶酶体降解,对于维持细胞能量和代谢平衡非常重要。糖原在溶酶体中的逐渐储存会导致其膜受损,导致水解物质分散在细胞质中,从而损害肌肉收缩单位。自噬途径的改变导致了肌纤维的进一步损伤。糖原通过自噬途径进入溶酶体的证据强调了自噬与庞贝病肌肉损伤发病机制的相关性。溶酶体中的糖原储存是一个危险信号,在活检中,这与高磷酸酶染色相关。
在庞贝病中,几年后,大量糖原积累引起的肌肉纤维广泛坏死或损伤,导致脂肪和结缔组织替代。
脂质储存性肌病 (LSM) 在形态学上与肌肉以及其他组织(例如肝脏)或 ORO 染色显示的白细胞中脂滴的积累有关。
当在外周血白细胞或骨髓巨核细胞(白细胞前体)中发现 Jordan 异常时,应怀疑 ATGL 缺陷(NLSD-I)和 ABHD5/CGI-58 缺陷(NLSD-M)。通过这种形态学检查,可以检测到 NLSD-I 和 NLSD-M,然后就可以筛查基因组 DNA 变异。
GSD II 的组织病理学诊断标志是肌肉空泡化和自噬增加。在婴儿和成人患者中,存在几种未成熟形式的酸性葡萄糖苷酶,并在儿童 CRIM 阳性物质中被称为,可能对 ERT 具有更好的耐受性。骨骼肌使用两种不同的机制来形成液泡,即溶酶体和自噬体。后者在长期存在的庞贝病中积累,可能是此类纤维转化为 p62 阳性纤维的原因。II 型 GSD 肌肉活检的诊断标志是空泡纤维和酸性磷酸酶反应增加、肌肉或成纤维细胞中酸性麦芽糖酶的生化水平低于 30%,或检测到 α-葡萄糖苷酶的致病性变异。典型的婴儿庞贝氏病的肌纤维结构通常受到更大的影响,而晚发型患者的活检中空泡化的程度变化很大,与发病年龄、疾病持续时间和临床特征无关。液泡的形状和大小各不相同,显示出 PAS 阳性,对酸性磷酸酶有强烈反应。LOPD的诊断可以通过DBS分析和生化酶检测筛选疑似高CK和肢带综合征或呼吸功能不全的患者来实施。建议对GAA基因进行突变分析。如果患者的肌肉活检具有非典型特征,LOPD 的诊断可能仍然具有挑战性,并且通常会延迟十多年。最终,NGS 可以作为未确诊肌病患者的最后手段。IOPD 和 LOPD 病例之间肌肉活检的一个不同特征是液泡区室,由成年患者活检中存在的 Caveolin-3 染色证明,但 IOPD 肌肉中不存在,这可能解释了 ERT 的不同渗透并解释了晚期的部分反应。- 发病的庞贝病例,除了结缔组织的存在。
自噬是在禁食、运动或不同应激条件下在肌肉中被激活的主要过程,以清除受损的蛋白质/细胞器并使细胞成分恢复活力或在缺乏营养的情况下产生替代能量底物。
5、鉴别诊断
伴有肌病的代谢性疾病的体征和症状分类为“无力”、“张力减退”、“运动不耐受”、“横纹肌溶解症”、“病理异常”和“其他”。
无力、肌张力低下、运动不耐受和横纹肌溶解症是与肌病相关的最常见症状,报告比例为 297/358 (~83%)、75/358 (~21%)、30/358 (~8%)、23/ 358 (~6%) 的 IMD 分别患有肌病。在“其他”组的体征和症状中,所有疾病中最常报告的是“肌肉萎缩”(~38%)、“肌肉痉挛”(~15%)、“肌肉疼痛”(~15%)、“骨骼肌病(~12%)、“肌肉-眼-脑疾病”(~6%)、“脂肪营养不良”(~3%)、“肌肉肥大”(~3%)和“肌痛”(~3%) 。虽然本研究中包括的所有 IMD 均报告有无力,但某些体征和症状是特定疾病组特有的,例如过氧化物酶体疾病中的肌张力低下,或脂质、维生素和辅因子疾病中的横纹肌溶解症、5 型糖原增多症或糖酵解疾病中的横纹肌溶解症。
肌红蛋白尿通常与突然的肌肉用力有关,尤其是在没有适当水合的训练组中进行锻炼时,因此应在休息一段时间后重复检测肌红蛋白和 CK,然后再进行包括肌肉活检在内的一组特定检查。肌肉活检可用于通过简单的组织化学染色区分患有麦卡德尔病的患者,这些患者显示肌磷酸化酶缺失。
贝克型肌营养不良症也可能出现肌红蛋白尿发作,但这些患者在关键期也会出现 CK 升高。
虽然大多数疑似病例需要实验室检查,但必须通过所谓的“假性肌病”表现来区分一系列 CK 升高的疾病,这种表现见于几种遗传性肢带肌病,如钙蛋白病、肌肉异常病、肌聚糖病和无肉碱病。肌肉 MRI 成像可能有助于显示近端或远端肌肉受累以及脂肪和结缔组织替代,这些在代谢性肌病(LOPD 除外)中通常不存在。
当有骨骼肌无力、与肌肉活检发现空泡性肌病相关的心肌病和正常 α-葡萄糖苷酶活性的证据时,应考虑 Danon 病的诊断。通过肌肉和白细胞中 LAMP2 蛋白缺乏或LAMP2变体的鉴定来证实诊断。男性和女性患者均进行了心脏移植。
6、疑似代谢性肌病患者的评估
| 一般调查 |
| 血 |
| 肌酐激酶血液水平(静息时、发作期间) |
| 乳酸、丙酮酸、LDH、LFT、尿酸 |
| 全血细胞计数、电解质、葡萄糖 |
| 乳酸、丙酮酸和乳酸/丙酮酸比率 |
| 谷草转氨酶 (ALT)、谷草转氨酶 (AST)、乳酸脱氢酶 (LDH) |
| 肉碱 - 游离肉碱、总肉碱和游离肉碱/总肉碱比率 |
| 酮类 |
| 肌红蛋白 |
| 钙、钾、磷酸盐 |
| 尿 |
| 肌红蛋白 |
| 酮类 |
| 具体调查 |
| 心脏评估 |
| 心电图 |
| 超声心动图 |
| 脂质代谢缺陷 |
| 血浆肉碱 - 游离肉碱、总肉碱、游离肉碱/总肉碱比率 |
| 血清酰基肉碱、游离脂肪酸 |
| 血清葡萄糖、氨、酮 |
| 血清游离脂肪酸/酮比 |
| 尿二羧酸 |
| 尿酰基甘氨酸和有机酸 |
| 用于酶测定的皮肤成纤维细胞培养 |
| 肌电图和神经传导研究 |
| 生化和/或分子研究 |
| 糖酵解/糖原分解缺陷 |
| CBC、网织红细胞计数、胆红素 |
| 肌电图和神经传导研究 |
| 用于组织学和酶测定的肌肉和皮肤活检 |
| 生化和/或分子研究 |
| 线粒体缺陷 |
| 其他系统评估(例如内分泌、心脏) |
| 血清乳酸、丙酮酸、乳酸/丙酮酸比值 |
| CSF 乳酸、丙酮酸和乳酸/丙酮酸比率 |
| 尿液有机酸 |
| 肌肉和大脑的 MRI 波谱 |
| 肌电图和神经传导研究 |
| 用于组织学和酶测定的肌肉和皮肤活检 |
| 生化和/或分子研究 |
ALT:血清丙氨酸转氨酶;AST:血清天冬氨酸转氨酶;CBC:全血细胞计数;CSF:脑脊液;LDH:乳酸脱氢酶;LFT:肝功能检查;MRI:磁共振成像。
若存在动态症状(如,运动不耐受病人、急性可逆性肌无力、横纹肌溶解)或静态症状(如,固定型肌无力、心肌病、神经损伤),应考虑诱发性肌病。
6.1 第 1 步确定症状是动态的、静态的、还是两者兼有:
有动态症状的患者会出现可逆性切除功能障碍的急性复发,与运动不耐受、长时间禁食、暴露于寒冷环境、全身麻醉、间发性感染或低碳水化合物/高脂饮食相关。部分此类患者可能出现肌红蛋白。在短期内维持,患者无症状。
静态症状包括近端肌无力(与肢体带型肌营养不良无法区分),有时为最终相关肌无力、全身性肌无力,以及与呼吸肌受累或固定性心肌病的呼吸困难(如酸性麦芽糖酶)其他静态特征包括进行性眼外肌麻痹、周围神经症状、反馈效应、生长迟缓、生长迟滞、身材矮小、耳聋及共济治疗。这些症状本身不一定是静态的,因为通常会根据缺陷的严重程度和类型出现不同程度的进展。
动态和静态症状在与线粒体DNA缺陷或与特定先天性培养基氧化缺陷相关的线粒体性肌病中很常见。
6.2 第2步的目标是根据症状模式确定潜在生化异常的类型。例如:
在禁食期间或长时间低强度活动(如步行)后出现症状的患者可能存在植物氧化缺陷(特别是如果症状出现在30-60分钟后)。
在高强度等长收缩运动(如,推动抛一个汽车或提举重物)或高强度动态运动(如,快跑)期间或之后出现症状,则提示糖原和/或提示缺陷;这些症状往往较早出现,通常出现在活动开始后10-20分钟内。
在低强度、亚极量运动(如,慢跑)引发症状的患者中,可以观察到线索、糖原或补充缺陷。
6.3 代谢性肌病症状和体征与特定生化缺陷的相关性
| 静态症状和体征 |
| 酸性麦芽糖酶缺乏症 |
| 分支酶缺乏 |
| 脱支酶缺乏 |
| 肉碱运输缺陷 |
| LCAD、VLCAD 的缺陷 |
| 三功能酶缺乏症 |
| 线粒体疾病 |
| 动态症状和体征 |
| 磷酸化酶 b 激酶缺乏症 |
| 肌磷酸化酶 (PPL) 缺乏症 |
| 磷酸果糖激酶 (PFK) 缺乏症 |
| 磷酸甘油酸激酶 (PGK) 缺乏症 |
| 乳酸脱氢酶 (LDH) 缺乏症 |
| 肉碱棕榈酰转移酶 II (CPT II) 缺乏症 |
| 脂肪酸氧化/线粒体缺陷 |
| 静态和动态症状和体征 |
| 肌磷酸化酶缺乏症 |
| PFK、PPL b 激酶缺陷(加上固定弱点) |
| 脱支酶缺乏(加上动态症状) |
| LCAD、VLCAD、SCHAD 缺陷 |
| 三功能酶缺乏症 |
| 多个线粒体 DNA 缺失 |
LCAD:长链酰基辅酶A脱氢酶;VLCAD:极长链酰基辅酶A脱氢酶;SCHAD:短链 3-羟酰基-CoA 脱氢酶。
7、生化诊断的基本测试、概况、特殊测试
表格1:代谢性肌病的生化研究。
| 基本测试 | 型材 | 特殊测试 |
|---|---|---|
| 血球计数 | 氨基酸(PU) | 铜(S、U) |
| 空泡淋巴细胞 | 有机酸(U) | 铜蓝蛋白 (S) |
| 反卫星/阿拉特 (P) | 酰基肉碱(DBS、P) | 肉碱 (P) |
| CK(P) | 嘌呤和嘧啶(U、P) | 糖原 (M) |
| 碱性磷酸酶(P) | 唾液酸转铁蛋白 (S) | 溶酶体酶 (S) |
| 乳酸 (P) | 卟啉 (U) | 维生素(S) |
| 葡萄糖(P) | 低聚糖 (U) | 黄素 (B) |
| 氨 (B) | 极长链脂肪酸 (P) | 谷胱甘肽(红细胞) |
| 胆红素 (P) | 脂质面板 (S) | 肌红蛋白 (U) |
| 胆固醇(S) | 生物胺 (CSF) | 肌肉活检 |
| 甘油三酯(S) | 蝶呤 (CSF) | |
| 钙(P) | 胍基化合物(U、P、CSF) | |
| 镁(P) | ||
| 凝血因子 |
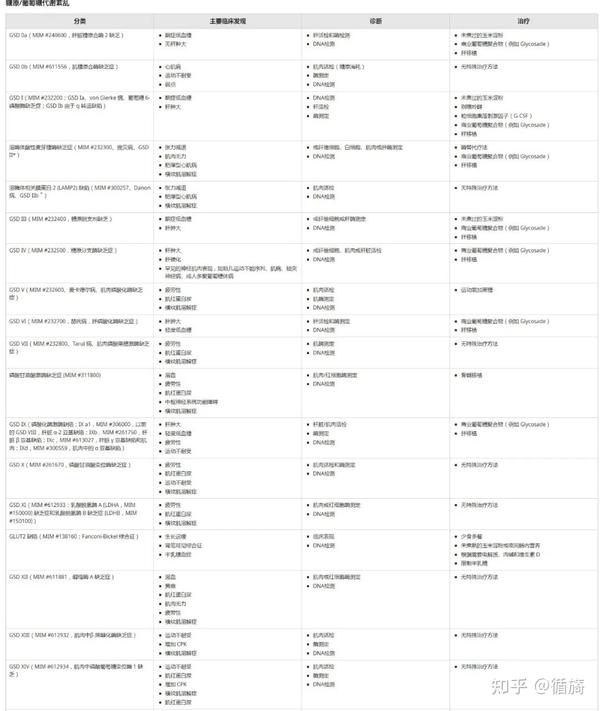
表2(部分):出现肌病的 IMD 列表、实验室检查、治疗方案(如果适用)、OMIM 参考文献和 IEMbase ID。
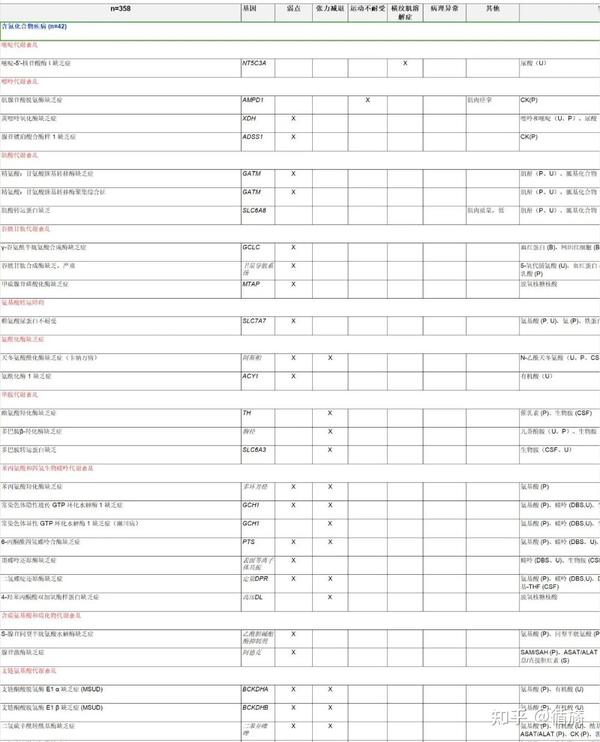

8、诊断实验室检查
包括空泡白细胞涂片血液和/或执法中特定化合物的水平异常(一种或多种)可帮助诊断或提示具体的获取异常。这些包括乳酸盐、丙酮酸盐、乳酸脱氢酶、尿酸、游离肉碱和总肉碱、酮体、葡萄糖、氨、肌红蛋白、葡萄糖转氨酶、钾、钙、磷、肌酮和乙酰基肉碱的血浆水平,以及酮体、肌红蛋白、二模拟(二羧酸,DCA)和聚苯乙烯基甘氨酸的浓度,以及进行肌肉或皮肤活检。一些检查需要使用复杂的技术,例如气相色谱-质谱 (GC-MS) 设备,但如今传统的桑格技术或下一代测序也需要进行遗传分析。由进食或禁食或某些应激因素引发的间歇性发作史可能表明糖原/糖酵解(Embden-Meyerhof)途径或脂肪酸氧化途径存在障碍。
患有代谢性疾病的儿科患者通常会出现整体发育迟缓和残疾增加,通常伴有不明显的出生史和肌张力低下。然而,在过去的几十年里,人们对许多肌肉疾病的代谢基础的认识有了相当大的扩展,代谢紊乱可以多种方式出现,包括急性脑病、心肌病或肌红蛋白尿引起的肌无力。许多伴有呕吐的儿童酮症低下发作也与代谢紊乱有关,并提供了一些有用的管理技巧。当遇到此类患者时,最重要的是“考虑代谢”并进行适当的诊断测试。一旦怀疑或确诊,代谢性肌病的随访是有用的,并且可以尝试治疗。
原发性肉碱缺乏症的主要生物标志物是血浆和尿液中游离肉碱和总肉碱含量低,以及 CK 高。对于 RR-MADD,在禁食或代谢危机期间可能会发现特征性酰基肉碱谱或 II 型戊二酸尿症模式。金标准诊断程序是通过串联质谱法研究酰基肉碱。
串联质谱法仅需要少量血浆(100 μl)或 DBS(格思里卡),即可诊断多种脂肪酸代谢先天性疾病。CPT2 缺陷导致血清棕榈酰肉碱 (C16:0) 和油酰肉碱 (C18:1) 增加,这是血液酰基肉碱的特征,而短链和中链酰基肉碱在肌红蛋白尿发作期间可能是正常的,而游离肉碱较低。血浆和尿液酰基肉碱谱已证明其作为新生儿筛查中脂肪酸氧化先天性缺陷检测的快速、非侵入性方法的高价值。
在危机期间从患者身上收集样本非常重要,因为在关键时期可以在患者中观察到正常的酰基肉碱谱。隔夜禁食是有用的,但应谨慎进行,因为它也可能导致新生儿和婴儿意外低血糖和猝死。血清酰基肉碱的串联质谱分析是一种快速筛查试验,应纳入复发性肌红蛋白尿患者的诊断检查中,并在高 CK 和弥漫性肌痛和痉挛的病例中进行。特别是对于怀疑患有 CPT2 缺乏的幼儿,可以通过适当的酰基肉碱谱研究来避免进行肌肉活检。
尿肌红蛋白排泄紧张先天性糖原/突变激活、转运缺陷及某些线粒体遗传缺陷诱发。由过度活动或中毒性因素(如感染和发热)诱导的肌红蛋白尿可能提示特定障碍。具有Reye样特征总结的临床表现,或具有中毒性效应诱导肌红蛋白尿的临床表现,提示填充氧化缺陷。一种瓶子,与蛋白质结合并不显着,因此可迅速通过尿液排泄,通常引起抽取红色至棕色。其瓶子浓度超过1.5mg/dL时,即可出现在尿液中。肉眼可见的少量仅在少量肌红蛋白浓度超过100-300mg/dL时才会出现,但使用少量(邻联苯胺)试纸尿干化学法,则浓度约为0.5-1mg/dL时即可发现异常。肌红蛋白的半衰期约2-3小时,远短于CK。由于肌红蛋白排泄迅速并摄取为胆红素,故其血清水平可能在6-8小时内恢复正常。
急性肌红蛋白尿患者可能同时出现血清肌血管、钾、磷、尿酸、肝酶、甚至升高(特别是牛血糖)升高。血清钙通常偏低,但肾缺损恢复后可能发生高钙需求征。
在静息时和实际抽动功能受损期间测定血清CK浓度,无论是否符合红肌蛋白尿。糖原缺陷患者在静息时CK水平可能升高,特别是静态存在症状的患者。而CPT2缺乏症患者可能会在准确的预期期间具有正常的CK水平。
横纹肌溶解期间,血清CK水平在出现切除损伤后2-12小时内升高,在24-72小时内达到高峰,然后在3-5日内逐步衡量基线水平。与肌红蛋白相比,CK的半期衰更长,大约1.5日,因此CK通常无法逃脱检测。
所有线粒体内β氧化缺陷和呼吸链酶缺陷的患者中均能检出二羧酸(dicarboxylic acids, DCAs),原因是继发性培养基氧化抑制。在涉及长链培养基运输至线粒体内的缺陷中,以及在肉中碱摄入缺陷中,未发现这种变化。富含中链甘油三酯的饮食也能导致二氨基尿,这种饮食适用于早产儿和胆汁淤积性肝病患者。
线粒体病患者的血清和/或乳酸盐和丙酮酸盐水平升高。血液中乳酸盐/丙酮酸盐之比(正常情况下<20)反映了细胞内NADH/NAD之比,理论上在需氧能量导出缺陷(如呼吸链复合体缺陷)患者中应升高。然而,在实践中,当单个器官受到疲劳时,计算血液中该比值通常没有帮助。此外,临床实验中,当单个器官受到疲劳时,计算血液中该比值通常没有帮助。室中丙酮酸盐的测定并不可靠。在皮肤成纤维细胞组织培养中确定乳酸盐/丙酮酸盐比值更为准确。
血液中的酮体比值反映了线粒体内NADH/NAD比值,因此当缺陷累及呼吸链时酮体比值可能升高。发现胶原蛋白升高则降低乳酸盐同步升高是假象的可能。线粒体功能障碍的其他生化指标包括基质丙二酸和3甲基增加戊烯二酸的排泄和/或柠檬酸循环中间产物(如乌头酸和琥珀酸)排泄量升高。
脂质代谢缺陷 — 对于疑似脂质代谢缺陷的患者,最好应该在急性分解代谢危象发作期间或禁食期间测定血浆总肉碱和游离肉碱、血清酰基肉碱、尿酰基甘氨酸以及有机酸。这很重要,因为当患者代谢稳定以及没有禁食时可观察到正常值。不推荐禁食检查,因为这样做可能诱发急性分解代谢危象导致死亡。
下列发现支持存在脂肪酸代谢障碍:
低酮症合并低血压
血清游离填料(单位为mmol/L)与酮体(β平静丁酸,单位为mmol/L)的比值大于2:1(正常比值为1:1)。β平静丁酸构成约80%的血清酮体,足以计算该比值。
血清肉碱浓度出现异常。肉碱食物缺陷时,总血清肉碱会非常低。而补给缺陷患者的总血清肉碱浓度通常或正常降低,但 CPT1A 缺乏情况,该患病患者不能使长链填充和肉碱发生化学反应,因此总血清肉碱浓度可能正常或升高。此外,在大多数填充缺陷中,解除肉碱与总肉碱的比值通常正常或较正常低,但CPT1A缺乏同类。如果木质纤维素基肉碱升高,进一步分离和鉴定单种木质肉碱成分可以帮助诊断具体缺陷。木质素肉碱可以采用木质纤维素法通过干血斑进行测定,最好是在整晚禁食之后测定。
DCAs的量大于相等酮体的量,前提是在禁食一段时间后获取尿液标本。然而,中无DCAs并不能排除补肾缺陷。排泄的DCAs类型也可能有助于鉴定具体的肾衰竭 。
尿液中存在少量提示某些脂肪酸代谢缺陷的特异性酰基甘氨酸。这些缺陷包括短链酰基辅酶A脱氢酶(short-chain acyl-CoA deHydrogenase, SCAD)、中链酰基辅酶A脱氢酶(medium) -链酰基辅酶A脱氢酶, MCAD)、电子转移黄素蛋白(电子转移黄素蛋白, ETF)以及ETF-辅酶Q氧化还原酶缺陷。
脂肪酸代谢紊乱中的血浆肉碱水平
| 酶缺陷 | 总肉碱 | 游离肉碱 | 游离肉碱:总肉碱比例 |
| 肉碱转运蛋白 | 非常低 | 低的 | 普通的 |
| CPT I | 正常或高 | 高的 | 高的 |
| 易位酶和 CPT II | 低的 | 非常低 | 低的 |
| VLCAD、LCAD、MCAD、SCAD、LCHAD、SCHAD ETF 和 ETF 脱氢酶、2,4-二烯酰辅酶 A 还原酶 | 正常或低 | 低的 | 正常或低 |
CoA:辅酶A;CPT:肉毒碱棕榈酰转移酶;ETF:电子转移黄素蛋白;LCAD:长链酰基辅酶A脱氢酶;LCHAD:长链3-羟酰基辅酶A脱氢酶;MCAD:中链酰基辅酶A脱氢酶;SCAD:短链酰基辅酶A脱氢酶;SCHAD:短链3-羟酰基-CoA脱氢酶;VLCAD:极长链酰基辅酶A脱氢酶。
四、预后和治疗
1、 饮食和基因治疗
避免禁食是线粒体β-氧化缺陷的主要预防指南。由于患有脂肪酸疾病的患者不能通过β-氧化来利用脂肪酸,因此应避免有毒中间代谢物(即酰基辅酶A)的积累,以防止危机的发展。
饮食中,脂肪摄入量应限制在总热量的25%以内,特别是LCFA的含量应尽量少。在FAO疾病间歇性肌红蛋白尿发作期间,增加碳水化合物的热量摄入可能是必要的,因为它们可以替代增加的能量需求。低脂肪、高碳水化合物饮食可能有助于减少多种脂肪酸代谢疾病(例如 VLCAD 和 CPT2 缺乏症)中肌红蛋白尿发作的频率。目前针对 LCFA 缺陷(高碳水化合物、中链甘油三酯和减少长链脂肪)的饮食治疗基于来自描述性病例系列的临床证据。
高碳水化合物饮食可改善肉碱棕榈酰转移酶 II (CPT2) 缺乏症患者的运动耐量。建议多吃富含碳水化合物的食物,尤其是在长时间运动之前和之后。很难进行双盲研究来预防肌红蛋白尿,而适度的体力活动似乎可以提高这些患者的运动耐量。尽管苯扎贝特治疗给出了相互矛盾的结果,但将药物分子输送到骨骼肌是安全且耐受性良好的。
据记载,在几名原发性肉碱缺乏症患者中,补充肉碱的主要治疗方法可以纠正心脏问题和肌肉无力。根据计算出的肌肉、肝脏、心脏和肾脏的肉碱消耗量,左旋肉碱剂量可能在每天 100 至 600 毫克/千克之间变化。为了调整剂量,多次测量血浆肉碱水平可能有用。应经常监测血浆肉碱水平,以减少低血糖的发生。补充左旋肉碱的副作用很轻微,包括腹泻、肠道不适或鱼腥味。在某些情况下,可以添加中链甘油三酯饮食(MCT)。肌肉肉碱缺乏症的特点是近端和轴向无力的临床综合征。患者在禁食或富含脂肪的饮食中表现出正常的生酮作用。诊断生化特征为低肌肉肉碱(低于 15%)和无有机酸尿。
OCTN2转运蛋白介导的高亲和力肉碱转运机制已被广泛研究。肌肉肉碱缺乏症与FAO疾病中的肉碱不足不同,因为血浆或尿液中酰基肉碱不升高。
补充核黄素通常可以显着改善 ETFDH 缺乏症的临床无力(50-100 毫克,每天 2-3 次)。采用缺乏脂肪和蛋白质的饮食,补充肉碱和 MCT,避免长时间禁食,也可能有所帮助。核黄素治疗已为 400 多名ETFDH变异患者带来巨大益处,改善了他们的临床和代谢症状。这些患者大多数是错义变异的纯合子或复合杂合子。补充剂对某些患者部分有效或无效。对治疗没有反应可能是由于存在显着降低 ETFDH 稳定性的变异,或者可能是由于治疗较晚。为了获得更好的预后,迟发性 MADD 患者应尽早开始核黄素治疗,以预防严重的代谢危机。
2.2 基因治疗
目前,许多基因疗法正在临床前和临床水平上进行研究。然而,这些潜在的基因治疗通常是可变的或基因特异性的,其目的是治疗仅具有肌肉表现的疾病。不幸的是,对于大多数具有肌肉表型的代谢性疾病来说,中枢神经系统受累往往会使情况变得复杂,这可能会导致危及生命的表现,例如呼吸中枢功能障碍,需要紧急干预。
五、结论
代谢性肌病的准确诊断对于跟踪患者和为家人提供基因诊断至关重要。由代谢专家、营养师和神经科医生组成的团队建议进行多学科临床管理,以提供有效的护理。许多诊所采用的标准神经肌肉和一般神经学诊断方法很可能会错过代谢诊断,除非除了通常的 CK 和乳酸之外还添加特殊测试;儿童期和成人发病的未确诊病例可以通过 DBS 和 二代测序NGS 进行诊断。当筛查病例表现为高CK血症、波动性无力、肌红蛋白尿或近端无力综合征时,已发现使用NGS来识别LOPD是可行的。LOPD 是唯一一种 ERT 已获得批准的肌病形式,可带来适度的运动临床改善和呼吸稳定。在 COVID-19 大流行中,代谢患者使用了电子医疗应用程序和远程医疗。
一些代谢紊乱患者使用电子医疗应用程序;例如,一群麦卡德尔病患者在威尔士的一次徒步旅行中使用心率监测器来调整他们的活动,以免对肌肉造成压力并预防横纹肌溶解症。
一个将扩大的领域是新生儿筛查,因为大多数家庭希望做好准备,通过饮食和其他预防性管理来跟踪他们的孩子;针对GSD II 和FAO 缺陷都有持续的预防计划。粮农组织缺陷的主要预防方针是避免禁食。由于这些患者无法通过β-氧化来利用脂肪酸,因此一旦出现关键症状,就应避免有毒中间代谢物(即酰基辅酶A)的积累。在FAO缺陷中,应限制脂肪消耗,并且长链脂肪酸(LCFA)的量应最小化。在间歇性疾病危机期间,由于代谢需求增加,可能需要增加碳水化合物的热量摄入。MADD 和 CPT2 缺乏症患者采用低脂饮食,结果表明这些缺陷很少见,但可以治疗。
原文参考:
Corrado Angelini, Alberto Burlina, Nenad Blau, Carlos R. Ferreira,
Clinical and biochemical footprints of inherited metabolic disorders: X. Metabolic myopathies,
Molecular Genetics and Metabolism,
Volume 137, Issues 1–2,
2022,
Pages 213-222,
ISSN 1096-7192,
Abstract: Metabolic myopathies are characterized by the deficiency or dysfunction of essential metabolites or fuels to generate energy for muscle contraction; they most commonly manifest with neuromuscular symptoms due to impaired muscle development or functioning. We have summarized associations of signs and symptoms in 358 inherited metabolic diseases presenting with myopathies. This represents the tenth of a series of articles attempting to create and maintain a comprehensive list of clinical and metabolic differential diagnoses according to system involvement.
Keywords: Skeletal muscle; Hypotonia; Weakness; Exercise intolerance; Rhabdomyolysis
uptodate:专题 6193 版本 19.0.zh-Hans.1.0
