
引起脑肌病、肌病的“线粒体呼吸链复合体 IV 缺乏症 ”COX 缺乏症

线粒体呼吸链复合物 IV 缺乏征(Complex IV Deficiency),也称细胞色素 C 氧化酶缺乏症(Cytochrome c oxidase deficiency, COX),是一种非常罕见的遗传性代谢性疾病,其特征是缺乏细胞色素 C 氧化酶 (COX) 或复合物 IV,这是一种在有助于调节能量产生(线粒体)的亚细胞结构中活跃的必需酶。COX 是线粒体氧化磷酸化 (OXPHOS) 系统的第四个复合物,其中顺序电子转移通过复合物 I-IV 与质子泵耦合。产生的电化学梯度最终被复合物 V(ATP 合酶)利用,从 ADP 和无机磷酸盐合成 ATP。COX 缺乏可能仅限于(局部)骨骼肌组织,也可能影响多种组织,例如心脏、肾脏、肝脏、大脑和/或结缔组织(成纤维细胞);在其他情况下,COX 缺乏可能是全身性的(系统性的)。
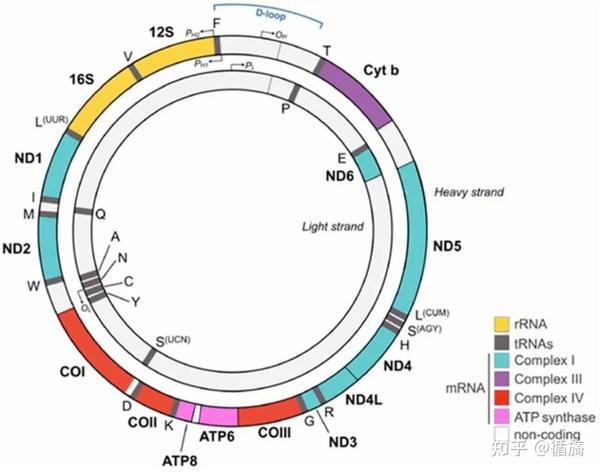

图线粒体 DNA (mtDNA) 示意图。每个蛋白质编码基因都用彩色条表示,并且编码同一复合物亚基的所有基因都用相同的颜色表示。rRNA 以黄色表示,tRNA 以灰色表示
线粒体疾病的合集:
一、临床表型
1、体征和症状
已鉴定出6种不同形式的细胞色素 C 氧化酶缺乏症。症状的范围和严重程度因情况而异。
第一种类型 称为 COX 缺乏症,即良性婴儿线粒体肌病。受影响的婴儿表现出许多与患有该疾病的更严重婴儿形式的婴儿相同的症状,可能在出生后不久开始,并伴有肌张力低下、虚弱、乳酸性酸中毒、红色纤维破碎、呼吸系统问题,由于此类型缺乏仅限于(局部)骨骼肌组织,但心脏和肾脏不受影响。患有这种疾病的婴儿可能会出现一些症状,其特征是血液中乳酸含量异常高(乳酸性酸中毒)。如果不及时治疗,可能会出现危及生命的并发症(例如呼吸衰竭)。通过适当的强化治疗,这种形式的 COX 缺乏症可能会在生命的最初几年内自发恢复。
第二种类型 称为 COX 缺乏症,致命性婴儿线粒体肌病,细胞色素 C 氧化酶的缺乏会影响骨骼肌组织以及其他一些组织,例如心脏、肾脏、肝脏、大脑和/或结缔组织。成纤维细胞)。与这种疾病相关的症状通常在出生后三到四周内出现,这些症状可能包括:全身骨骼肌普遍无力(肌强直),心脏和肾脏异常,受影响的婴儿也可能无法以预期的速度增加体重(无法成长)和/或表现出微弱的哭声;吸吮、吞咽和/或呼吸困难;和/或“松弛”或肌张力差(张力减退)。也可能会出现乳酸性酸中毒,可能导致呼吸和肾功能受损。其他症状可能是由导致托尼-范科尼-德布雷综合征的肾脏特定缺陷引起的,这种疾病会导致肾功能障碍,并涉及葡萄糖、磷酸盐、氨基酸、碳酸氢盐、钙和水的过量尿液排泄。de Toni-Fanconi-Debre 综合征引起的症状可能包括过度口渴(烦渴)和过度排尿(多尿)。
第三种类型 称为COX 缺乏与致命性婴儿心脑肌病 [CEMCOX] 相关(由SCO2、COX15、COA5或COA6基因突变引起)细胞色素 C 氧化酶缺乏 [CEMCOX] 引起的心脑肌病的主要特征是心肌病,可在子宫内或出生后几天内出现。以下神经症状也很常见:呼吸异常、眼球震颤和回旋异常。这种疾病在婴儿早期通常是致命的。那些SCO2基因突变的人往往患有更严重的疾病。
第四种类型 称为Leigh’s disease(亚急性坏死性脑脊髓病),被认为是 COX 缺乏症的全身(脑肌病)形式,由SURF1、COX15、TACO1或PET100突变引起。利氏病的特点是大脑进行性退化和身体其他器官(包括心脏、肾脏、肌肉和肝脏)功能障碍。症状通常在三个月到两岁之间开始,利氏病最主要的症状涉及大脑和脊髓(中枢神经系统)。在大多数受影响的婴儿中,第一个明显的迹象可能包括丧失先前获得的运动技能或丧失头部控制和吸吮能力差。这些症状可能伴有严重的食欲不振、呕吐、烦躁、持续哭泣和/或可能的癫痫发作。如果发病时间较晚(即 2 岁),受影响的儿童可能会出现发音困难(构音障碍)和协调行走或跑步等随意运动(共济失调)。先前获得的智力技能可能会减弱,也可能出现智力障碍。
第五种类型 称为COX缺乏症法国-加拿大型,由LRPPRC基因突变引起,细胞色素 C 氧化酶缺乏会影响骨骼肌组织、结缔组织(成纤维细胞),特别是脑组织(利氏病)和肝脏。然而,肾脏和心脏组织表现出接近正常的细胞色素 C 氧化酶活性。受影响的婴儿和儿童可能表现出发育迟缓、肌张力减退(张力减退)、轻微面部异常(轻度面部畸形)、利氏病、斜视(斜视)、控制随意运动的能力受损(共济失调)、体内脂肪堆积肝脏退化(微泡性脂肪变性)和/或乳酸酸中毒发作,可能导致危及生命的并发症,例如呼吸和肾衰竭。
第六种类型 称为成人形式,在某些罕见的情况下,COX 缺乏症的症状可能要到青春期或成年后才会出现。成人形式的特征是全身肌肉疼痛、肌张力减退以及肌肉组织偶尔抽搐和僵硬。这些患者可能有糖尿病、急性听力损失、高脂血症、高尿酸血症、动脉高血压、多关节病、性腺功能减退症和甲状腺功能减退症病史。家族史中可能存在虚弱、肌痛、CK升高和糖尿病。临床研究可能显示姿势性震颤、腱反射减弱和血清 CK 水平升高。肌肉活检是非特异性的,但肌肉匀浆的生物化学可能揭示出孤立的复杂 IV 缺陷和辅酶 Q (CoQ) 量减少。
2、病因
已发现 20 多个基因突变会导致细胞色素C氧化酶缺乏。大多数基因存在于细胞核中的DNA (核DNA)中。然而,一些基因存在于称为线粒体的特殊细胞结构的 DNA 中。这种类型的 DNA 被称为线粒体 DNA (mtDNA)。大多数细胞色素C氧化酶缺乏症是由核 DNA 中的基因突变引起的。然而,在极少数情况下,线粒体 DNA 内的基因突变会导致这种情况。
与细胞色素c氧化酶缺乏相关的基因通过氧化磷酸化过程参与线粒体的能量产生。这些基因的突变会影响一种称为细胞色素c氧化酶的酶复合物,该酶负责氧化磷酸化的最后步骤之一。细胞色素c氧化酶由称为全酶的两大酶(复合物)组成,每个酶都由多个蛋白质部分(亚基)组成。许多其他蛋白质参与将这些亚基组装成全酶。
在大多数情况下,细胞色素C氧化酶缺乏症是由改变组装全酶的蛋白质的突变引起的。结果,全酶要么部分组装,要么根本不组装。没有完整的全酶,细胞色素c氧化酶就无法形成。不太常见的是,突变会改变全酶亚基,导致细胞色素c氧化酶无功能。无论细胞色素c氧化酶是否未形成或不起作用,这种缺失的酶复合物都会破坏氧化磷酸化的最后一步,导致能量产生减少。
研究人员认为,氧化磷酸化受损会减少细胞中可用的能量,从而导致细胞死亡。某些需要大量能量的组织,例如大脑、肌肉和心脏,似乎对能量的减少特别敏感。这些和其他敏感组织中的细胞死亡可能导致细胞色素C氧化酶缺乏的特征
3、遗传模式
细胞色素c氧化酶缺乏症根据所涉及的基因可能有不同的遗传模式。
当这种情况是由核 DNA 内的基因突变引起时,它会以常染色体隐性模式遗传,这意味着每个细胞中该基因的两个副本都有突变。常染色体隐性遗传病患者的父母各携带一份突变基因,但他们通常不会表现出该病的体征和症状。
当这种情况是由线粒体 DNA 内的基因突变引起时,它会以线粒体模式遗传,这也称为母系遗传。这种遗传模式适用于 mtDNA 中包含的基因。由于卵细胞(而不是精子细胞)为发育中的胚胎提供线粒体,因此儿童只能从母亲那里继承线粒体 DNA 突变导致的疾病。这些疾病可能出现在家庭的每一代人中,并且可能影响男性和女性,但父亲不会将与线粒体 DNA 变化相关的特征遗传给孩子。
人类特征,包括经典的遗传疾病,是两种基因相互作用的产物,一种来自父亲,另一种来自父亲。母亲。
当一个人继承了同一性状的异常基因的两份拷贝(父母各一份)时,就会发生隐性遗传病。如果一个人接收了一种正常基因和一种疾病基因,则该人将成为该疾病的携带者,但通常不会表现出症状。两个携带者父母每次怀孕都会传递缺陷基因并生出受影响孩子的风险为 25%。每次怀孕生出像父母一样是携带者的孩子的风险是 50%。孩子从父母双方获得正常基因并在该特定性状上保持正常基因的几率为 25%。男性和女性的风险相同。
所有个体都携带4-5个异常基因。近亲(近亲)的父母比无血缘关系的父母更有可能携带相同的异常基因,这增加了生出患有隐性遗传疾病的孩子的风险。
极少数情况下,COX 缺乏症是由线粒体基因的新突变或遗传突变引起的。人体大多数细胞中都有数百个线粒体,它们调节细胞能量的产生,并在其独特的 DNA 中携带此过程的遗传蓝图。细胞色素 C 氧化酶由 13 个亚基组成,其中三个亚基被认为由线粒体 DNA (mtDNA) 编码,而其余亚基由细胞核 DNA 编码。
影响线粒体基因 (mtDNA) 的突变是从母亲遗传的。精子细胞中的线粒体 DNA 通常会在受精过程中丢失。因此,人类所有的线粒体DNA都来自于母亲。受影响的母亲会将突变遗传给她所有的孩子,但只有她的女儿会将突变遗传给她们的孩子。一些受影响的个体有一种新的线粒体 DNA 突变,该突变不是遗传的。
当细胞分裂时,正常线粒体DNA和突变线粒体DNA的数量以不可预测的方式分布在不同组织中。因此,突变的线粒体DNA在同一个体的不同组织中以不同的速率积累。因此,具有相同线粒体DNA突变的家庭成员可能在不同时间和不同严重程度表现出各种不同的症状和体征。
复合体 IV(细胞色素 C 氧化酶缺乏症)是呼吸链的末端酶,由 14 个亚基组成,其中 3 个亚基(名为 COX1、COX2 和 COX3)由线粒体 DNA 编码。引起 COX 缺陷的致病突变大多见于核 DNA 编码基因,与常染色体隐性遗传有关(APOPT1、C12ORF62、COA3、COA5、COA6、COX10、COX14、COX15、COX20、COX6B1、FARS2 FASTKD2、LRPPRC、PET100、POLG、 SCO1、SCO2、SURF1、TACO1)。极少数情况下,COX 缺乏症可能是由线粒体 DNA 基因突变引起的,显示母系遗传(MTCO1、MTCO2、MTCO3、MTTL1、MTTS1)。这种情况主要影响骨骼肌,但也可能是全身性的,因此也会影响心脏、大脑、肾脏、结缔组织和肝脏。
COX 缺陷可能是孤立的(当由上述任何基因的突变引起时)或作为染色体疾病的一部分(当由涉及相邻基因的大缺失引起时 – 参见例如染色体 19q13.11 的纯合性缺失 )
4、受影响人群
细胞色素 C 氧化酶 (COX) 缺乏症是一种非常罕见的代谢性疾病,影响男性和女性的人数似乎相同。各种形式的疾病(即婴儿线粒体肌病和利氏病)的总体发病率尚不清楚。然而,在魁北克省东北部 Saguenay-Lac-Saint-Jean 地区的法裔加拿大人中,已有报道称法裔加拿大人患有 COX 缺乏症,估计每 2,473 名新生儿中就有 1 人患有 COX 缺乏症。
在大多数婴儿线粒体肌病形式的这种疾病中,发病发生在出生后第一个月内。利氏病通常在三个月到两岁之间变得明显。法国-加拿大型 COX 缺乏症也往往在婴儿期或儿童期变得明显。然而,在某些罕见的情况下,COX 缺乏症的症状可能要到青春期或成年后才会出现。
5、具有类似症状的疾病
以下疾病的症状可能与细胞色素 C 氧化酶缺乏症的症状相似。比较可能有助于鉴别诊断:
MELAS 综合征是一组称为线粒体脑肌病的罕见疾病之一,其特征是反复发作的中风样发作,其中突然头痛,随后呕吐和癫痫发作。通常会出现身材矮小、血液中乳酸积聚(乳酸性酸中毒)和身体一侧肌肉无力(偏瘫)的情况。视觉症状可能包括视力受损或一半视野失明(偏盲)和/或因与视觉有关的大脑区域病变而失明(皮质失明)。MELAS 综合征被认为是由于线粒体 (mtDNA) 内遗传物质(突变)的异常变化而遗传的。MELAS具体介绍可以参见:遗传代谢线粒体肌病脑病伴高乳酸血症及卒中样发作MELAS:乳酸中毒和碳酸氢盐治疗
Kearns-Sayre 综合征是一种罕见的神经肌肉疾病,具有以下三个主要表现:某些眼部肌肉进行性麻痹(慢性进行性外眼肌麻痹 [CPEO]);有色(色素)物质在眼睛内层富含神经的膜上异常积聚(非典型色素性视网膜炎),导致慢性炎症、进行性退化和某些眼睛结构的磨损(视网膜色素变性);和心脏病(心肌病),例如心脏传导阻滞。其他发现可能包括肌肉无力、身材矮小、听力损失和/或由于影响大脑部分(小脑)的问题而导致自主运动协调能力受损(共济失调)。卡恩斯-塞尔综合征(部分)属于一组罕见的神经肌肉疾病,称为线粒体脑肌病。在这些疾病中,存在异常大量的有缺陷的线粒体。在大约 80% 的卡恩斯-塞尔综合征病例中,测试将揭示涉及线粒体中独特 DNA (mtDNA) 的遗传物质缺失(缺失)。
MERRF 综合征(与参差不齐的红色纤维相关的肌阵挛癫痫)是一组罕见的神经肌肉疾病(称为线粒体脑肌病)之一。MERRF 综合征最典型的症状是肌阵挛发作,通常是突然、短暂、抽搐的痉挛,可能影响手臂和腿(四肢)或整个身体。受影响的个体也可能存在协调运动能力受损(共济失调)以及血液中乳酸积聚(乳酸性酸中毒)。也可能出现说话困难(构音障碍)、视神经萎缩、身材矮小、听力丧失、痴呆和眼睛不自主抽动(眼球震颤)。MERRF 综合征被认为是由于线粒体 (mtDNA) 内遗传物质(突变)的异常变化而遗传的。
可导致心力衰竭的扩张型心肌病 (DCM) 也与 COX 缺乏有关。由于心脏的高能量需求,COX 在线粒体能量供应中的作用对于心脏功能至关重要。尽管 DCM 具有多种病因,但早在 1990 年就报道了 COX 缺乏的证据。后来的研究描述了与 DCM 相关的功能上重要的 COX 基因突变。DCM 患者的心脏活检发现 COX 表达(IV 亚基 mRNA 和蛋白质)减少,这与心脏功能下降相关。然而,COX 缺乏在 DCM 领域的相关性和重要性还有待进一步更明确的研究。
二、诊断
细胞色素 C 氧化酶 (COX) 缺乏症可在出生后(产后)根据全面的临床评估、特征性发现、详细的患者病史和各种专门测试来诊断。根据医学文献,对于出现乳酸性酸中毒的婴儿或儿童,应考虑诊断 COX 缺乏症。
可以进行专门的实验室研究来帮助确认这样的诊断,包括结缔组织细胞(成纤维细胞)的酶测试(化验),这可能揭示细胞色素 C 氧化酶的活性降低。此外,肌肉活检研究可能会揭示“参差不齐的红色纤维”(一种引人注目的、独特的肌肉组织异常,在显微镜下观察时很明显),表明 COX 活性水平显着降低以及线粒体结构的改变或异常。其他实验室测试可能包括专门的染色技术,以揭示 COX 酶的哪些亚基受到影响。
对于患有 COX 缺乏型婴儿线粒体肌病且同时表现出 de Toni-Fanconi-Debre 综合征的婴儿,实验室研究可能会揭示肾功能障碍的迹象,包括血液中葡萄糖、磷酸盐、氨基酸、碳酸氢盐、钙和水含量异常高。
先进的成像技术可能有助于诊断 COX 缺乏症的leigh病。大脑的计算机断层扫描 (CT) 扫描或磁共振成像 (MRI) 可能会发现大脑某些区域(例如脑干、小脑、基底神经节)的异常。在 CT 扫描过程中,计算机和 X 射线用于制作显示大脑组织结构横截面图像的胶片。在 MRI 过程中,磁场和无线电波用于创建大脑的横截面图像。此外,实验室研究可能揭示大脑、骨骼肌、结缔组织(成纤维细胞)、心脏、肝脏和肾脏等组织细胞内细胞色素 C 氧化酶活性普遍降低。实验室测试还可能表明血液中酸性废物含量较高(乳酸性酸中毒)。
在法裔加拿大型 COX 缺乏症患者中,实验室研究和先进的影像学检查可能会揭示 COX 缺乏症 Leigh 病形式的特征性发现。此外,酶测定可以证明细胞色素C氧化酶活性降低的严重程度在不同组织细胞中差异很大。例如,虽然检测可能显示心脏和肾脏中 COX 活性几乎正常水平,但骨骼肌和结缔组织细胞(成纤维细胞)内的 COX 酶活性可能约为正常水平的 50%,而脑和肝组织细胞中的 COX 酶活性严重降低。此外,影像学研究或其他测试可能会揭示肝脏内异常的脂肪堆积和退化(微泡性脂肪变性)。
分子遗传学检测可识别与 COX 缺乏相关的一些核和线粒体基因突变。
通过全外显子组测序进行的基因检测可以揭示已知基因中的致病性突变,或检测尚未未知的候选基因中可能致病的突变。
Breda Genetics建议针对这种情况进行小组测试:
线粒体复合体 IV 缺陷 - 细胞色素 C 氧化酶缺陷(APOPT1、C12ORF62、COA3、COA5、COA6、COX10、COX14、COX15、COX20、COX6B1、FARS2、FASTKD2、LRPPRC、MTCO1、MTCO2、MTCO3、MTTL1、MTTS1、PET100、POLG、 SCO1、SCO2、SURF1、TACO1)
三、标准疗法
1、总体疗法
所有形式的 COX 缺乏症的治疗均针对每个个体明显的特定症状。治疗可能需要专家团队的协调努力,他们可能需要系统且全面地规划受影响儿童的治疗。这些专家可能包括儿科医生;诊断和治疗肾脏异常(肾脏科医生)、肌肉骨骼系统异常(骨科医生)、心脏异常(心脏病专家)、肺异常(肺科医生)、神经系统异常(神经科医生)和/或肝脏异常(肝科医生)的医生;和/或其他医疗保健专业人员。对于良性婴儿线粒体肌病,重要的是早期诊断和强化治疗直至实现自然康复。
通常建议患有线粒体疾病(例如 COX 缺乏症)的个体避免禁食。如果患者无法口服液体,则可以通过静脉输液治疗因呕吐或疾病引起的脱水。癫痫发作通常用抗惊厥药控制。一些受影响的人可能会受益于专门针对他们的需求而定制的物理、职业和言语治疗。包括某些维生素和辅因子在内的膳食补充剂,如:核黄素、硫胺素、生物素、辅酶 Q10、肉碱在个别情况下显示出不同程度的益处。
建议受影响的个人及其家人进行遗传咨询。其他治疗是对症治疗和支持治疗。
线粒体疾病管理:
2、成人类型疗法
对于成人患者,辅酶 Q补充剂、生酮饮食和无麸质饮食可能至少对某些表现有有益的影响。
生酮饮食:
无麸麦饮食:
四、SURF1 缺陷
SURF1 缺陷是一种单基因线粒体疾病,是细胞色素c氧化酶 (COX) 缺陷 Leigh 综合征 (LS)的最常见原因。COX 全酶的复杂生物发生需要许多组装因子,并且对脱氮副球菌和酿酒酵母的研究已确定 SURF1 是 COX 早期组装的关键参与者,由于其临床和遗传异质性,线粒体疾病通常是一个诊断挑战。LS 是一种致命的亚急性坏死性脑脊髓病,是一种遗传异质性线粒体疾病,可能与任何线粒体氧化磷酸化(OXPHOS )缺乏有关。自1998 年SURF1突变与 COX 缺乏和 LS 相关以来 ,已有孤立的病例报告和突变系列报道。清楚地了解该疾病的临床连续性对于改善这种疾病的诊断非常重要。受影响的家庭经常询问预后,但文献中提供预后信息的数据有限。记录齐全的自然历史研究对于规划未来的临床试验也是非常宝贵的帮助。
1、最初症状
首次出现症状的中位年龄为 9.5 个月(范围 0-60 个月),大多数患者在第一年就诊(32/44,73%)。最常见的初始症状是进食/呕吐不良(通常归因于胃食管反流)和体重增加不良。除两名有喂养问题的患者和一名肌张力低下的患者外,大多数患者(41/44,93%)的新生儿期平安无事。发育退化(定义为认知或运动技能丧失)是 3/44 (7%) 患者的首发症状。大多数患者(26/44,59%)同时出现胃肠道症状、体重增加缓慢和肌张力低下。三名在头两个月接受全身麻醉的患者在恢复期间遇到了重大问题。一名患者(病例 30)在第 9 天接受了大动脉转位手术,出现持续呕吐并需要肠内喂养。第二名患者(病例 26)在幽门狭窄手术后第 8 周出现低渗。第三名患者(病例4)在接受上消化道内窥镜检查后出现运动障碍。
表:44例SURF1缺陷患者的初始症状
| 最初症状 | 患者人数(%) | 初次就诊的年龄范围(月) |
|---|---|---|
| 喂养不良/呕吐 | 20 (46) | 0-24 |
| 体重增加不佳 | 19 (43) | 1.5-20 |
| 发育迟缓 | 10 (23) | 9-51 |
| 肌张力减退 | 9 (21) | 0-10 |
| 运动障碍 | 3(7) | 10-24日 |
| 发育倒退 | 3(7) | 10-18日 |
| 共济失调 | 2(5) | 14-60 |
2、主要临床特征
对 44 例 SURF1 缺陷病例的回顾显示,32 例患者符合 Leigh 综合征的标准 ,12 例患者由于放射学特征不典型或正常或无法进行神经影像学检查而被归类为“Leigh 样”。临床特征随时间的发展 。体重增加不良和肌张力低下是最常见的症状,并且往往先出现胃肠道症状,例如进食/呕吐不良。21/44 (48%) 需要长期肠内喂养。27/38 (71%) 的病例出现发育退化(中位发病年龄 19 个月),并由 12 名患者并发病毒感染引发,18/44 (41%) 的患者报告有多毛症。多毛症与乳酸性酸中毒之间没有显着相关性。眼球震颤和眼肌麻痹分别见于 25/42 (60%) 和 22/42 (52%) 的患者,且发生时间较晚(中位发病年龄 29 个月)。22/42 (52%) 患者(中位发病年龄 2 岁)出现运动障碍,其中 13 名患者出现孤立性意向性震颤,3 名患者出现舞蹈样手足徐动,2 名患者出现肌张力障碍,而 4 名患者则表现出这些症状的组合异常动作。只有 6/44 (14%) 患者报告癫痫发作(全身强直阵挛 5 次,肌阵挛 1 次),其中 4 名患者来自澳大利亚。其他不太常见的临床特征包括 10/44 (23%) 的视神经萎缩、9/44 (20%) 的脑病和 1/44 (2%) 的肥厚性心肌病。据报道,没有患者患有色素性视网膜病变或感音神经性耳聋。
3、生化数据
所有进行此项测量的受试者的脑脊液乳酸均升高(平均值 4.3,范围 2.5-8.6 mmol/L,正常 <2 mmol/L)。21/33 (64%) 患者存在代谢性酸中毒(平均碳酸氢盐水平 14.7,范围 10-17 mmol/L)。31/38 (81%) 患者血乳酸升高(平均 4.4,范围 2.3-7.3 mmol/L)。发现所有接受测试的受试者中成纤维细胞 COX 活性较低(25/25,100%),其水平范围从低于检测限的水平到参考范围下限的 57%(参考范围 30- 90 nmol/毫克蛋白质/分钟)。
表: SURF1 缺陷的实验室和磁共振成像 (MRI) 结果
| 实验室或神经影像学发现 | 患者人数 | % |
|---|---|---|
| 血 | ||
| 代谢性酸中毒 | 21/33 | 64 |
| 乳酸升高 | 31/38 | 81 |
| 脑脊液 | ||
| 乳酸升高 | 30/30 | 100 |
| 肌肉 | ||
| COX 组织化学减少/缺失 | 23/33 | 70 |
| I 型纤维优势 | 8/33 | 24 |
| 肌肉脂质含量升高 | 16/33 | 48 |
| 肌肉 COX 活性降低 | 25/26 | 96 |
| 成纤维细胞 | ||
| 成纤维细胞 COX 活性降低 | 25/25 | 100 |
| 神经传导研究 | ||
| 周围神经病变 | 13/16 | 81 |
| MRI病变 | ||
| 中脑 | 12/33 | 36 |
| 庞斯 | 10/33 | 30 |
| 髓质 | 15/33 | 45 |
| 壳门 | 16/33 | 48 |
| 苍白球 | 14/33 | 42 |
| 尾状核 | 12/33 | 36 |
| 丘脑底核 | 4/33 | 12 |
| 导水管周围灰质 | 4/33 | 12 |
| 橄榄核 | 3/33 | 9 |
| 红核 | 2/33 | 6 |
| 小脑白质/灰质 | 6/33 | 18 |
| 小脑萎缩 | 3/33 | 9 |
| 齿状核 | 13/33 | 39 |
| 小脑脚 | 8/33 | 24 |
| 白质脑病 | 2/33 | 6 |
4、肌肉活检
我们分析了所有进行肌肉活检的患者 (n = 33) 的组织学和 RCE 结果。15/33 (46%) 的活检组织中脂质含量增加,8 个活检组织中发现 1 型纤维占主导地位。COX 组织化学显示 9 例 (27%) 活检中不存在 COX,14 例 (42%) 活检中 COX 染色减少。据报道,十个 (30%) 的活检组织具有正常的 COX 组织化学,所有纤维均染色均匀。在 28 个活组织检查中测量了 RCE 活性。两次尸检活检被排除,因为尸检延迟可能导致 RCE 活性人为损失。除一项活检外,所有活检均发现 COX 活性单独降低,其中 COX 活性处于参考范围的下限(COX 活性/柠檬酸合酶比率为 0.016(参考范围 0.014-0.034)。
5、神经影像学
39 名受试者可获得磁共振成像 (MRI) 和计算机断层扫描 (CT) 信息(33 幅 MRI 和 6 幅 CT 扫描)。大多数 MRI 显示 Leigh 综合征特征(28/33,85%),脑干和/或基底神经节 T2 加权成像上有对称性高信号病变。两名患者 1 岁时进行的 MRI 扫描正常,但无法进行后续成像。两名患者患有白质脑病(定义为主要或专门影响大脑白质的疾病),而一名患者患有小脑萎缩并累及齿状核。6 例患者进行 CT 扫描,其中 3 例正常,3 例双侧基底节对称性低密度。
6、外周神经系统
十六名患者进行了神经传导研究。13/16 (81%) 的受试者发现周围神经传导异常,其中大多数 (7) 表现出脱髓鞘性神经病变,而 2 名受试者则表现为轴突神经病变。4 名受试者的神经病变类型未明确。许多(9/13,69%)受试者表现出混合的感觉运动受累,而 2 名受试者患有纯粹的感觉神经病变。
7、SURF1基因突变
报告在 57 名患者中发现了SURF1突变;16/57(28%)有纯合的c.312_320del10insAT插入/缺失,15/57(26%)有复合杂合子,12/57(21%)有纯合剪接位点突变,10/57(18%)有纯合子缺失,2/57(4%)有纯合插入,1/57(2%)有纯合错义突变,1/57(2%)有纯合无义突变。
队列中最常见的突变是 c.312_321del10insAT (p.Leu105X)(16 个纯合子和 11 个复合杂合子)。其次最常见的突变是 c.792_793delAG,仅发生在孟加拉国孟加拉受试者(n = 5,3 个谱系)中,剪接突变 c.240+1G>T 在 5 个个体中观察到,全部为欧洲白人。c.792_793delAG 突变此前仅在一名日本裔患者中被描述过 。巴基斯坦血统的个体具有新的 c.799_800delCT 突变或 c.324-11T>G 突变。c.516-2A>G 突变在两个不同的印度古吉拉特血统中分离,之前仅在一名未明确的混血白人亚裔父母中被描述过 。c.324-11T>G 基因型在来自 2 个不同巴基斯坦谱系的 4 名个体和一名印度古吉拉特裔个体中观察到。来自澳大利亚队列的所有五名患者都至少有一种 c.312_321del10insAT (p.Leu105X) 突变。没有观察到特定的基因型表型相关性。存活超过 10 年的患者具有不同的SURF1基因型:一名是 c.312_321del10insAT 突变纯合子,两名是复合杂合子,两名是纯合插入,一名是剪接位点突变。
8、新的致病突变
在我们的队列中,我们在 57 名患者中发现了 19 个SURF1突变,其中包括 5 个新的致病性突变(c.468_469delTC、c.799_800delCT、c.575G>A (p.Arg192Gln)、c.751+5G>A. 和 c. .752-2A>G)。
在发现的新突变中,纯合子 c.799_800delCT 是在一个巴基斯坦家庭中发现的,该家庭的患者(病例 29)从出生起就患有早发性疾病,并在 2 岁时死亡。在一名复合杂合子受试者(病例 3)中发现了新的剪接位点 c.751+5G>A 突变,该突变产生异常转录物,将内含子 7 的 31 个碱基对掺入一定比例的成熟 mRNA 中。312_321del10insAT,c.751+5G>A)。她的临床病程非典型,较温和,生存期较长。她的第一个令人担忧的症状是在 16 个月大时,当时她被发现有轻度运动迟缓和共济失调,但回想起来,从出生起就存在轻度肌张力低下。她随后出现眼球震颤和眼外肌麻痹。19 岁的她目前正在学习商业研究。在一名患有共济失调的 12 岁女性(病例 2)和一名患有发育迟缓、共济失调和肥厚性心肌病的 4 岁男性(病例 22)中发现了新的错义突变(c.575G>A,p.Arg192Gln)。这是该队列中唯一患有心肌病的患者。该女性患者随后出现眼球震颤并伴有眼肌麻痹,但目前她能够骑自行车和拉小提琴。剪接位点突变 c.752-2A>G 在一名澳大利亚籍患者(病例 37)中发现,该患者自出生以来就表现出烦躁和喂养不良。不幸的是,我们无法获得带有纯合 c.468_469delTC 突变的患者的临床信息。
9、生存
在 44 名拥有详细临床数据的患者中,有 5 名患者在撰写本文时还活着(年龄范围 2-19 岁),而 3 名患者目前的生命状况未知。在36名已知死因的死亡患者中,29/36(80%)的死因是中枢性呼吸衰竭。7名患者存活超过10岁。其中,6人出现共济失调和运动发育迟缓等神经系统症状;值得注意的是,胃肠道症状并不是这些病例的突出表现特征。此外,这六名患者也没有出现发育退化。
文献检索确定了 98 个 SURF1 缺陷病例以及可用的生存数据,将其与 44 个病例的数据汇总在一起。在 Kaplan-Meier 分析中,将这 142 例 SURF1 缺陷病例的生存经历与另外两组因核基因突变而患有 LS 的病例进行了比较。报告将 SURF1 缺陷患者的生存期与 56 名 LRPPRC 缺陷患者 和 63 名核编码复合物 I 缺陷 LS/“Leigh 样”病患者的生存期进行了比较 , SURF1 缺陷患者的中位生存期较长(中位生存期较长)。5.4,第 25 个百分位 3.0,第 75 个百分位 10 年)比 LRPPRC 缺陷患者(中位 1.8,第 25 个百分位 1.0,第 75 个百分位 4 年)和核编码复杂 I 缺陷 LS 患者(中位 1.6,第 25 个百分位 1.0,第 75 个百分位 4 年)高10 年)(组间差异 p < 0.001;对数秩检验)。
10、文献中的其他案例
对文献的系统回顾揭示了已发表的 129 名 SURF1 缺陷患者的临床数据和另外 28 名患者的突变数据。在这些患者中发现但在我们的队列中不存在的特征包括未明确的肝脏受累(3 例)、肾小管病变(4 例)和面部畸形(5 例)。
11、讨论
SURF1 缺陷是英国人群中 Leigh 综合征最常见的单一原因,对临床特征的仔细记录可能会提高对受影响儿童的认识和更快的诊断。当受影响的家庭寻求产前诊断时,早期基因诊断的需求尤其迫切。改进诊断过程依赖于公认的临床特征,但通常没有足够数量的患有线粒体能量代谢单基因疾病的患者来表征表型。报告收集了 57 名 SURF1 缺陷患者的队列,其中包括 44 名患者的全面表型数据。发现SURF1缺陷的患者具有相对同质的表型,通常在婴儿晚期开始出现胃肠道症状,随后出现阵发性神经退行、眼肌麻痹、运动障碍,最后导致呼吸衰竭死亡。尽管存在一致的临床表型,但没有临床特征可以将 SURF1 缺陷与 LS 的其他原因(例如复合物 I 缺陷或复合物 V 缺陷)区分开来。
心肌病很少见(一名患者),这一观察结果与文献相似,文献中只有 2/129 例患有心肌病 。癫痫症似乎也不常见,主要见于澳大利亚患者,这可能表明环境因素会诱发癫痫发作。高温不太可能是诱发因素,因为在澳大利亚的病例中没有观察到缉获量的季节性变化。癫痫发作的低发生率与之前描述的病例相当,据报道只有 5% (7/129) 有癫痫发作。在这个 SURF1 缺陷队列中没有发现感音神经性耳聋,这与文献中的低发生率一致,即之前只有两例病例患有感音神经性耳聋 。先前报道有 3 例 (2%) 病例患有视神经萎缩 ,而报告的 10 例 (23%) 患者被发现患有视神经萎缩。视觉和听觉体征在儿科实践中很难确定,并且在许多未进行正式测试的情况下可能被诊断不足。三名接受早期全身麻醉的患者出现神经功能恶化;在考虑对 SURF1 缺陷患者进行全身麻醉时必须谨慎。
尽管 SURF1 缺陷的长期生存并不常见,但令人惊讶的是,我们发现与复杂 I 缺陷 LS 或 LRPPRC 缺陷 LS 相比,SURF1 缺陷 LS 具有更有利的生存结果。与 SURF1 疾病不同,LRPPRC 缺陷会引发急性代谢危机,从而显着增加死亡率 。
逐步的神经功能退化通常由疾病和其他高能量需求状态引起,被认为是 LS 自然史的一部分。在报告的队列中,存活超过 10 年的 6 名患者并未经历发育退化,这表明开发有效的治疗策略来预防和治疗阵发性失代偿可以改善 SURF1 缺陷患者的预后。然而,在无法识别明显促发因素的情况下,预防失代偿可能具有挑战性。由于没有足够的治疗数据可供该队列得出任何有意义的结论,因此很难评论支持疗法对 SURF1 缺陷的效果。最近的候选疗法如 EPI-743 正在接受评估,一项开放标签 2A 期研究招募的 2 名 SURF1 缺陷患者已证明其生活质量和运动功能评分有所改善。
除了迄今为止在SURF1基因中发现的 78 个突变之外,我们还描述了 5 个新突变。SURF1 缺陷中发现的大约 80% 的突变是由异常剪接、移码缺失或无义突变引起的截短突变。我们的研究表明某些种族群体内特定突变的分离。基因型分布表明一些突变的创始人效应,例如欧洲白人中的 c.311_312insATdel10 和 3 个不同的孟加拉国孟加拉血统中显示的 c.790_800delAG。此前的报道表明,c.604G>C突变仅在中国个体中报道过。尽管早期报告 ,表明错义SURF1突变有利于更好的预后,但报告没有发现独特的基因型-表型关联。在我们的队列中不可能证实这一假设,因为只有三名受试者有错义突变(1 名纯合子,2 名复合杂合子)。然而,其中两名患者目前还活着,分别为 12 岁和 17 岁。描述的 7 名表型较轻的患者具有不同的基因型,包括常见的 c.312_320del10insAT 纯合性,表明还有其他因素可能影响生存。
报告证明 SURF1 缺陷具有独特的生化表型,包括脑脊液乳酸持续升高以及成纤维细胞和肌肉中 COX 活性降低。因此,当遇到LS的典型临床表现时,如果发现成纤维细胞COX活性较低,则应进行SURF1基因的靶向测序。如果患者病情迅速恶化,则应谨慎进行肌肉活检并进行初步调查,因为 SURF1 缺乏症没有明显的临床特征,无法与 LS 的其他原因进行区分。肌肉组织化学显示 COX 活性的整体均匀减少,但这可能非常微妙,并且报告为正常,因为许多中心使用延长的 COX 组织化学孵育时间,以最大程度地区分 COX 阳性和阴性纤维。肌肉组织学显示非特异性异常,例如脂质积累或纤维类型不成比例(I 型纤维占优势)。参差不齐的红色纤维通常表明线粒体 DNA 突变,仅在之前的两个 SURF1 缺陷病例中被发现 ,并且在队列中的任何患者中都没有发现。SURF1 缺乏症的临床表现是 LS 的典型表现,只有具有支持性生化表现,例如成纤维细胞和/或肌肉 COX 活性低,才能怀疑 SURF1 缺乏症。
之前的综述表明,SURF1 疾病的 MRI 一致发现丘脑底核受累 。在报告的研究中,只有 4 例 (12%) 的丘脑底核有病变。相比之下,16/33 (48%) 的患者壳核有病变。SURF1 缺陷中这些病变的特异性需要通过与其他 LS 组进行比较来验证。大多数 (85%) MRI 显示 LS 的特征性病变。白质脑病改变是一种非典型表现 (n = 2),但在这些病例中不应忽视 SURF1 缺陷。2 名 1 岁受试者的 MRI 正常。疾病早期正常的神经放射学可能随后在重复成像时演变为异常。
文献之前的病例中已经注意到脱髓鞘性周围神经病的存在,在这里证明,虽然在大多数病例中神经病确实具有脱髓鞘性,但也可能存在轴突神经病。
研究有一些局限性。尽管研究数据是使用标准化问卷收集的,但这是一项回顾性研究,其中临床信息是通过审查患者的医疗记录/病史获得的,其中许多人现已去世。然而,不太可能针对这种罕见疾病进行大规模前瞻性研究。核磁共振成像是在不同的中心进行的,并由不同的神经放射科医生进行审查,这使得很难归因于特定的疾病模式。在大多数情况下,MRI 结果基于单次扫描,仅代表神经放射学变化的快照,而不代表疾病进展。通常不可能捕获这些变化,因为幼儿的重复成像通常涉及全身麻醉,并伴有代谢失代偿的风险。
原文参考:
TEXTBOOKS
Bennett JC, Plum F, eds. Cecil Textbook of Medicine, 20th ed. W.B. Saunders Co.;1996:2167.
Behrman RE, ed.; Nelson Textbook of Pediatrics, 15th Ed. W.B. Saunders Company;1996:1754.
Lyon G, et al., eds. Neurology of Hereditary Metabolic Diseases in Childhood, 2nd Ed. McGraw-Hill;1996:27.
Scriver CR, et al., eds. The Metabolic and Molecular Bases of Inherited Disease, 7th Ed. McGraw-Hill, Inc.;1995:1537-38,1615-16.
Buyse ML, editor-in-chief. Birth Defects Encyclopedia. Blackwell Scientific Publications;1990:1202-03.
JOURNAL ARTICLES
Vogt, Sebastian, Volker Ruppert, Sabine Pankuweit, JÞrgen pj Paletta, and Petra Weber. Reduced Cytochrome C Oxidase Subunit Iv in Patients with Dilated Cardiomyopathy. Exp Clin Card 2014;20: 1009-1028.
Hüttemann M, Klewer S, Lee I, Pecinova A, Pecina P, Liu J, Lee M, Doan JW, Larson D, Slack E, Maghsoodi B, Erickson RP, Grossman LI, Mice deleted for heart-type cytochrome c oxidase subunit 7a1 develop dilated cardiomyopathy. Mitochondrion 2012;12:294-304.
Huigsloot M, Nijtmans LG, Szklarczyk R, Baars MJ, van den Brand MA, Hendriksfranssen MG, van den Heuvel LP, Smeitink JA, Huynen MA, Rodenburg RJ, A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488-9.
Vogt S, Portig I, Irqsusi M, Ruppert V, Weber P, Ramzan R, Heat shock protein expression and change of cytochrome c oxidase activity: presence of two phylogenic old systems to protect tissues in ischemia and reperfusion. J Bioenerg Biomembr 2011;43:425-35.
Arbustini E, Diegoli M, Fasani R, et al.. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol 1998;153:1501-1510.
DiMauro S, et al. Cytochrome C oxidase deficiency. Pediatr Res. 1990;28(5):536-41.
Von Kleist-Retzow JC, et al. A high rate (20% – 30%) of parental consanguinity in cytochrome-oxidase deficiency. Am J Hum Genet. 1998;63(2):428-35.
Isobe K, et al. Nuclear-recessive mutations of factors involved in mitochondrial translation are responsible for age-related respiration deficiency of human skin fibroblasts. J Biol Chem. 1998;273(8):4601-06.
Possekel S, et al. Immunohistochemical analysis of muscle cytochrome C oxidase deficiency in children. Histochem Cell Biol. 1995;103(1):59-68.
Tiranti V, et al. Nuclear DNA origin of cytochrome C oxidase deficiency in Leigh’s syndrome: genetic evidence based on patient’s-derived RHO degrees transformants. Hum Mol Genet. 1995;4(11):2017-23.
Saunier P, et al. Cytochrome C oxidase deficiency presenting as recurrent neonatal myoglobinuria. Neuromuscul Disord. 1995;5(4):285-89.
Morin C, et al. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am J Hum Genet. 1993;53(2):488-96.
Merante F, et al. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am J Hum Genet. 1993;53(2):481-87.
DiMauro S, et al. Mitochondrial encephalomyopathies. Arch Neurol.1993;50(11):1197-208.
Salo MK, et al. Reversible mitochondrial myopathy with cytochrome C oxidase deficiency. Arch Dis Child. 1992;67(8):1033-35.
Keppler K, et al. Variable presentation of cytochrome C oxidase deficiency. Am J Dis Child. 1992;146(11):1349-52.
Eshel G, et al. Autosomal recessive lethal infantile cytochrome C deficiency. Am J Dis Child. 1991;145(6):661-64.
Lutz R et al.An atypical case of cytochrome C oxidase deficiency with biochemical heterogeneity in fibroblasts. Neurology. 1991;41(12):1957-60.
Lombes A, et al. Biochemical and molecular analysis of cytochrome C oxidase deficiency in Leigh’s syndrome. Neurology. 1991;41(4):491-98.
Tritschler HJ, et al. Differential diagnosis of fatal and benign cytochrome C oxidase-deficient myopathies of infancy: an immunohistochemical approach. Neurology. 1991;41(2 Pt 1):300-05.
Adamovich K, et al. Cytochrome C oxidase deficiency. Orv Hetil. 1990;131(32):1761-63.
Buchwald A, Till H, Unterberg C, Oberschmidt R, et al. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J 1990; 11:509- 516.
Nozaki H, et al. Cytochrome C oxidase deficiency with acute onset and rapid recovery. Pediatr Neurol. 1990;6(5):330-32.
Wedatilake, Y., Brown, R.M., McFarland, R. et al. SURF1 deficiency: a multi-centre natural history study. Orphanet J Rare Dis 8, 96 (2013). https://doi.org/10.1186/1750-1172-8-96
