
慢性进行性眼外肌麻痹 (CPEO):引起眼睑下垂、眼球运动受限、延髓肌无力和近端肢体无力的线粒体疾病

慢性进行性眼外肌麻痹 (CPEO)是一种线粒体 DNA 缺失综合征,临床特征为双侧眼睑下垂、眼球运动受限,有时伴有肢体和延髓肌受累。这种疾病可由点突变、单个大规模线粒体 DNA (mtDNA) 缺失、重复或继发于核突变(如ANT1、POLG1、POLG2、OPA1、C10orf2和SLC25A4基因)的多个mtDNA 缺失引起。CPEO疾病症状通常出现在 18 岁至 40 岁之间的成年人中。尽管疾病的确切病程因人而异,但随着时间的推移,症状往往会恶化,进行性外眼肌麻痹的第一个症状通常是眼睑下垂(下垂),这可能会影响一个或两个眼睑。随着上睑下垂恶化,受影响的人可能会使用前额肌肉尝试抬起眼睑,或者抬起下巴以便看清东西。进行性外部眼肌麻痹的另一个特征是移动眼睛的肌肉无力或麻痹(眼肌麻痹)。受影响的人必须转动头部才能看到不同的方向,尤其是当眼肌麻痹恶化时。患有进行性外眼肌麻痹的人也可能出现用于运动的肌肉普遍无力(肌病),特别是颈部、手臂或腿部的肌肉无力。这种弱点在运动期间可能特别明显(运动不耐受)。肌肉无力也可能导致吞咽困难(吞咽困难)。
当受影响个体的肌肉细胞被染色并在显微镜下观察时,这些细胞通常会出现异常。这些异常的肌肉细胞含有过量的称为线粒体的细胞结构,被称为参差不齐的红色纤维。
虽然肌肉无力是进行性外眼肌麻痹的主要症状,但这种情况可能伴有其他体征和症状。在这些情况下,这种情况被称为进行性外眼肌麻痹综合征(PEO+)。其他体征和症状可能包括内耳神经损伤引起的听力损失(感音神经性听力损失)、神经损伤导致的四肢无力和感觉丧失(神经病)、肌肉协调受损(共济失调)、运动异常模式称为帕金森症和抑郁症。
进行性眼外肌麻痹是一系列具有重叠体征和症状的疾病的一部分。类似的疾病包括共济失调神经病谱和卡恩斯-塞尔综合征(KSS)。与进行性外眼肌麻痹一样,该范围内的其他病症也可能涉及眼部肌肉无力。然而,这些病症还有许多其他特征是大多数进行性外眼肌麻痹患者所不具备的。
治疗可以解决一些症状,患有孤立性 CPEO 的个体通常具有正常的预期寿命。
一、病因
进行性外部眼肌麻痹是一种由线粒体缺陷引起的疾病,线粒体是细胞内的结构,利用氧气将食物中的能量转化为细胞可以使用的形式。这个过程称为氧化磷酸化。尽管大多数DNA被包装在细胞核内的染色体中(核DNA),但线粒体也有少量自己的DNA,称为线粒体DNA或mtDNA。该 DNA 含有氧化磷酸化所必需的基因。
进行性外眼肌麻痹可能是由几种不同基因之一的突变引起的。在某些情况下,核DNA突变是造成这种情况的原因,包括POLG、TWNK、RRM2B和SLC25A4基因等的突变。这些基因对于线粒体DNA的产生和维持至关重要。尽管机制尚不清楚,但这些基因的突变导致肌肉细胞中线粒体 DNA 的大片段缺失。删除区域的大小范围为 2,000 至 10,000 个 DNA 构建块(核苷酸)。
在其他情况下,这种情况是由线粒体 DNA 的单个大缺失引起的,该缺失与核 DNA 基因的突变无关。
不太常见的是,改变 mtDNA 基因(例如MT-TL1基因)中单核苷酸的突变会导致进行性外眼肌麻痹。这些突变发生在为制造称为转移 RNA 的分子提供指令的基因中。转移 RNA 有助于将蛋白质构件(氨基酸)组装成功能蛋白质。与进行性外眼肌麻痹相关的转移 RNA 存在于线粒体中,有助于组装执行氧化磷酸化步骤的蛋白质。
研究人员尚未确定 mtDNA 缺失或 mtDNA 基因突变如何导致进行性外眼肌麻痹的具体体征和症状,尽管这种情况的特征可能与氧化磷酸化受损有关。有人认为,眼部肌肉通常受到线粒体缺陷的影响,因为它们特别依赖氧化磷酸化来获取能量。
二、遗传模式
根据所涉及的基因,进行性外眼肌麻痹可能具有不同的遗传模式。
当涉及核基因POLG、TWNK、RRM2B或SLC25A4时,进行性外眼肌麻痹通常以常染色体显性遗传模式遗传,这意味着每个细胞中改变的基因的一个拷贝足以引起该疾病。
POLG或RRM2B基因中的某些突变也可能导致一种以常染色体隐性遗传模式遗传的疾病,这意味着每个细胞中该基因的两个拷贝都有突变。常染色体隐性遗传病患者的父母各携带一份突变基因,但他们通常不会表现出该病的体征和症状。
当这种情况是由MT-TL1基因和其他线粒体转移 RNA 基因突变引起时,它会以线粒体模式遗传,这也称为母系遗传。这种遗传模式适用于 mtDNA 中包含的基因。由于卵细胞(而不是精子细胞)为发育中的胚胎提供线粒体,因此儿童只能从母亲那里遗传由线粒体 DNA 突变引起的疾病。这些疾病可能出现在家庭的每一代人中,并且可能影响男性和女性,但父亲不会将与线粒体 DNA 变化相关的特征遗传给孩子。
线粒体 DNA 的单个大缺失通常不会遗传,而是发生在母亲卵细胞的形成过程中或胚胎的早期发育过程中。携带这些突变的个体通常没有家族遗传病史。
大多数 CPEO 病例是由线粒体 DNA 缺失引起的。大多数是散发性的,没有其他家庭成员的参与,并且存在传播给后代的风险。这些缺失可能是由与 CPEO 相关的核基因突变引起的,包括:POLG、POLG2、TK2、OPA1、RRM2B、TWNK、SLC25A、RRM1、TOP3A、C1QBP、DNA2、C10ORF2、DGUOK、MPV17、MGME1、SPG7、AFG3L2、RNASEH1、GMPR
不太常见的是,CPEO 可能是由线粒体基因的单点突变引起的,例如MT-TL1 m.3243A>G 变异,它也会导致线粒体脑肌病乳酸性酸中毒中风样发作(MELAS)。
这些突变可能是遗传的,也可能是在没有 CPEO 家族史的个体中偶然出现的。单个 mtDNA 缺失通常会自发出现,但有单个 mtDNA 症状的女性会将突变遗传给 4% 的孩子。相比之下,线粒体DNA的点突变通常会从母亲遗传给所有的孩子。核基因突变根据特定基因以常染色体显性、隐性或 X 连锁模式传播。
三、临床诊断
CPEO 患者通常表现为双侧、对称、无痛、瞳孔不受影响的上睑下垂和眼肌麻痹。CPEO 会在数年的时间内缓慢进展,这使其与引起急性/亚急性或静态眼肌麻痹的其他病因不同。由于双眼对称受累,即使眼球运动严重缺陷,患者也可无症状而无复视。患者还会通过移动头部来补偿眼球运动的不足。此外,由于病程是渐进的,除非由第三方提出,否则患者可能不会注意到眼睑下垂。其他不太常见的眼科表现包括色素性视网膜病变和视神经萎缩。同样,有些病例可能没有上睑下垂或不对称。
疼痛、眼球突出、眶周肿胀、眼睑滞后/回缩和瞳孔受累不是 CPEO 的症状,而是表明不同的病因。单侧或快速进展的症状也不典型,应提示进行额外的评估,包括神经影像学检查。详细的家族史对于识别可能的遗传性疾病非常重要。
其他非眼科表现包括感音神经性听力损失和吞咽困难。
如果 CPEO 与另一种综合征相关,则被视为 CPEO 附加综合征。
1、鉴别诊断
1.1 卡恩斯-塞尔综合症KSS
与通常在三四十岁时出现的孤立性 CPEO 不同,KSS 患者通常在 20 岁之前出现症状。KSS 患者还患有有趣的双侧色素性视网膜病变,这是一种视网膜退行性病变,会损害视网膜的功能。色素上皮细胞和光感受器。患者的周边视力和夜视力逐渐丧失。因此,对于 CPEO 病例,应进行散瞳眼底检查,以观察任何色素沉着不足或过度的区域,表明视网膜色素上皮 (RPE) 紊乱,可能提示 KSS。非典型 RPE 脱色被描述为具有“椒盐状”或“虫蛀状”外观。KSS 的其他相关发现包括心血管传导缺陷、脑脊液 (CSF) 中蛋白质升高和小脑性共济失调。因此,KSS 患者应考虑进行心脏评估、腰椎穿刺和彻底的神经系统评估。
1.2 眼咽肌营养不良症
眼咽肌营养不良症 (OPMD) 是一种常染色体显性遗传性疾病,其多聚丙氨酸结合蛋白 1 (PABP1) 基因中存在病理性 GCG 三核苷酸重复扩增。突变的 PAPB1 蛋白聚集为核内管状丝,并可通过尚不清楚的机制导致肌肉再生失败。与其他一些 CPEO 综合征不同,OPMD 不是线粒体肌病。这种疾病在法裔加拿大人中最为普遍。除了 CPEO 之外,在 50 岁时出现的症状还包括延髓症状,例如吞咽困难(咽肌无力)、眼轮匝肌无力和近端肢体无力。
1.3 强直性肌营养不良
强直性肌营养不良有先天性、儿童期和经典性三种形式,分别在出生时、儿童期和成年期发病。这种综合征可以表现为多种眼部和全身症状。眼部症状包括 CPEO、眼睑滞后、缓慢眼跳和白内障。远端肌肉无力很常见,患者报告需要手部精细运动控制的活动有困难。肌强直,或收缩后肌肉延迟松弛,最常涉及手部肌肉,在体检时用反射锤敲击鱼际肌时可观察到“敲击性肌强直”。面部肌肉、舌头和其他延髓肌的肌强直会导致面部表情、咀嚼、说话和吞咽障碍。
1.4 先天性眼外肌纤维化
先天性眼外肌纤维化 (CFEOM) 是一种严重的斜视,伴有眼球运动障碍。患有这种先天性、非进行性疾病的患者有限制性眼肌麻痹和眼睛错位,伴有严重的先天性上睑下垂和由此导致的突出的引体向上头部位置。垂直眼球运动缺陷,尤其是向上凝视,是该病的一个标志,患者的眼睛经常陷入视线障碍。水平眼球运动缺陷变化更大,从完全水平运动到几乎完全眼肌麻痹。主要位置的眼睛排列可以是外向性、内向性或直线
三种形式
CFEOM1=常染色体显性遗传、双侧上睑下垂、抬高不足(通常低于 20-30 度)、水平运动受限代偿性引体向上头部位置。结果对称,没有其他神经功能缺陷。
CFEOM2=常染色体隐性遗传,双侧眼肌麻痹和上睑下垂。经常为外斜和垂直中线。水平和垂直眼球运动均受到限制。其他:瞳孔小、反应迟钝、视力低下,符合视网膜功能障碍。
CFEOM3=常染色体显性遗传、孤立性或综合征性、表型可变,从轻微到相当严重,甚至在同一家族内也有不同的范围。单侧或不对称受累、+/-上睑下垂、抬高眼睛的能力较差。水平运动的缺陷也变化很大。CFEOM3 还可能与多种其他神经系统异常相关,例如相关的面瘫或虚弱。其他:无隔离;综合症:颅骨和脊髓周围神经病变、发育迟缓、智力和社交障碍以及脑畸形;面部无力和声带麻痹;面部畸形、卡尔曼综合征(性腺功能减退症伴嗅觉丧失),并可能出现轴突周围神经病变和周期性呕吐;突变与特定的脑畸形存在相关性,包括前连合和胼胝体薄至缺失、基底节畸形、脑干发育不全、嗅沟、嗅球和面神经发育不全或缺失。
1.5 其他
其他综合征(通常发生在儿童期或 20 岁之前)包括皮尔逊综合征、阿尔珀斯综合征、Leigh 综合征、感觉性共济失调神经病、构音障碍、眼肌轻瘫;线粒体脑肌病、乳酸性酸中毒和中风样发作(MELAS)以及线粒体神经胃肠道脑肌病
2、体检
全面的体检,重点关注眼科和神经科的组成部分,对于识别 CPEO 及其相关综合征的表型至关重要。眼肌麻痹的严重程度可以通过测量单眼注视场和诱导场来量化。Goldmann 视野计可用于绘制各个注视视野中眼外运动 (EOM) 的范围。例如,在右眼中,基轴为 0 o(外直肌)、67 o(上直肌)、141 o(下斜肌)、180 o(医学直肌)、216 o(上斜肌)和 293 o(下直肌)。在检查过程中,一只眼睛被遮挡,患者沿着每个轴跟随照明目标,直到失去对目标的中心固定。在一项研究中,与对照组相比,CPEO 患者的 EOM 总体平均范围降低了 73%。上睑下垂的程度可以通过垂直裂高度(VFH)、提肌功能(LF)和边缘反射距离(MRD)来测量。
3、实验室研究
虽然 CPEO 是一种临床诊断,但实验室研究可以帮助确认诊断并排除其他诊断。CPEO 中血清乳酸、肌酐激酶和脑脊液乳酸水平可能升高,但这一发现既不敏感也不特异。当仅凭病史还不够时,缺乏抗乙酰胆碱酯酶抗体和甲状腺自身抗体可以分别帮助评估重症肌无力和甲状腺相关眼病。肌肉活检的特征性发现还可以区分不同的潜在肌病。KSS 将在 Gomori 毛状体染色上显示细胞色素 C 氧化酶阴性纤维和参差不齐的红色纤维。眼咽肌营养不良症表现为嗜碱性边缘空泡和丝状核内包涵体。强直性肌营养不良患者的肌肉活检显示内部细胞核、环状纤维、肌浆团和早期 I 型纤维萎缩。如果临床或家族史表明某种特定的综合征,基因检测对于确定预后和需要进行的其他研究可能非常有价值。考虑到所涉及的潜在核和线粒体 DNA 的广泛清单,可能需要经验丰富的实验室来进行遗传分析。
4、影像学
眼眶磁共振成像 (MRI) 经常显示 CPEO 患者 EOM 萎缩。一项研究表明,与对照组相比,CPEO 患者的 EOM 横截面积减少了 43%。脑部MRI检查结果众多且非特异性,包括白质高信号、皮质萎缩、小脑萎缩和脑干高信号。光学相干断层扫描可能显示视网膜外层和黄斑中央凹的厚度减少,以及视神经乳头和边缘的体积减少。
5、鉴别诊断
- 重症肌无力
- 甲状腺相关眼病
- 先天性眼外肌纤维化
- 眼结节病
- 肉毒中毒
- CN 3 麻痹
四、疾病管理
虽然 CPEO 或其相关综合征没有明确的治愈方法,但控制症状可以显着改善患者的生活质量。需要转诊治疗伴随的神经系统或心脏病。大约三分之一的患者会出现持续性或间歇性复视。在对眼睛偏差的方向和幅度进行全面的正交评估后,棱镜镜片可有效控制复视。棱镜通过折射光线来补偿偏斜的眼睛,使图像落在每只眼睛的黄斑上。虽然一些患者选择手术矫正水平直肌,但由于 CPEO 的进行性,复视或斜视可能会随着时间的推移再次出现。临床上明显的上睑下垂可以通过手术矫正。如果提上睑肌功能保持中等或良好,可以考虑推进或切除上睑提肌,以最大限度地提高眼睑肌肉的高度。然而,通常提肌功能会逐渐恶化,导致提肌偏移最小。如果提上睑肌功能较差,应考虑使用自体或合成吊带材料将眼睑悬吊在额肌上。由专业的眼整形外科医生进行仔细的术前评估和手术至关重要。通过手术过度矫正上睑下垂可能会导致眼球突出和角膜暴露,从而导致暴露性角膜病、角膜溃疡或眼穿孔等潜在的致盲并发症。一些患者使用带有硅胶棒的额肌吊带来矫正 CPEO 的上睑下垂由于角膜病变是 CPEO 最常见的并发症之一,因此修复曝光是关键。有一种新的方法可以通过睑裂转移(PFT)来手术治疗 CPEO,在没有垫片的情况下抬高下眼睑,成功覆盖角膜巩膜镜可能是一种可行的非手术替代方案,并具有舒适性和眼表保护的额外好处。
患者视角:
我和 CPEO 一起生活了 20 多年。我很早就学会了如何控制症状,为自己提供最高质量的生活。在 CPEO 中,眼睑无法完全闭合且无力。这会导致眼球得不到适当的保护,进而导致视力障碍(视力模糊、复视、严重干眼)。好消息是,通过适当的日常护理,一个人可以改善和帮助他们的眼睛。在我知道并开始我的团之前,我的角膜就出现了溃疡。自从采取主动措施以来,我什么都没有。以下是与 CPEO 一起生活时有助于保护眼睛环境的建议:
- 泪管塞——有助于保持眼睛湿润,眼科医生只需一分钟即可将其放入。
- 每天晚上用胶带把眼睛闭上——早上醒来时眼睛不再干燥、发痒。睡觉前将 1 英寸 3MM 手术泡沫胶带贴在每只眼睛上。首先在每只眼睛中涂抹眼部润滑剂。然后从胶带卷上撕下约 3 英寸,并将眼睑向下固定(就像将其向下拉并固定到位一样)。3 英寸的胶带应水平放置。
- 每日使用眼部润滑滴剂。
- 每天使用 Restasis RX 滴眼液。
- 早上醒来时热敷每只眼睛。
- 当眼睛特别干燥时,需要加厚的眼部润滑剂或凝胶滴眼液。
- 额肌吊带手术请咨询专门从事眼睑提升术的眼整形外科医生,看看您是否适合。抬起眼睑可以改变游戏规则,从而获得更好的视线。
- 外出时戴防护眼镜,以遮挡风吹日晒。
- 通过 UMDF 检查肌肉无力(线粒体肌病)的临床试验。
以上不是医疗建议,而是患者多年来学到的帮助管理 CPEO 症状的方法。对于该患者的肌肉无力,每周进行三次物理治疗。
*请注意,这是一位患者的观点,而不是医疗建议。
五、慢性进行性眼外肌麻痹伴炎性肌病患者报告
2010年12月8日,一名44岁的中国男性因双侧上睑下垂、复视二十年首次就诊。他的眼球运动逐渐受到限制。没有明显的四肢无力,但有一点运动不耐受。他没有白内障、色素性视网膜炎。41岁时,病情突然加重。患者表现为严重的延髓麻痹。存在构音障碍、吞咽困难和饮水时窒息。首先发现颈部肌肉无力,尤其是躺在床上时,头抬不起来。没有皮疹或肌痛。他没有家族史。神经系统检查显示面部肌病、双侧上睑下垂、眼球固定完全性眼肌麻痹。水平凝视中呈现双重视觉。注意到面部肌肉无力和面部表情减少。颈屈肌为 MRC 2 级。近肢肌肉轻度受累,MRC 5 级。深腱反射正常。实验室检查显示血清CK正常(146.9 U/L,正常:20-200 U/L),乳酸(2.82 mmol/L;正常:0.7-2.1 mmol/L)和丙酮酸(110.4 umol/L;正常:0.7-2.1 mmol/L)轻度升高。正常:10-100 umol/L)。甲状腺功能和血清自身抗体正常。针肌电图显示肌病性改变。神经传导和重复神经刺激试验均正常。心电图(ECG)也正常。
1、肌肉的组织学变化
进行了第一次肌肉活检。H&E 染色显示肌纤维尺寸的变化,并且纤维经常表现出肥大、再生、分裂和核内化。同时观察到成群的肌纤维变性、坏死、吞噬,肌周炎细胞浸润明显。值得注意的是,发现大量淋巴细胞浸润在小血管周围。束周围没有萎缩纤维的分布。MGT染色显示约15%的RRF,与COX染色中的阴性反应相对应。SDH 染色显示纤维的酶活性增加。免疫组织化学染色显示大量 CD4+ 细胞侵入血管周围区域和纤维肌膜。针对 CD8、MHC-I 和 C5b9 膜攻击复合物的抗体微弱地沉积在肌纤维之间。CD68 强表达存在于血管周围区域和纤维肌膜中。超微结构研究显示大量肌膜下晶状包涵体和糖原颗粒。
2、基因改变
遗传分析结果表明,长 PCR 扩增的患者片段比对照片段短,表明存在缺失。2363 bp 和 570 bp 的 PCR 产物分别显示患者肌肉中存在 4977 bp 缺失。mtDNA分析显示,患者肌肉中缺失的mtDNA呈异质性,缺失的mtDNA比例约为84%。发现该缺失与之前报道的 4977 bp 缺失(常见缺失)相同,已与 CPEO 相关联。断点发生在位置 8470-13446 或 8483-13459,其两侧是正常线粒体基因组中 bp 8470-8482 和 13447-13459 处的完美 13 bp 同向重复序列。由于该 13 bp 序列可能来自侧翼区域的任一侧或部分由两侧贡献,因此无法精确确定缺失的断点。为了验证高通量测序确定的缺失边界,使用3730 DNA分析仪对缺失区域的570 bp PCR产物进行测序。直接桑格测序显示,该序列与剑桥参考序列一致,直到 bp 8469,然后出现 13 bp 直接重复序列(5'-ACCTCCCTCACCA-3'),并且该序列在 bp 13460 处继续。结果与使用 Miseq 测序获得的结果完全相同。
3、治疗和结果
根据临床表现、肌肉病理及基因分析,诊断为单纯CPEO并发炎症性肌病。因此,他接受了口服辅酶 Q10、维生素 E 和泼尼松治疗。泼尼松剂量初始为50mg/d,持续2周,逐渐减量每周5mg,直至剂量为15mg/d,维持1个月。然后每周减少该药5mg,直至5mg/d,持续3个月。
服药6个多月后,吞咽困难、构音障碍得到很大改善。喝水时的哽咽现象从未出现过,颈肌无力也稍有改善。与此同时,进行了第二次肌肉活检。它显示没有血管周围或肌束膜炎症细胞侵袭,CD4、CD8、CD68、MHC-I、C5b9免疫组化染色未见阳性反应。仍然观察到 RRF 和 COX 阴性纤维。于是他停止服用泼尼松,但辅酶Q10和维生素E仍在使用。2013年3月24日,患者复诊。双侧眼睑下垂、眼球固定如前,四肢近端肌无力稍有进行性,MRC 4 级,但延髓肌无力几乎消失。
4、讨论
由于双侧上睑下垂和眼球运动受限病史超过 20 年,肌肉活检中 RRF 和 COX 阴性纤维,以及 m.8470_13446del4977 的 mtDNA 缺失,该患者 CPEO 的诊断是明确的。
但值得注意的是,当患者41岁时,病情出现了亚急性且显着的进展。存在延髓麻痹,颈伸肌受累,导致明显的吞咽困难、构音困难以及躺在床上时抬头困难。这种亚急性加重与CPEO的病程并不一致。尽管一些 CPEO 患者有口咽无力,但这些表现往往是隐匿性的。本例患者的吞咽困难症状不是慢性进行性的,而是亚急性的。CPEO 很少累及轴向肌肉,但该患者有严重的颈部无力。CPEO的诊断无法解释亚急性进展和新的临床表现,更像是肌炎等获得性肌病。在肌炎中,颈部伸肌可能受累,导致头部难以抬起(头下垂),在肌炎的晚期病例和罕见的急性病例中,会出现吞咽困难伴窒息发作和呼吸肌无力。为了确认诊断,进行了肌肉活检。有趣的是,活检不仅显示了 RRF 和 COX 阴性纤维,还显示了血管周围和肌束膜区域的炎症。此外,免疫组织化学染色显示标本上CD4和CD68呈阳性。因此病理表明患者同时患有炎症性肌病和线粒体功能障碍。随后对患者进行了泼尼松治疗,效果很好,延髓麻痹明显好转,颈部肌肉无力也较之前有所好转。治疗后,第二次活检显示炎症和免疫组化染色阳性区域消失。因此,根据临床、病理和治疗效果的所有证据,证明该患者不仅患有CPEO,而且患有炎症性肌病。但它不是典型的皮肌炎或多发性肌炎,应被鉴定为炎性肌病。
其他文献报道了一名患有3251A>G mtDNA突变的CPEO成年男性患者,该患者出现突发性呼吸衰竭。肌肉活检显示炎症变化。报道了一例m.3243A>G突变的线粒体脑肌病、乳酸性酸中毒和中风样发作(MELAS),其组织学显示非特异性肌炎。在我们的案例中,他是一名 CPEO 患者,但也患有炎症性肌病。那么这就需要考虑到,是否有一些由线粒体紊乱引起的因素引发了炎症反应呢?虽然在实验动物中,线粒体DNA的变化会增加对自发产生的免疫综合征的易感性,包括胶原诱导的关节炎、糖尿病、肾炎和胰腺炎,或免疫后,例如胶原诱导的关节炎和实验性自身免疫性脑脊髓炎,但线粒体疾病使之复杂化。伴有炎症的情况确实很少见。所以没有足够的证据,这些案例更像是巧合,但仍需要未来的研究。但另一方面,据报道,在免疫和炎症性肌病中观察到线粒体异常。COX 染色减少表明线粒体疾病与线粒体 DNA (mtDNA) 缺陷相关,这种缺陷可以在散发性包涵体肌病、多发性肌炎和皮肌炎中观察到。但这被认为与线粒体DNA氧化损伤、核苷酸库改变或线粒体DNA聚合酶错误率增加有关。
总之,虽然3251A>G mtDNA突变并发炎症性肌病的CPEO已有报道,但线粒体DNA缺失综合征中最常见的m.8470_13446del4977 mtDNA缺失的CPEO从未报道过 。从这个病例可以看出,反复的肌肉活检和及时的治疗对于诊断和患者本人都非常重要,所以对于一些慢性进展性病程的遗传性肌病,如果出现急性或亚急性进展并伴有一些不典型的症状,临床应该考虑并发其他疾病的可能性。
六、慢性进行性外眼肌麻痹 (CPEO) 患者的脑桥中风
1、病例报告
一名 70 岁的非洲裔美国男性因右侧面部无力和构音障碍急性发作五个小时而被送往急诊室。经过进一步询问,他患有非波动性进行性上睑下垂大约 10 年,并且在过去几个月里液体和固体吞咽困难不断恶化。他的父亲和祖父有双侧上睑下垂的家族史。详细检查显示对称且双侧反应性瞳孔,严重双侧上睑下垂和眼肌麻痹,完全垂直凝视麻痹和水平眼球运动严重受限,以及涉及眼轮匝肌和口部的右侧面部无力。收敛性受到损害。腭抬高和吞咽功能受损。完全垂直凝视麻痹。上肢或下肢无肌肉无力。除了不存在踝关节抽搐外,深部腱反射正常。患者否认疼痛。
根据症状的严重程度,考虑为急性中风。在急诊科进行了脑部 CT 扫描,结果显示出血呈阴性。第二天的脑部 MRI 显示,右脑桥背侧第七脑神经核和束水平发生急性缺血性中风。该患者出现在溶栓窗口之外,因此通过二级预防措施来治疗缺血性脑卒中。初步相关实验室工作显示肌酸激酶 (CK) 升高至 6,080 U/L(一周内降至 477 U/L),临界高低密度脂蛋白 (LDL) (131 mg/dL),且正常糖化血红蛋白 (5.6%)。
一项神经传导研究,结合踝反射缺失,提示可能存在长度依赖性感觉轴突神经病。复合肌肉动作电位(CMAP)为单峰。对左面神经(在鼻肌处记录)和左脊副神经(在斜方肌处记录)的重复神经刺激没有显示出 CMAP 振幅的任何衰减。口服吡斯的明和冰袋试验均未改善眼球症状。抗乙酰胆碱受体和肌肉特异性酪氨酸激酶的血清抗体呈阴性。
基于对线粒体疾病的怀疑以及缺乏用于生化和组织化学分析的可行组织活检,在血液中完成了基因检测。与多基因组或靶向下一代测序方法相反,全外显子组测序(WES)作为疑似线粒体疾病和其他模拟神经肌肉疾病(例如先天性肌无力)的首选且更有效的诊断工具而受到追捧。与其他方法相比,WES 的成功源于线粒体疾病和神经肌肉疾病的显着遗传和临床异质性,这些疾病可能具有重叠的临床特征。
WES 证明了PEO1 / TWNK (C10ORF2, NM_021830.4/5) 基因中的一个变异 (c.1510G > A (p.Ala504Thr)) ,该基因编码一种称为“twinkle”的线粒体蛋白,这是一种参与维持 mtDNA 的 DNA 解旋酶。使用几种计算机致病性预测工具时,p.Ala504Thr 替换可能是有害的:从耐受中排序不耐受 (SIFT)、PolyPhen2、对齐梯度验证梯度偏差 (GVGD)和 Rare外显子组变异集成学习器 (REVEL) 。 因此,该突变代表了与 CPEO 相关的新突变。
患者出院后失访;由于患者的任何受影响的家庭成员均已去世,因此无法完成基因检测。
2、讨论和结论
在临床上,该患者的急性脑桥卒中和 CPEO 表现提出了几个有趣的学习点。首先,脑桥中风引起孤立性 CN VII 麻痹的情况很少见。如果患者没有基线眼肌麻痹,患者的中风会因右外展肌麻痹而导致右眼外展受损,因右侧内侧纵束受累而导致右眼内收受损,以及因旁桥网状结构受累而导致左眼内收受损。即一只半综合症(是一种罕见的眼球运动无力)。除了一只半综合征外,中风还会因CN VII受累而导致右侧面神经麻痹。然而,患者与中风相关的动眼神经症状被他潜在的眼肌麻痹所掩盖。其次,中风或中风样发作是线粒体疾病的潜在表现,通常是线粒体脑肌病、乳酸性酸中毒和中风样症状 (MELAS)。我们的患者没有 MELAS 表型或与 MELAS 相关的已知突变,并且中风的位置对于与 MELAS 相关的中风来说是不典型的,通常是皮质或丘脑,可能不符合任何已知的血管区域 。另一方面,有人提出,除 MELAS 之外的线粒体疾病也可能诱发中风 。考虑到他的年龄、脑成像中存在小血管疾病和高血压血管病以及入院时血压升高的范围,他的脑桥腔隙性中风的病因很可能是小血管疾病。头颈部计算机断层扫描血管造影(CTA)显示弥漫性动脉粥样硬化性疾病,包括颈内动脉、椎动脉和双侧大脑中动脉的多灶性狭窄。根据时间进程,出现症状的星座根据敏锐度进行区分。患者突发右半面瘫和构音障碍。该患者上下面部无力提示下运动神经元病变,累及面神经核、束或神经。考虑到患者的急性发病和年龄,考虑到累及颅神经 VII 核或其束的 CVA。
该患者进行性对称性眼球肌无力的鉴别诊断包括:
1)神经肌肉接头(NMJ)疾病,特别是重症肌无力和先天性肌无力综合征;
2)遗传性肌病,如CPEO、眼咽肌营养不良症和强直性肌营养不良症。根据重复神经刺激、冰袋试验和重症肌无力血清学的阴性结果,认为该患者不太可能患有神经肌肉接头疾病。CMAP 的正常配置反对慢通道综合症。考虑到整体临床表现、CK 显着升高以及三角肌中存在肌病性运动单位募集,EMG 中存在弥漫性颤动和正波提示潜在的肌病(而不是弥漫性去神经支配)。
线粒体肌病不仅可以解释慢性眼球症状,还可以解释远端神经病变和横纹肌溶解症的亚临床发作。其他遗传性家族性肌病,如眼咽肌营养不良症和 I 型强直性肌营养不良症,均通过基因检测排除。尽管本例没有进行肌肉活检,但临床特征非常提示 CPEO 的诊断,并且与PEO1 / TWNK基因中类似突变的影响一致。
PEO1 ( TWNK ) 是编码闪烁蛋白的核基因,该基因的突变与 CPEO 相关。PEO1的常染色体显性突变已被证明会引起一系列症状,包括上睑下垂、外眼肌麻痹和肌病。PEO1的常染色体隐性突变与婴儿期发病的脊髓小脑共济失调有关。临床表现和家族史(患者父亲和祖父有类似症状)均符合常染色体显性遗传模式;因此,我们没有进行线粒体 DNA 测序。twinkle 蛋白由 684 个氨基酸组成,分子量为 77kD。研究人员指出该蛋白质具有三个功能域:5'引物酶结构域、连接子结构域和3'解旋酶结构域。大多数人类突变发生在接头和解旋酶结构域中,PEO1中的致病性突变聚集在残基 Arg303 和 Tyr508 之间。该患者的新突变 (p.Ala504Thr) 位于解旋酶结构域中,影响预计与稳定寡聚结构相关的残基 。
我们的案例研究有几个局限性:
1)我们无法获得肌肉活检,这可能进一步支持线粒体肌病的诊断,
2)无法在患者的家庭成员中进行基因检测来确认家族遗传和突变的致病性质。
总而言之,我们介绍了一名 70 岁男性,其急性发作的右半面部无力,伴有慢性进行性双侧上睑下垂、眼肌麻痹和吞咽困难。症状的时间进程和正确的解剖定位对于识别多种病理的存在至关重要:急性发作的脑桥中风、孤立性第 VII 脑神经麻痹、潜在的 CPEO、慢性进行性眼延髓肌无力的线粒体肌病。基因检测发现 PEO1 基因的致病“热点”存在新变异,支持 CPEO 的诊断。
七、头皮动脉粥样硬化:单个线粒体 DNA 缺失导致的慢性进行性眼外肌麻痹的新特征
1、案例展示
患者为76岁白人女性,身高174cm,体重65kg,自15岁起逐渐出现进行性双侧眼睑下垂,随后逐渐出现眼睑麻痹,无复视。她在 38 岁时被诊断出患有多结节性甲状腺肿和甲状腺功能减退症,这就是她开始服用左甲状腺素的原因。42岁时,她的肺活量减少了一半。48岁时首次发现肝功能参数升高、肝肿大和脂肪变性肝炎。此外,她从56岁起就注意到上肢缓慢进行性肌肉无力。此外,她还经历过睡眠过度(10-11每天睡眠时间)、右脸抽动、左侧视神经萎缩、双侧白内障需要手术、青光眼、慢性胃炎、胆囊结石需要胆囊切除、憩室病、反复腹泻、易疲劳、多汗症、牛皮癣、头皮多发性粥样硬化、自发性腰椎病2号椎骨骨折、肌酸激酶(CK)轻度升高、高脂血症和高尿酸血症。根据临床表现,怀疑患有线粒体疾病 (MID),并于 49 岁时开始检查。MID 家族史呈阴性。
表:一般情况和指标患者 CPEO-plus 的表型特征
| 特征 | 先前报道过的研究 | 索引 | KSS |
| 下垂症 | [1] | 是的 | 是的 |
| 眼肌麻痹 | [1] | 是的 | 是的 |
| 白内障 | [8] | 是的 | 是的 |
| 视神经萎缩 | [9] | 是的 | 不 |
| 四肢瘫痪 | [10] | 是的 | 是的 |
| 甲状腺功能减退症 | [11] | 是的 | 是的 |
| 高脂血症 | [12] | 是的 | 不 |
| 心肌病 | [13] | 是的 | 是的 |
| 沮丧 | [14] | 是的 | 不 |
| 运动不耐受 | [14] | 是的 | 是的 |
| 全局ID | [14] | 是的 | 不 |
| 帕金森症 | [14] | 不 | 不 |
| 糖尿病 | [14] | 不 | 是的 |
| 认知障碍 | [14] | 不 | 是的 |
| 共济失调 | [14] | 不 | 是的 |
| 视网膜变性 | [15,16] | 不 | 是的 |
| 偏头痛 | [17] | 不 | 是的 |
| 癫痫 | [18] | 不 | 是的 |
| 神经病 | [19] | 不 | 不 |
| 肾衰竭 | [14] | 不 | 是的 |
| 嗜睡症 | 不 | 是的 | 是的 |
| 面部抽动 | 不 | 是的 | 不 |
| 青光眼 | 不 | 是的 | 是的 |
| 巴格达公司 | 不 | 是的 | 是的 |
| 多节瘤 | 不 | 是的 | 不 |
| 肝肿大 | 不 | 是的 | 是的 |
| 脂肪肝 | 不 | 是的 | 是的 |
| 憩室病 | 不 | 是的 | 不 |
| 慢性胃炎 | 不 | 是的 | 不 |
| 多汗症 | 不 | 是的 | 不 |
| 动脉粥样硬化 | 不 | 是的 | 不 |
| 银屑病 | 不 | 是的 | 不 |
BGC:基底神经节钙化,GID:胃肠动力障碍,KSS:卡恩斯-塞尔综合征
临床神经系统检查显示抑郁情绪、明显双侧上睑下垂、眼肌麻痹伴球部发散以及四肢瘫痪 M5-。此外,还存在轻度高CK血症(113U/l,n,<70U/l)、高脂血症和维生素D缺乏。乳酸应激测试结果出现高度异常。右肱二头肌针肌电图正常。视觉诱发电位显示 P100 潜伏期显着延长,左侧为 186 毫秒,右侧为 172 毫秒。脑CT显示轻度基底神经节钙化以及多发性头皮粥样斑块。脑部 MRI 也证实了头皮粥样斑块,但结果尚无定论。颈椎 MRI 发现 C5/6 和 C6/7 椎体狭窄,但没有脊髓病。颈动脉超声和经颅双功能超声检查尚无结论。超声心动图也显示仅轻度左心室肥厚,但收缩功能正常。此外,重复的 24 小时心电图仅显示偶尔的室上性异位搏动。
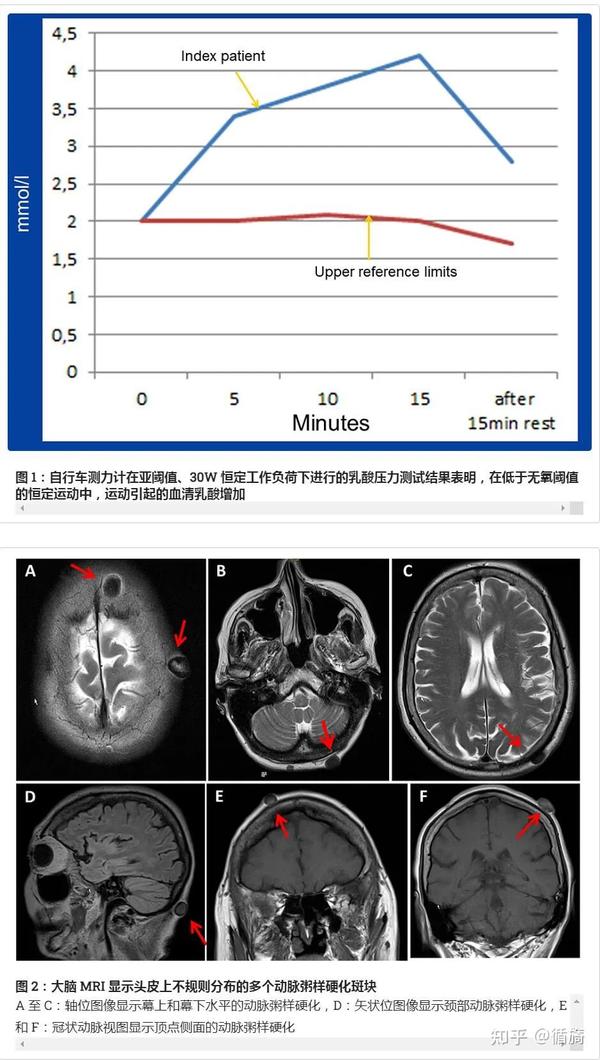

49 岁时,肌肉活检显示大量参差不齐的红色纤维、大量脂滴和畸形线粒体。没有 COX 阴性肌纤维。59 岁时,眼外肌活检证实了之前的发现。57 岁时,首次使用 Southern 印迹对淋巴细胞中 mtDNA 点突变和 mtDNA 缺失进行了基因检测,但没有提供任何信息。通过长程聚合酶链反应和 Southern blot 对肌肉 mtDNA 进行的第二次基因检查显示,骨骼肌和眼外眼肌中存在单个 4977bp mtDNA 缺失(常见缺失),异质率分别为 60% 和 30%。同样,在血液淋巴细胞中未发现 mtDNA 缺失。一级亲属没有接受线粒体 DNA 缺失检测。
59岁时,她因眼肌麻痹接受了斜视手术。此外,她还做过两次上睑下垂矫正手术,但都只能暂时缓解症状。她目前使用的药物是L-甲状腺素、环拉西酮、三他嗪酮和噻吗洛尔滴眼液。抗氧化剂、辅助因子和其他补充剂没有效果。
2、案例讨论
该病例表明,由单个mtDNA缺失引起的CPEO可以在40年以上稳定病程,在发展为CPEO-plus之前,眼睑下垂和眼睑麻痹是唯一的表现。CPEO-plus不仅可能表现为上睑下垂、眼肌麻痹、共济失调、偏头痛、癫痫、视神经萎缩、白内障、视网膜变性、四肢瘫痪、甲状腺功能减退、高脂血症、心肌病和神经病,还可能表现为以前未报道的特征,例如嗜睡、面部抽动、青光眼、基底节钙化、甲状腺肿、肝病、憩室病、慢性胃炎、多汗症、头皮动脉粥样硬化和牛皮癣。此外,CPEO-plus 可以被认为是纯 CPEO 和 Kearns-Sayre 综合征 (KSS) 之间的桥梁,因为许多 CPEO-plus 特征也存在于 KSS 患者中。
目前还不清楚是否所有未报告的表型特征都真正归因于 mtDNA 缺失,因为之前没有报道过一些与 CPEO-plus 相关的表型特征,可能是由于其他原因造成的,因此是一个巧合的发现。与此同时,支持这些新特征与 mtDNA 缺失之间关系的论点包括这样的事实:至少其中一些特性之前已在除 CPEO 之外的 MID 中检测到,并且除了 mtDNA 缺失之外,没有其他明显的可能原因。
据报道,白天过度嗜睡会导致线粒体耗竭综合征以及线粒体脑病、乳酸性酸中毒和中风样发作 (MELAS) 综合征。最近在中国患者中发现了线粒体tRNA基因变异的抽动障碍。青光眼已被宣布为多种 MID,包括综合征型和非综合征型。无论有或没有甲状旁腺功能减退症,基底神经节钙化是各种 MID 共有的另一个表型特征。内分泌疾病,包括甲状腺肿和甲状腺功能减退症,在 MID 中很常见。POLG1 突变会导致多种线粒体耗竭综合征中伴有或不伴有脂肪变性的肝病。胃肠道损害在 MID 中很常见,甚至可能是主要特征,如线粒体神经胃肠脑病 (MNGIE)。TARS-2 变异与出汗增加(多汗症)有关,这可能与或不与自主神经功能障碍有关。尽管银屑病与线粒体功能障碍有关,但在综合征型或非综合征型 MID 中尚未有报道。颅骨动脉粥样硬化是指标患者的另一个特征,在之前的 CPEO+ 病例、KSS 患者或其他 MID 中未显示过。头皮动脉粥样硬化与 MID 肿瘤发生率的增加有关。目前尚不清楚患者将来是否会经历额外的报告或未报告的 CPEO 特征,但考虑到之前的病程,表型很可能会进展,可能发展为类似 KSS 的特征。
仅偶尔有报道称 CPEO 进展为 CPEO-plus。研究人员对 CPEO 患者进行的一项研究中。一名患者在 5 岁时出现肾上腺功能不全,另一名患者在 14 岁时出现肢体肌肉无力。在 19 年的观察期内,228 名携带单个 mtDNA 缺失的患者中,除 CPEO 之外的表型表现数量有所增加。
3、结论
评估的病例表明 CPEO-plus 的表型谱比以前想象的要广泛得多,牛皮癣和头皮粥样硬化是 CPEO-plus 的独特特征,CPEO 经过多年演变为 CPEO-plus,并且单个 mtDNA 缺失可能是在血液淋巴细胞中缺失,但在肌肉细胞中检测到。
本文参考
Chen T, Pu C, Shi Q, Wang Q, Cong L, Liu J, Luo H, Fei L, Tang W, Yu S. Chronic progressive external ophthalmoplegia with inflammatory myopathy. Int J Clin Exp Pathol. 2014 Dec 1;7(12):8887-92. PMID: 25674260; PMCID: PMC4314000.
Chronic Progressive External Ophthalmoplegia (CPEO)
DiseasesDB
29124
ICD-10
H49.4
MeSH
D017246
Eliyan, Y., Rezania, K., Gomez, C.M. et al. Pontine stroke in a patient with Chronic Progressive External Ophthalmoplegia (CPEO): a case report. BMC Neurol 23, 231 (2023). http://doi.org/10.1186/s12883-023-03249-9
Finsterer J (December 23, 2021) Atheromatosis of the Scalp: A Novel Feature of Chronic Progressive External Ophthalmoplegia Plus Due to a Single Mitochondrial DNA Deletion. Cureus 13(12): e20641. doi:10.7759/cureus.20641
