Cytolysins and Actinoporins
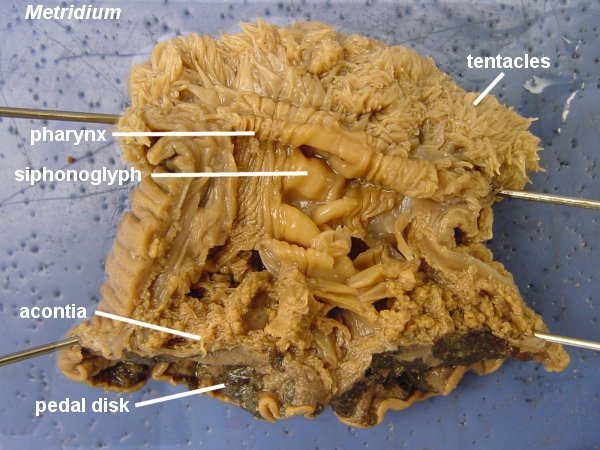
It’s kinda funny that it took me five posts before I started writing about actual anemone toxins, but I thought proving context was important before I dive into the really juicy bits. Compared to other cnidarians (e.g. Jellyfish), anemones have more variety in the types of toxins they use.
Here is a short (but by no means comprehensive) list of all the goodies in anemone venom: cytolysins, actinoporins, potassium channel toxins, sodium channel toxins, phosolipases, metalloproteases, serine protease inhibitors/Kunitz peptides, ASIC (acid sensing ion channel) inhibitors, TRPV1 inhibitors and small cysteine rich peptides (SCRiPs). In this post, I’ll focus on the ‘ripper toxins’ AKA your cytolysins and actinoporins.
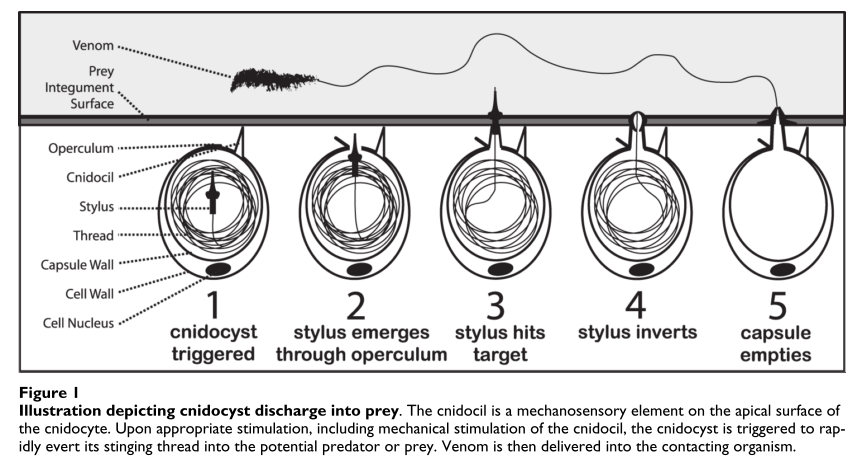
Figure 1: “Ripper” toxins in action
Almost every cnidarian species has a “ripper toxin” or a psychotic molecular chainsaw that rips through the cell membrane and causes water to flood into the cell, until it swells and bursts due to osmotic pressure. There are two contradictory parts to our pore forming toxin: one part is hydrophilic (or water loving/water soluble) and it binds to the receptors of our poor cell. The second part is the membrane bound structure which is more hydrophobic since it plays a part in pore formation. There are two main categories of pore forming toxins: actinoporins and cytolysins.
Actinoporins (~20 kDa) have a really simple structure which really shows when you realize that they don’t even have any disulphide bridges. Some actinoporins will only bind to sphingomyelin while others can bind to phosphatidylcholine in membranes (E.g. Sticholysin II). Actinoporins plan of attack involves using their aromatic loops to bind to the membrane, aligning themselves in the perfect position for cellular murder, oligomerising and then shoving the N-terminus of their alpha helix into the membrane like a tiny drill. Some studies suggest that the amphiphilic alpha helix actually detaches from the main body of toxin before drilling into the membrane. Since actinoporins can causes cardiovascular and respiratory problems, you can usually use a hemolytic assay to experimentally figure out if you have actinoporins or cytolysins in samples.
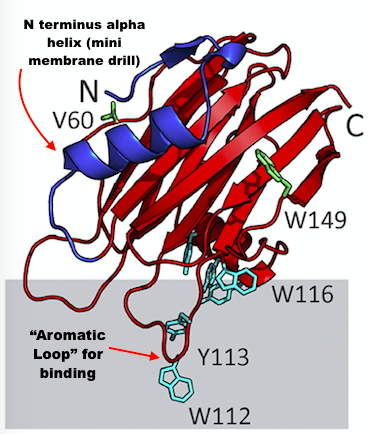
Figure 2: A diagram of pore forming toxin with our two key components
One of the appeals of studying anemone toxins, is using toxins to mess with ion channels or cell membranes in order to get an understanding of how cell membranes and ion channels work. A lot of times in Biology we learn about how things work by screwing up specific parts of proteins, cells, genomes etc. Cytolysins have been used in studies to study cell membrane dynamics and structure. Since biologists are a bunch of pedantic nerds who have to classify everything they categorize cytolysins based on molecular weights and other commonalities.
Figure 3: 3D stcrure of EQUINATOXIN II
- Type I, cytolysins tend to have a molecular weight of
5–8 kDa peptides and they tend to form pores in phosphatidylcholine containing membranes. They have antihistamine
activity (plays a part in the nervous and cardiovascular system). Some type I
cytolysins have no cysteine residues and they tend to have high as hell isoelectric
points.
- Type II (~20 kDa) cytolysins are really common and well-studied.
They tend to target phospholipid domains,
get inhibited by sphingomyelin and they oligomerise to form pores. One of the
key structural features is the cation selective hydrophobic core which can
cause haemolysis.
- Type III (30–40 kDa) cytolysins can have PLA (phosolipases
A) activity in some cases.
- The largest type of cytolysins are Type IV
cytolysins (~80 kDa) and they are thiol-activated.
Why do you need to know about these types of cytolysins?
FOR FUN! And also, because, knowing the size of your toxin helps with isolation and identification of specific toxins. For example, you get your crude anemone venom (through some magical miracle) and do SDS PAGE to get a rough venom profile. If you know the approximate sizes you can guess what kind of toxins are in the anemone’s venom.
This post is a brief introduction to actinoporins and cytolysins. I could showcase specific cytolysins or dive into the structure-function relationships in future posts. (Even more foreshadowing….)
Citations
- Norton, R. S. (2009). Structures of sea anemone toxins. Toxicon, 54(8),
1075–1088. http://doi.org/10.1016/j.toxicon.2009.02.035
- Norton, R. S. (2006). Sea Anemone Venom Peptides. Handbook of
Biologically Active Peptides. Elsevier Inc. http://doi.org/10.1016/B978-012369442-3/50056-8
- Jouiaei, M., Yanagihara, A. A., Madio, B., Nevalainen, T. J., Alewood,
P. F., & Fry, B. G. (2015). Ancient venom systems: A review on cnidaria
toxins. Toxins, 7(6), 2251–2271. http://doi.org/10.3390/toxins7062251
- Rojko, N., Dalla Serra, M., Maček, P. &
Anderluh, G. Pore formation by actinoporins, cytolysins from sea anemones. Biochim.
Biophys. Acta - Biomembr.1858, 446–456 (2016).