
Abstract
The fungus Trichoderma arundinaceum exhibits biological control activity against crop diseases caused by other fungi. Two mechanisms that likely contribute to this activity are upregulation of plant defenses and production of two types of antifungal secondary metabolites: the sesquiterpenoid harzianum A (HA) and the polyketide-derived aspinolides. The goal of the current study was to identify aspinolide biosynthetic genes as part of an effort to understand how these metabolites contribute to the biological control activity of T. arundinaceum. Comparative genomics identified two polyketide synthase genes (asp1 and asp2) that occur in T. arundinaceum and Aspergillus ochraceus, which also produces aspinolides. Gene deletion and biochemical analyses in T. arundinaceum indicated that both genes are required for aspinolide production: asp2 for formation of a 10-member lactone ring and asp1 for formation of a butenoyl subsituent at position 8 of the lactone ring. Gene expression and comparative genomics analyses indicated that asp1 and asp2 are located within a gene cluster that occurs in both T. arundinaceum and A. ochraceus. A survey of genome sequences representing 35 phylogenetically diverse Trichoderma species revealed that intact homologs of the cluster occurred in only two other species, which also produced aspinolides. An asp2 mutant inhibited fungal growth more than the wild type, but an asp1 mutant did not, and the greater inhibition by the asp2 mutant coincided with increased HA production. These findings indicate that asp1 and asp2 are aspinolide biosynthetic genes and that loss of either aspinolide or HA production in T. arundinaceum can be accompanied by increased production of the other metabolite(s).
Key points
• Two polyketide synthase genes are required for aspinolide biosynthesis.
• Blocking aspinolide production increases production of the terpenoid harzianum A.
• Aspinolides and harzianum A act redundantly in antibiosis of T. arundinaceum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple species of the fungus Trichoderma are used for biological control of plant pathogenic fungi, and secretion of antimicrobial secondary metabolites (antibiosis) contributes to the biological control (Cardoza et al. 2011; Degenkolb et al. 2008; Hermosa et al. 2014; Nielsen et al. 2005; Sivasithamparam and Ghisalberti 1998). For example, the ability of Trichoderma arundinaceum to limit disease caused by the plant pathogenic fungi Botrytis cinerea and Rhizoctonia solani has been attributed, in part, to production of harzianum A (HA), a secondary metabolite (SM) that is a member of the trichothecence family of toxic sesquiterpenoids (Malmierca et al. 2012, 2013). However, HA-nonproducing mutants of the fungus retain varying but significant levels of antifungal activity. Retention of the activity has been attributed to the production of aspinolides, a family of antimicrobial polyketide-derived SMs that are produced at higher levels in HA-nonproducing mutants than in wild-type T. arundinaceum (Lindo et al. 2018, 2019a, 2019b; Malmierca et al. 2015, 2013, 2012).
Although recent studies report aspinolide production by T. arundinaceum (phylum Ascomycota, class Sordariomycetes), production was first reported in the relatively distantly related fungus Aspergillus ochraceus (phylum Ascomycota, class Eurotiomycetes) (Fuchser and Zeeck 1997). This first report included precursor feeding experiments with labeled acetate and O2 gas. From these experiments and the deduced structure of several aspinolide analogs, the authors proposed an aspinolide biosynthetic pathway in which both the aspinolide backbone, a 10-member lactone ring, and a butenoyl substituent are polyketides (Fuchser and Zeeck 1997).
Nine aspinolide analogs (AspB-AspJ) have been identified in cultures of wild-type and HA-nonproducing strains of T. arundinaceum, but not all strains produced all analogs (Fig. 1A, Figure S1) (Izquierdo-Bueno et al. 2018; Malmierca et al. 2015). Aspinolide production by T. arundinaceum is dependent on culture conditions and is generally inversely proportional to HA production. Although only low levels of AspB and AspC are produced at early time points in cultures of wild-type T. arundinaceum, both are produced at markedly higher levels in cultures of HA-nonproducing mutants (Izquierdo-Bueno et al. 2018; Lindo et al. 2018). Interestingly, the biological activity of AspC is similar to that of HA in that both metabolites exhibit antifungal activity against B. cinerea and in vitro phytotoxicity against tomato. In contrast, some other aspinolide analogs (e.g., AspB and AspF) lack these activities (Malmierca et al. 2015). In addition, both HA and AspC can upregulate the expression of plant defense-related genes in tomato (Malmierca et al. 2015).

A Chemical structure of aspinolides B and C, the two major aspinolides produced by Trichoderma arundinaceum (Malmierca et al. 2015; Izquierdo-Bueno et al. 2018). B Comparison of the putative aspinolide biosynthetic gene clusters in T. arundinaceum IBT 40837 and A. ochraceus NRRL 35121. The predicted functions of the genes included in the clusters in T. arundinaceum and A. ochraceus are indicated in Table 1 and Table S3, respectively. Gray shading indicates gene synteny shared by the two clusters. The pink and green arrows indicate the proposed genes, asp1 and asp2, respectively.
There is little information available on the genetics of aspinolide biosynthesis in T. arundinaceum. By contrast, there is substantial information on the genetics of HA biosynthesis, and this information has provided important insights into the role of HA in the ecology of the fungus (Gutiérrez et al. 2021; Proctor et al. 2020). In T. arundinaceum, trichothecene biosynthetic genes (tri genes) occur at three loci: the large tri cluster, small tri cluster, and tri5 locus. Most of the genes at these loci have closely related homologs in other trichothecene-producing species of Trichoderma and/or other fungal genera, such as Fusarium, Myrothecium, and Trichothecium (Gutiérrez et al. 2021; Proctor et al. 2020). The chemical structure common to all trichothecene analogs (12,13-epoxytrichothec-9-ene, EPT) consists of two 6-member rings, one 5-member ring, and an epoxide group. The tri5-encoded terpene synthase catalyzes the conversion of the primary metabolite farnesyl diphosphate (FDP) to trichodiene, the parent compound of all trichothecene analogs. Enzymes encoded by three genes in the large tri cluster catalyze the conversion of trichodiene to trichodermin (4-acetyl EPT), and enzymes encoded by the three genes in the small tri cluster catalyze the conversion of trichodermin to HA (4-octatrienedioyl EPT).
The objective of the current study was to identify aspinolide biosynthetic genes in order to investigate the role of aspinolides in the biocontrol activity of T. arundinaceum. To accomplish this goal, we used a three-step approach. First, we identified an aspinolide-producing strain of Aspergillus; second, we used comparative genomics to identify closely related polyketide synthase (PKS) gene homologs in T. arundinaceum and an aspinolide-producing Aspergillus strain; and third, we used gene deletion and biochemical analyses to determine whether the PKS genes are required for aspinolide production. From this approach, we identified two PKS genes as well as a putative aspinolide biosynthetic gene cluster in both T. arundinaceum and A. ochraceus. We then used mutant strains of T. arundinaceum in which the PKS genes were deleted to examine how aspinolides impact antifungal activity of T. arundinaceum. Finally, we proposed an updated aspinolide biosynthetic pathway. Together, the results of this study provide a basis for further investigations into the complex nature of antifungal activity of T. arundinaceum.
Material and methods
Strains used and culture conditions
Trichoderma arundinaceum IBT 40837 (= CBS 119573 = Ta37) was used as the wild-type progenitor strain for the gene deletion studies and for the genomic, metabolomic, and transcriptomic analyses. Tylophoron protrudens CBS 121320, T. rodmanii CBS 121553, and T. turrialbense CBS 112445, also members of the Brevicompactum clade, were used to assess the production of aspinolides in species other that T. arundinaceum. Trichoderma strains were maintained on potato-dextrose agar medium (PDA), which was prepared from PDB broth (Becton Dickinson Co., Sparks, MD) amended with 2.5% agar (Oxoid, Ltd.). Sporulation occurred during growth on PDA at 28 °C in the dark for 7 days.
Aspinolide production was assessed in three strains of Aspergillus ochraceus: NRRL 35158, NRRL 35121, and NRRL 62072, and six strains of Aspergillus westerdijkiae: NRRL 35061, NRRL 35072, NRRL 3174, NRRL 6160, NRRL 35460, and NRRL 35050 obtained from the ARS Culture Collection (U.S. Department of Agriculture, Agriculture Research Service). Aspergillus strains were grown, and sporulation was induced under the conditions described above for Trichoderma except that the incubation temperature was 30 °C.
The filamentous fungus Rhizoctonia solani strain R43 and the yeast Kluyveromyces marxianus strain CECT 1018 were used in antifungal assays. Rhizoctonia solani was grown on PDA medium at 28 °C for 5–7 days. Kluyveromyces marxianus CECT 1018 was grown on malt extract agar (MEA) medium (2% malt extract, 0.1% peptone, 2% glucose, 2% agar) at 30 °C for 2–3 days.
Constructions of gene deletion plasmids pΔasp1 and pΔasp2
For the construction of asp1 deletion plasmid pΔasp1, DNA regions adjacent to T. arundinaceum asp1 were PCR amplified using oligonucleotides Asp1-5r_F_BamHI/Asp1_5r_SmaI and Asp1_3r_F_SmaI/ Asp1_3r_R_XhoI (Table S1). The fragments 5′ (1,117 bp) and 3′ (1,218 bp) were digested with BamHI-SmaI and SmaI-XhoI, respectively, and after gel purification were ligated into pBluescript KS + (Stratagene, La Jolla, CA) previously digested with BamHI and XhoI. The resulting plasmid, pBasp1-5r3r (5,252 bp) was linearized with SmaI, dephosphorylated with alkaline phosphatase (Thermo Scientific), and ligated to a 2,710 bp fragment containing the hyg resistance cassette, which was isolated from pAN7-1 (Punt et al. 1987) by digestion with Ecl136II-HindIII, and treated with Klenow fragment (Thermo Scientific, Waltham, MA), to finally obtain plasmid pΔasp1 (7,960 bp) (Figure S2A).
Similarly, for construction of asp2 deletion plasmid pΔasp2, DNA regions adjacent to T. arundinaceum asp2 were PCR amplified using primer pairs Asp2_5r_F_XbaI/Asp1_5r_R_SmaI and Asp1_3r_F_SmaI/Asp1_3r_R_XhoI (Table S1). The fragments 5′ (1227 bp) and 3′ (1142 bp) were digested with XbaI-SmaI and SmaI-XhoI, respectively, and after gel purification were ligated into pBluescript KS + vector (Stratagene) previously digested with XbaI and XhoI, creating pBasp2-5r3r (5,279 bp). Similar to above, the hyg resistance cassette was then cloned into pBasp2-5r3r creating pΔasp2 (7,985 bp) (Figure S2B).
Trichoderma arundinaceum transformation and selection of transformants
Ten micrograms of either plasmid pΔasp1 or pΔasp2, linearized with XhoI, were transformed into protoplasts of T. arundinaceum Ta37. Putative transformants were selected for resistance to hygromycin (250 μg/mL), as previously described (Cardoza et al. 2006; Proctor et al. 1999).
Metabolomics characterization and antifungal activity
Quantification of HA by HPLC and determination of the antifungal activity on plate assays (antibiogram)
To assess HA production, a 500-mL flask containing 100 mL of malt complex (CM) medium (0.5% malt extract, 0.5% yeast extract, 0.5% glucose) was inoculated with 2 × 106 spores/mL and incubated for 48 h at 28 °C and 250 rpm in an orbital shaker. Twenty milliliters of this pre-culture was then transferred to a new 500-mL flask containing 100 mL of potato-dextrose broth (PDB) medium (Difco, Becton Dickinson) as described previously (Cardoza et al. 2011) and incubated in an orbital shaker as described above. After 48 h, mycelia were harvested by filtration through sterile Nytal filters (Sefar Maissa S.A., Barcelona, Spain), and the resulting culture filtrates stored at – 80 °C until metabolomics analysis. HA was purified and quantified by HPLC from liquid PDB cultures of T. arundinaceum strains as previously described (Cardoza et al. 2011).
Antibiograms against Kluyveromyces marxianus CECT 1018 were carried out as previously described (Cardoza et al. 2011), and antifungal assays against Rhizoctonia solani R43 on cellophane membranes were performed as described by Gutiérrez et al. (2021).
Gas chromatography-mass spectrometry (GC-MS) analysis for aspinolide detection
To assess aspinolide production, 20 mL of YEPD (0.1% yeast extract, 0.1% peptone, 2% glucose) medium in a 50-mL Erlenmeyer flask were inoculated with spores from one plate of V8 agar medium (Difco, Becton Dickinson) and incubated at 28 °C, 200 rpm. After 7 days of growth, cultures were extracted with 8 mL ethyl acetate. Extracts were dried under a stream of nitrogen and the residue was resuspended in 1 mL of ethyl acetate. GC-MS analyses of the extracts were performed on a Hewlett Packard 6890 gas chromatograph fitted with a HP-5MS column (30 m, 0.25 mm, 0.25 μm) and a 5973-mass detector. The carrier gas was helium with a 20:1 split ratio and a 20 mL/min split flow. The column was held at 150 °C for 1 min following injection, heated to 280 °C at 30 °C/min, and held for 7.7 min. Compound identifications were based on GC-MS comparisons with purified standards of aspinolides B and C. In the GC-MS analysis, the presence of aspinolide analogs was detected using a fragment ion with m/z 69, which is derived from the 8-butenoyl substituent of aspinolides (Malmierca et al. 2015).
Isolation of aspinolides and NMR analysis
To generate sufficient quantity of aspinolides for 1H-nuclear magnetic resonance (NMR) analysis, 1-cm diameter mycelia plugs, collected from 5 days old PDA plates, were used to inoculate 40 Petri dishes (150-mm diameter) containing 100 mL MEA and incubated at 25 °C in presence of white artificial light for 12 days. The mycelium was scraped from the agar surface and discarded. The agar medium was then fragmented and extracted with ethyl acetate in 500-mL flasks in an ultrasonic bath for 15 min. The extract was filtered to remove the agar debris and dried using a rotary evaporator at low pressure.
The extracts were fractionated by silica gel column chromatography (CC), eluting with mixtures containing increasing percentages of ethyl acetate/hexane (10–100%) and methanol as the mobile phase. TLC and extensive spectroscopic analyses by 1H NMR and 13C NMR were used to detect the presence of the various metabolites in each fraction. Final purification of metabolites from selected fractions was carried out by HPLC. Metabolites were eluted with a gradient of ethyl acetate in hexane (10–100%) at a flow rate of 1 mL/min. The column was then rinsed with 100% methanol.
Purification by semipreparative and analytical HPLC was performed with a Hitachi/Merck L-7100 apparatus equipped with a differential refractometer detector (RI-7490) and either a LiChrospher® Si 60 (5 μm) LiChroCart® (250 mm × 4 mm) or a LiChrospher® Si 60 (10 μm) LiChroCart® (250 mm × 10 mm) column. Silica gel (Merk) was used for column chromatography. TLC was performed using a 0.25-mm-thick Merk Kieselgel 60 F254 plate.
1H and 13C NMR measurements were recorded on Agilent 400 MHz and Agilent 500 MHz spectrometers with SiMe4 as the internal reference. Chemical shifts were referenced to CDCl3 (δH 7.25, δC 77.0). NMR assignments were made using a combination of 1D and 2D techniques. Multiplicities are described using the following abbreviations: s, singlet; d, doublet; t, triplet; q, quarter; quint, quintuplet; sext, sextuplet; m, multiplet; br; broad. High-resolution mass spectroscopy (HRMS) was recorded with a QTOF mass spectrometer in positive ion electrospray mode with a cone voltage of 20 V.
Quantification of ergosterol and squalene
Total intracellular sterols were extracted from the mycelia harvested from the same cultures used for HA quantification. Ergosterol and squalene contents were calculated as reported previously (Cardoza et al. 2007; Ghimire et al. 2009).
Genome sequence of A. ochraceus NRRL 35121 and Δasp1.9 and Δasp2.3 mutants and bioinformatic analysis
Genome sequences were obtained with an Illumina MiSeq platform (Illumina, Inc.). To prepare DNA for genome sequencing, A. ochraceus NRRL 35121 wild-type strain, and Δasp1 and Δasp2 mutant strains were grown in YEPD medium for 7 days, at room temperature with shaking at 200 rpm. Mycelia were harvested by filtration, lyophilized, ground to a powder, and genomic DNA was extracted using the ZR Fungal/Bacterial DNA MiniPrep kit (Zymo Research). Libraries for DNA sequencing were prepared using the Nextera XT DNA library Preparation Kit as specified by the manufacturer (Illumina). Sequence reads were processed and assembled using CLC Genomics Workbench (Qiagen Inc.). The resulting assembly of A. ochraceus NRRL 35121 genomic sequence was submitted to the GenBank/National Center for Biotechnology Information (NCBI) database as accession JANYMM000000000.
RNAseq analysis
RNA was extracted from mycelia of wild-type T. arundinaceum and the tri6 mutant strain Δtri6.66 grown for 48 h in PDB medium as previously described (Cardoza et al. 2015). cDNA synthesis, library construction, sequencing, and analysis were carried out as described previously (Lindo et al. 2018). Briefly, relative transcription levels for each gene model were reported as TPM (transcripts per million) (Li et al. 2010), which was computed as RPKMi × 106/∑j RPKMj, where RPKM (reads per kilobase per million mapped reads) (Mortazavi et al. 2008) corresponds to the total exon reads/mapped reads (millions) for a particular gene (i) or for all the T. arundinaceum genes (j). A total of 8,396,204 trimmed reads (average length = avl = 122.8 bp) were obtained for the wild-type samples, and 11,900,790 (avl = 127.3 bp) trimmed reads were obtained for the Δtri6 mutant. Where noted, RNAseq data corresponding to accession SRP156794 were retrieved from the GenBank Sequence Read Archive (SRA) database (Lindo et al. 2018).
Antifungal assays
Antibiograms against Kluyveromyces marxianus CECT 1018 (Cardoza et al. 2011) and antifungal assays on membranes against Rhizoctonia solani R43 (Gutiérrez et al. 2021) were carried out as described previously. The degree of radial growth inhibition of R. solani was calculated as previously described (Cardoza et al. 2007; Royse and Ries 1978).
Phylogenetic analysis
The full-length predicted amino acid sequences of ASP1, ASP2, and 159 fungal PKSs with known functions as well as the fatty acid synthase (FAS) from Gallus gallus, which was added as an outgroup (Brown et al. 2022), were aligned using Muscle as implemented in MEGA X (Kumar et al. 2018). The evolutionary history of the PKSs was then inferred using the Neighbor-Joining and Maximum Parsimony methods in MEGA X (Nei and Kumar 2000). The percentage of trees in which the associated taxa clustered together in the bootstrap test (500 replicates) is shown next to the branches.
Homologs of asp1 and asp2 in other fungi were identified by BLASTx analysis in which the DNA sequence of the T. arundinaceum genes TARUN_4144 and TARUN_4155 were used as queries against the GenBank/NCBI nonredundant fungal database. The BLASTx analysis using the asp1 and asp2 as queries was then repeated using databases that were limited to the fungal species corresponding to the 10 best hits for each query in the initial analysis. This facilitated identification of fungi that had homologs of both asp1 and asp2. Genome sequence data in the NCBI/GenBank database were used to determine whether the asp1 and asp2 homologs were located near one another and, therefore, potentially in the same gene cluster. The predicted amino acid sequences of the asp1 and asp2 homologs identified from the BLASTx analysis were aligned with sequences from A. ochraceus and Trichoderma homologs using Muscle, as described above. The resulting alignments were then subjected to maximum likelihood analysis with ultrafast bootstrapping as implemented in the program IQ-Tree version 1.6.7 (Minh et al. 2013; Nguyen et al. 2015).
Results
Comparative analyses to identify candidate aspinolide biosynthetic PKS genes
Rationale for aspinolide PKS gene identification strategy
The 10-member lactone ring of aspinolides and the butenoate substituent esterified at carbon atom 8 (C-8) of the lactone ring have a pentaketide and diketide origin, respectively (Fuchser and Zeeck 1997; Malmierca et al. 2015). Based on biochemical considerations and comparison to lovastatin biosynthesis, which requires a nonaketide and a diketide synthesized by two different PKS enzymes, the pentaketide and diketide precursors of aspinolide biosynthesis are likely synthesized by two different PKS enzymes (Fig. 1A).
Species of Aspergillus and Trichoderma are reported to produce aspinolides. Although these genera are both in the phylum Ascomycota, they are in different classes (Eurotiomycetes and Sordariomycetes) and, therefore, distantly related. Given this distant relationship, SM biosynthetic genes in the two genera should also be, in general, distantly related. However, if aspinolide biosynthesis requires two PKS genes, aspinolide-producing species of Aspergillus and Trichoderma should have closely related homologs of the two PKS genes. Furthermore, because enzyme-encoding genes involved in synthesis of the same SM tend to be clustered in fungi, the two putative aspinolide PKS genes should be located near one another in the genome sequences of the aspinolide-producing Aspergillus and Trichoderma species.
Aspinolide-producing strains
We selected T. arundinaceum strain IBT 40837 for this study based on its availability and its demonstrated ability to produce aspinolides (Malmierca et al. 2015). Because the A. ochraceus strain with a demonstrated ability to produce aspinolides was not available, we examined aspinolide production in multiple strains of both A. ochraceus and A. westerdijkiae. The strains of A. westerdijkiae were included because A. ochraceus was recently resolved into two species, A. ochraceus (sensu stricto), and A. westerdijkiae (Frisvad et al. 2014), and it is not clear which of these two species was previously examined for aspinolide production (Fuchser and Zeeck 1997). Of the nine Aspergillus strains examined, aspinolide production was detected in one A. ochraceus strain (NRRL 35121) and two A. westerdijkiae strains (NRRL 35050 and NRRL 35072). Only one aspinolide analog (aspinolide B) was detected in the three strains. Based on these results, A. ochraceus NRRL 35121 was selected for further analysis.
Comparison of predicted PKS genes in A. ochraceus and T. arundinaceum
To identify the two putative aspinolide PKS genes, we used tBLASTn analysis in which the genome sequence of A. ochraceus NRRL 35121 was queried with the amino acid sequences of the 23 predicted PKSs from T. arundinaceum. In the tBLASTn results, the Expect-values (E-values) for most of the hits in the A. ochraceus genome sequence were between 1.25 × 10−7 and 3.30 × 10−79. However, a hit on A. ochraceus contig 382 to the T. arundinaceum PKS protein associated with locus tag TARUN_4144 had an E-value of 0.00. Contig 382 also contained a second hit, located ~ 12 Kb from the first hit, with an E-value of 1.07 × 10−33 to the T. arundinaceum PKS protein TARUN_4155. In T. arundinaceum, PKS genes TARUN_4144 and TARUN_4155 were located 33 kb apart on the same contig (Table S2). To help determine whether the two PKS genes could be part of the same SM biosynthetic gene cluster, we examined the results of antiSMASH analysis of the A. ochraceus and T. arundinaceum genome sequences. In both fungi, the two PKS genes were in one predicted SM biosynthetic gene cluster (Fig. 1B, Table 1, Table S3). The putative aspinolide clusters in the two fungi also include closely related homologs of seven other genes, and the relative organization of these nine genes was partially conserved (Fig. 1B). Hereafter, the two PKS genes are referred to as asp1 (= TARUN_4144 and AOCHR_5364) and asp2 (= TARUN_4155 and AOCHR_5357).
Deletion of asp1 and asp2 genes
asp1 gene disruption
Transformation of the wild-type T. arundinaceum strain with deletion plasmid pΔasp1 (Figure S2A) yielded 67 hygromycin-resistant transformants. Initial PCR analyses yielded 15 transformants with amplicon patterns consistent with deletion of asp1. That is, a 466-bp amplicon with the primer pair (Db741 and Db742) designed to amplify an internal 466-bp hph fragment and no amplicon with the primer pair (Asp1R and Asp1F) designed to amplify an internal 1,028-bp asp1 fragment. Subsequent PCR analyses yielded one transformant, Δasp1.9, with amplicon patterns consistent with deletion of the asp1 coding region via integration of the asp1 deletion cassette from pΔasp1 into the target region by double homologous recombination. That is, a 1,491-bp amplicon with the primer pair (Asp1_5rr and TtrpC-d) designed to detect integration of the deletion cassette into the 5’ flanking region of asp1 and a 1,410-bp amplicon with the primer pair (Asp1_3rr and PgpdA_d) designed to detect integration of the deletion cassette into the 3’ flanking region of asp1 (Figure S3A, Figure S4). Sanger sequence analysis of the latter two amplicons provided additional evidence that the recombination process occurred as designed. Genome sequence analysis confirmed deletion of asp1 in transformant Δasp1.9 (Figure S3B).
asp2 gene deletion
Transformation of wild-type T. arundinaceum with deletion plasmid pΔasp2 (Figure S2B) yielded 35 hygromycin-resistant transformants. PCR analysis yielded four transformants with amplicon patterns consistent with the deletion of the asp2 coding region via integration of the asp2 deletion cassette by double homologous recombination. That is, the transformants yielded a 1,603-bp amplicon with the primer pair (Asp2-5rr and TtrpC-d) designed to detect integration of the deletion cassette in the 5’ flanking region of asp2 and a 1,338-bp amplicon with the primer pair (PgpdA-d and Asp2-3rr) designed to detect integration of the cassette in the 3’ flanking region of asp2 (Table S1) (Figure S5A, Figure S6). Sanger sequence analysis of the latter amplicons from two of the transformants (Δasp2.3 and Δasp2.18) provided additional evidence that the recombination process occurred as designed. Transformant Δasp2.3 was selected for further analyses. Genome sequence analysis which confirmed deletion of asp2 in transformant Δasp2.3 (Figure S5B).
Targeted metabolomics of Δasp1- and Δasp2 mutants
Lack of aspinolide GC–MS signals in Δasp1 mutant
Initial GC-MS analysis, focused on m/z (mass-to-charge) 69 ion specific for detection of aspinolides, was carried out with Δasp1 mutant and the wild-type strain. Four aspinolide analogs were detected in cultures of the wild-type strain, but none of the analogs was detected in cultures of the Δasp1 mutant. Thus, asp1 deletion abolished the wild-type aspinolide production phenotype (Figure S7).
Production of known aspinolide analogs
To further examine aspinolide production, large-scale chromatographic separations of extracts from cultures of Δasp1, Δasp2, and wild-type strains were conducted. Aspinolides B and C were present at 24.4 and 14.3 mg/L, respectively, in extracts of the wild-type strain but were not detected in extracts of the Δasp1 and Δasp2 mutants (Table 2).
Production of novel aspinolide analog
In addition to data presented above, the analysis of the large-scale chromatographic fractions collected from extracts of Δasp1 mutant cultures indicated production of a major metabolite (compound 1) in one of the fractions. Total purification by HPLC (tr= 37 min, Hex:Ea 50%, Flux of 1 mL/min, analytical silica gel column) and subsequent spectroscopic analysis characterized 1 as a novel aspinolide analog called 8-hydroxy aspinolide A (Fig. 2). Furthermore, 1 was detected in culture extracts of the Δasp1 mutant but was absent in culture extracts of the Δasp2 mutant and wild-type strain (Table 2).

Chemical structure of compounds 1 (8-hydroxy aspinolide A) and 2 (3,11-diepiisotrichotriol) isolated from culture broths of Δasp1.9 mutant
To determine the structure, purified 1 was subjected to NMR spectroscopy as described in the “Materials and methods” section. The resulting 1H NMR and 13C NMR spectroscopic data show a pattern of signals characteristic of aspinolides but with significant differences (Table 3). Specifically, the 1H NMR spectrum resembled that of aspinolide D (Figure S1) but lacked signals corresponding to C-4 ester at C-8, indicating its underlying carbon skeleton similar in structure to the aspinolide lactone ring. The associated H-8 resonance of compound 1 now appeared at δ 3.88 ppm and the characteristic signals of the proton geminal to the hydroxyl group at C-5 (δ 68.3 ppm) carbon appeared at δ 4.49 (d(br), J = 6.0 Hz). The 13C- NMR spectrum displayed signals corresponding to the double bond at C-6 (δ 130.1 ppm) and C-7 (δ 135.8 ppm). We also detected heteronuclear multiple bond correlation (HMBC) signals between H-9 and C-1, C-8; H-2 and C-1, C-3, and C-4; and between H-5 and C-3, C-4, C-6, and C-7. The position of the double bond was confirmed by signals between H-6 and C-5, C-7 and between H-7 and C-5, C-8, and C-9. This relationship to aspinolide D was supported by COSY and HSQC experiments confirming the proposed structure for compound 1 as 8-hydroxy aspinolide A (Fig. 2).
The accumulation of 1 in cultures of the Δasp1 mutant combined with its absence in cultures of the Δasp2 mutant and wild-type strain indicated that the Δasp1 mutant can produce the 10-member lactone ring that forms the aspinolide backbone but not the 8-butenoyl substituent (Fig. 1A). This is consistent with the asp1-encoded PKS (ASP1) catalyzing synthesis of the diketide precursor of the 8-butenoyl substituent. Furthermore, the absence of both 1 and previously described aspinolides in culture extracts of the Δasp2 mutant is consistent with the asp2-encoded PKS (ASP2) catalyzing synthesis of the polyketide precursor of the aspinolide backbone.
Production of harzianum A
To examine whether the change in aspinolide production in the Δasp1 and Δasp2 mutants affected HA production, we quantified the amount of HA present in 48 h PDB cultures of the mutants and wild-type strain. The Δasp2 mutant produced significantly more HA (296.3 ± 17.0 μg/mL) than the wild type (220.1 ± 11.1 μg/mL), which represents an increase of 34% (Fig. 3A, B). The amount of HA produced by the Δasp1 mutant (222.3 ± 24.0 μg/mL) was similar to what was produced by the wild-type strain (Fig. 3A, B).

A HPLC chromatograms showing the HA peak detected in 48 h PDB cultures of Ta37 (wild type), Δasp1.9, Δasp2.3, and Δtri6.66*. B Quantification of HA in Ta37 and Δasp1 and Δasp2 mutants (n = 2). C Antibiogram of ethyl acetate extracts of 48 h PDB cultures of wild-type T. arundinaceum (Ta37) and the mutant strains Δasp1.9 and Δasp2.3 against Kluyveromyces marxianus CECT 1018. *Δtri6.66 is a mutant deleted on tri6** gene that produces trace amounts of HA (Lindo et al., 2018) and was used in this study for comparative purposes. **tri6 gene encodes a Cys2His2-Zn finger transcription factor that regulates expression of tri genes and is thereby essential for trichothecene biosynthesis
Production of a novel isotrichotriol epimer
Chromatographic fractions from culture extracts of the Δasp1 and Δasp2 mutants contained a second unknown metabolite (compound 2). Purification by HPLC and subsequent spectroscopic analysis indicated that 2 is a C-3, C-11 diepimer of isotrichotriol. 2 was not detected in culture extracts of the wild-type strain (Table 2, Fig. 2).
To determine the structure, purified 2 was subjected to high-resolution mass spectrometry (HRMS) and NMR. These data indicated that 2 has a molecular formula of C15H24O4. The 1H NMR signals resembled those of a sesquiterpene (Table S4) and included signals characteristic of an oxiranic methylene at δ 2.99 (H-13a) and 2.80 (H-13b) (d, J = 4.1 Hz) and three geminal protons to hydroxyl groups, indicating that 2 was a trihydroxylated sesquiterpene with an epoxide. Although the 1H and 13C NMR signals of 2 were similar to those of tricho-9-ene-2α,3α,11α-triol (isotrichotriol) (McCormick et al. 1989) and included the characteristic signals of a double bond at C-9:C-10 and the three protons germinal to hydroxyl groups at C-2, C-3, and C-11, but there were some significant differences. The signal assigned to H-11 was shielded at δ 3.58 while the signal of H-3 was deshielded at δ 4.40, with the H-2 signal appearing at δ 3.66 (Table S4). The stereochemistry was assigned based on nuclear Overhauser effect (NOE) experiments. Irradiation of H-2 enhanced signals corresponding to H-13a and H-4β, while α configuration for H-11 and H-3 was established by irradiation of H-15, which produced NOE effects at H7α, H-8α, H-11, and H-3. On the other hand, irradiation of H-3 enhanced signals corresponding to H-11, H-15, and H-4α. Consequently, the compound was assigned the structure 2. All the carbon and their associated proton signals were assigned using the COSY, HSQC, and HMBC spectra leading to the structure and stereochemistry of compound 2, which was named 3,11-diepiisotrichotriol (2). HRMS(ESI+) for compound 2: calcd. for C15H24O4Na [M + Na]+ 291.1572, found 291.1571. 2 (or 3,11-diepiisotrichotriol) is an epimer of isotrichotriol, an intermediate in biosynthesis of 3-oxygenated trichothecenes produced by Fusarium spp., but not an intermediate in biosynthesis of HA (Gutiérrez et al. 2020; Proctor et al. 2020).
Production of primary metabolites
In previous works, changes in the levels of squalene and ergosterol were observed in T. arundinaceum strains blocked in trichothecene biosynthesis (Malmierca et al. 2013). In the current study, we quantified the levels of squalene and ergosterol produced in cultures of the Δasp1 and Δasp2 mutants and the wild-type strain to determine if blocking aspinolide biosynthesis affected their levels. We found no significant changes in levels of either squalene or ergosterol in the Δasp1 and Δasp2 mutants as compared to the wild type (Table S5). In addition, the analysis of the low polarity chromatographic fractions collected from the extracts of the culture filtrate of these mutants indicated that they contained high levels of fatty acids (Table 2). No difference in growth was observed in the two mutants compared with the wild-type strain.
Effect of Δasp1 and Δasp2 gene deletion on the antifungal activity
The contribution of aspinolide production to the antifungal activity of T. arundinaceum was assessed using the yeast K. marxianus and the filamentous fungus R. solani. The antifungal activity of pure AspB and AspC was first evaluated against these two fungi. AspC inhibited both fungi, while AspB failed to inhibit either fungus (Figure S8).
In subsequent assays with K. marxianus, ethyl acetate extracts of 48 h PDB cultures of Δasp1 and Δasp2 mutants and the wild-type progenitor strain were examined. Extracts of the Δasp2 mutant inhibited growth of K. marxianus more than extracts of the Δasp1 mutant and the wild type. There were no differences in the inhibition caused by culture extracts of the Δasp1 mutant and wild type (Fig. 3C). The greater inhibition observed by extracts of the Δasp2 mutant is consistent with the higher levels of HA in the extracts compared to extracts of Δasp1 mutant or the wild type (Fig. 3A, B).
In assays with R. solani, plates were incubated for 7 and 10 days after pathogen placement in the center of the plate, after removal of the cellophane membranes where the T. arundinaceum strains were previously grown for 24 h. Mean percent radial inhibition caused by the Δasp2 mutant was significantly higher than that caused by the Δasp1 mutant and wild type at both 7 and 10 days. By contrast, inhibition caused by Δasp1 mutant and wild type was not significantly different at either day (Table S6). These results indicate that the Δasp2 mutant inhibits the growth of R. solani more than the Δasp1 mutant and the wild-type strain (Figure S9).
Transcription analysis of the genomic region with asp1 and asp2
In the current analysis, we used RNAseq to assess expression of the 32 genes (locus tags TARUN_4134 to TARUN_4165) in the genomic region in which asp1-asp2 are located in the wild-type T. arundinaceum and a tri6-deletion mutant strain Δtri6.66). The 32 genes include asp1 and asp2, 10 genes located between them, 10 genes downstream of asp1, and 10 genes downstream of asp2 (Table S7). The rationale for including the tri6 mutant in this analysis was that in a previous study (Lindo et al., 2018) it produced higher levels of aspinolides than its wild-type progenitor strain (Ta37) and, therefore, transcription of aspinolide biosynthetic genes could be higher in the mutant than in the wild type. The RNAseq analysis in the current study revealed that asp1, asp2, and the 10 genes in between them exhibited similar patterns of expression in that they were more highly expressed in the tri6 mutant than in the wild type, even though there were marked differences in the levels of expression among some genes (Fig. 4, Table S8). In contrast, the 10 genes downstream of asp1 and the 10 genes downstream of asp2 exhibited low levels of expression, and their expression levels in the tri6 mutant were not consistently higher than in the wild type. The greater expression of asp1 and asp2 in the tri6 mutant relative to the wild type was consistent with the previously reported higher levels of aspinolides produced by the mutant (Lindo et al., 2018). Together, these data provide evidence that asp1, asp2, and the 10 genes located between them (i.e., TARUN_4144 and TARUN_4155) are coregulated and, therefore, could be an aspinolide biosynthetic gene cluster.

Upper panel. Expression of genes flanking asp1 (TARUN_4144) and asp2 (TARUN_4155) in a wild-type (blue bars) and a tri6 mutant (orange bars) strain of T. arundinaceum grown for 48 h in PDB medium. nTPMs, transcripts per million (TPM) of total reads of each gene normalized against TPM of the actin gene (housekeeping) of each sample. Lower panel. Graphic representation of the genomic region showing positions of genes analyzed in the upper panel. Genes and their direction of transcription are indicated by arrows. The positions of asp1 and asp2 are highlighted using green text. Predicted function for each gene is indicated by arrow color and their associated codes (Table 1, Table S3). Predicted functions for all genes included in this genomic region are indicated in Table S7. Numeric data used to generate this histogram are included in Table S8
Identification of aspinolide gene clusters orthologs and production of aspinolides in other Trichoderma species
BLASTp analysis was performed using the T. arundinaceum ASP1 (TARUN_4144) and ASP2 (TARUN_4155) homologs as queries against an in-house database containing protein sequences predicted to be encoded by genes in the genome of 35 evolutionary diverse Trichoderma species (Gutiérrez et al. 2021; Kubicek et al. 2019).
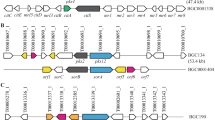
Putative asp1 homologs were identified in three species belonging to the Brevicompactum clade: T. rodmanii, T. protrudens, and T. turrialbense. Putative asp2 homologs were identified in these three species as well as two other species in the Brevicompactum clade: T. aurantioeffusum and T. brevicompactum. Analysis of the genomic region flanking the asp1 and asp2 gene homologs in two species, T. protrudens and T. turrialbense, indicated that they shared a high degree of sequence identity and synteny to the SM cluster predicted by antiSMASH that included asp1 and asp2 in T. arundinaceum (Fig. 5). In contrast, the putative asp1 and asp2 homologs in T. rodmanii were located on two different contigs. Analysis of the regions flanking asp1 and asp2 revealed putative homologs of most genes flanking asp1 and asp2 in T. arundinaceum except that homologs to TARUN_4148 and TARUN_4156 were missing and the homolog of TARUN_4146 is likely nonfunctional (Fig. 5). Analysis of genomic region flanking asp2 in T. aurantioeffusum and T. brevicompactum indicated that they share a high degree of similarity spanning one and five predicted genes, respectively, with genes flanking asp2 in T. arundinaceum. Finally, no genes belonging to this cluster were found in T. margaretense, another species belonging to the Brevicompactum clade (Fig. 5) Thus, based on sequence similarity, shared synteny between these Trichoderma species, and co-expression in T. arundinaceum, we propose that the asp1 and asp2 are part of a gene cluster that includes 12 genes.

Occurrence of intact and partial homologs of the proposed aspinolide biosynthetic gene cluster in Trichoderma species in the Brevicompactum clade. Predicted functions of proteins encoded by these genes are indicated at the right. Ψ indicates a pseudogene. Color codes used to illustrate predicted function of each gene were as described in the legend for Fig. 4
Production of aspinolides by Trichoderma species in the Brevicompactum clade
To determine whether the presence of ASP clusters in T. rodmanii, T. protrudens, and T. turrialbense correlates with aspinolide production, all three strains were grown for 7 days, at either 20 °C or 28 °C. GC-MS analysis of culture extracts indicated that both T. protrudens and T. turrialbense can produced aspinolides (B or C) at both assayed temperatures with the highest production observed at 20 °C. In addition, the sesquiterpene intermediate metabolite trichodermol was also detected in the T. turrialbense 28 °C extracts (Fig. 6). The detection of the aspinolides was fully consistent with the presence of an intact aspinolide gene cluster in both strains. On the other hand, no aspinolides were detected in T. rodmanii extracts. Furthermore, a significant amount of the trichothecene analog trichodermin was detected in T. rodmanii extracts at both temperatures (Fig. 6). The failure of T. rodmanii to produce aspinolide suggests that the genetic differences observed between the aspinolide clusters in T. rodmanii versus T. arundinaceum, T. protrudens, and T. turrialbense are critical for aspinolide production. These differences can be summarized as follows: (i) pseudogenization of a cytochrome P450-monooxygenase gene (labeled as Ψ in Fig. 5) and (ii) absence of an amidotransferase-like gene (labeled as 8 in Fig. 5). In addition, it is noteworthy that T. rodmanii is missing a gene encoding a Zn2Cys6 transcriptional regulator that is adjacent to asp2 (labeled A in Fig. 5). The close proximity of gene A to asp2 suggests a potential role in aspinolide synthesis.

GC-MS analysis of extracts and identification of aspinolides and trichothecene analogs produced by T. rodmanii, T. protrudens, and T. turrialbense, in cultures grown at 20 and 28 °C. Note the scale at the Y-axis is different on each graphic
Phylogenetic relationships and distribution of asp1 and asp2
Phylogenetic analysis of predicted ASP1, ASP2, and 159 fungal PKSs with known functions were resolved into three previously described clades of fungal PKSs: non-reducing PKSs (NR-PKSs), reducing PKSs (R-PKSs), and partial reducing PKSs (PR-PKSs) (Fig. 7). The R-PKSs were further resolved into three smaller clades: R-PKS I, R-PKS II, and R-PKS III. ASP1 was nested within the R-PKS I clade, and ASP2 was nested within the R-PKS III clade (Fig. 7).

Phylogenetic analysis resolved ASP1, ASP2, and 159 predicted fungal PKS proteins with known functions (Brown et al. 2022) into three previously described groups represented by five clades. The three groups are non-reducing PKSs (NR-PKSs), reducing PKSs (R-PKSs), and partial reducing (PR-PKSs). The R-PKS group has been further divided into three subgroups based on domain functionality and phylogenetic relationships: R-PKS I, R-PKS II, and R-PKS III. The fatty acid synthase from Gallus gallus served as an outgroup
BLASTx analysis using the T. arundinaceum asp1 and asp2 homologs as queries against the GenBank/NCBI nonredundant fungal database identified homologs of both genes in other fungi (Figure S10). We focused our analysis on fungi corresponding to the 10 best hits in the BLASTx analysis in which T. arundinaceum asp1 was the query (E-value 0.0; 50–61% amino acid sequence identity over 97–98% of the query sequence) because each of these hits were for fungi that also had asp2 homologs with 48–61% amino acid sequence identity to T. arundinaceum ASP2. In contrast, when T. arundinaceum asp2 was used as a query sequence in BLASTx, the top 10 hits were for fungi that only had relatively distantly related homologs of T. arundinaceum ASP1 (< 36% amino acid sequence identity). The fungi corresponding to the 10 best hits in the asp1 BLASTx analysis were from two orders within class Sordariomycetes: Sordariales and Xylariales. The one exception to this was Aspergillus melleus, which like A. ochraceus is in class Eurotiomycetes and order Eurotiales. In five of the fungi, the asp1 and asp2 homologs were located near one another on the same contig and, therefore, could be in the same biosynthetic gene cluster. In the other five fungi, the two genes were located on different contigs. However, in some of the latter fungi (e.g., Madurella mycetomatis), the genes were located on small contigs (< 50 kb) and/or at the ends of contigs. In these fungi, it is possible that the asp1 and asp2 homologs are located near one another on a chromosome.
In trees inferred by maximum likelihood analysis of the predicted amino acid sequences of ASP1 and ASP2 homologs, Trichoderma sequences formed a well-supported and exclusive clade (Figure S10). Aspergillus ochraceus and A. melleus sequences also formed a well-supported and exclusive clade in both trees. In the ASP2 tree, the Aspergillus sequences were nested within a well-supported clade that excluded the Trichoderma sequences (Figure S10). In the ASP1 tree, however, the relationships of the Aspergillus sequences to the Trichoderma sequences were not resolved except that sequences from each of these two genera were resolved as exclusive clades (Figure S10). Nevertheless, in both trees, the Aspergillus sequences were nested within a well-supported clade (bootstrap values = 100) that otherwise consisted of sequences from fungi in class Sordariomycetes.
Discussion
Evidence for role of asp1 and asp2 in aspinolide biosynthesis
Aspinolide production in T. arundinaceum was first discovered in studies in which the antifungal activity of the fungus was retained following loss of production of the antifungal metabolite HA (Lindo et al. 2019a, 2019b; Malmierca et al. 2015). These studies also provided evidence that HA and AspC may have redundant roles in the ecology of the fungus in that they both have antifungal activity and can induce expression of plant defense-related genes. We initiated the current study to identify aspinolide biosynthetic genes as a means to further investigate the role of aspinolide production in the ability of T. arundinaceum to inhibit growth of other fungi. We obtained multiple lines of evidence that the PKS genes asp1 and asp2 are part of an aspinolide biosynthetic gene cluster. First, evidence from previous studies indicates that aspinolide synthesis requires two PKS genes, and asp1 and asp2 are the only two T. arundinaceum PKS genes that have closely related homologs in A. ochraceus, a distantly related, aspinolide-producing fungus. Second, deletion of asp1 and asp2 in T. arundinaceum blocked wild-type aspinolide production in T. arundinaceum. In-depth biochemical analyses indicated that the asp2 mutant does not produce any aspinolides while the asp1 mutant produced a novel aspinolide analog that lacks the 8-butenoyl substituent. Third, in both T. arundinaceum and A. ochraceus, asp1 and asp2 are located within an antiSMASH-predicted gene cluster. The gene content of the cluster in the two species differs, but in addition to asp1 and asp2, some genes that are common to the two homolog clusters are predicted to encode enzymes with the types of activities predicted to be necessary for aspinolide biosynthesis (see below for proposed biosynthetic pathway). Finally, two other Trichoderma species that have homologs of the T. arundinaceum 12-gene cluster produced aspinolides, while a third species with only a partial cluster did not (Fig. 5, Fig. 6).
The finding that ASP1 and ASP2 are required for aspinolide biosynthesis adds to the growing list of fungal SMs whose synthesis requires two PKSs. For example, the synthesis of zearalenone by Fusarium species (Gaffoor and Trail 2006), botcinic acid by Botrytis cinerea (Dalmais et al. 2011), and lovastatin by Aspergillus terreus (Kennedy et al. 1999) all require two PKSs. Furthermore, a recent genus-wide analysis of PKSs indicated that approximately 8% of polyketide-derived SMs produced by Fusarium likely require two PKSs for their formation (Brown et al. 2022).
Changes in aspinolide and HA production
The increase in the levels of HA produced by the Δasp2 mutant in the current study (Fig. 3) is reminiscent of increases in aspinolide production in previous studies on T. arundinaceum strains in which HA production was blocked by deletion of tri genes. In one study, AspB and AspC were not detected in cultures of wild-type T. arundinaceum but were present at high levels in cultures of the Δtri5 mutant, which does not produce HA or HA biosynthetic intermediates (Malmierca et al. 2015). In another study, levels of AspC were over 10 times higher in cultures of Δtri18 mutants, which are blocked in HA production but produce the trichothecene biosynthetic intermediate trichodermin, than in cultures of wild-type T. arundinaceum (Lindo et al. 2019a). The 34% increase in HA levels produced by the Δasp2 mutant constitutes a relatively small increase compared to the marked increases (> 10 times) in aspinolide production observed in at least some HA-nonproducing Δtri mutants (Lindo et al. 2019a; Malmierca et al. 2015).
In contrast to the Δasp2 mutant, the Δasp1 mutant did not exhibit a detectable increase in HA production compared to wild-type T. arundinaceum (Fig. 3). Because the Δasp1 mutant produced 8-hydroxy aspinolide A, a presumed aspinolide biosynthetic intermediate, the latter result raises the possibility that the increased HA production requires a complete loss of production of all aspinolide analogs. This contrasts the situation with Δtri mutants, where deletion of tri18, which results in the formation of trichothecene analog trichodermin, caused marked increases in AspC production (Lindo et al. 2019a). The genetic and physiological mechanisms by which a loss of HA production leads to increased aspinolide production and loss of aspinolide production leads to increased HA production remain to be determined.
Antifungal activity of Δasp1 and Δasp2 mutants
Both the Δasp1 and Δasp2 mutants retained antifungal activity against K. marxianus and R. solani (Fig. 3C, Figure S9). In fact, the Δasp2 mutant had slightly higher than wild-type levels of antifungal activity against both K. marxianus and R. solani. This increased antifungal activity coincided with a 34% increase in levels of HA produced by the Δasp2 mutant. Retention of antifungal activity of T. arundinaceum mutants blocked in production of HA was attributed to increased production of aspinolides. Thus, it is possible that retention of antifungal activity of the Δasp2 mutant resulted from an increase in the production of HA by the mutant. These data add to existing information indicating that HA and aspinolides have redundant bioactive roles in a manner analogous to the redundant roles of botcinic acid and botrydial analogs in the biology of B. cinerea (Dalmais et al. 2011). However, we cannot rule out the possibility that another metabolite(s) contributes to the antifungal activity of T. arundinaceum mutants that are blocked in aspinolide or HA production. In order to address this possibility, we are currently attempting to generate double Δasp2-Δtri5 mutants, which should not produce aspinolides, HA, or any other trichothecene analogs.
Our finding that the level of antifungal activity of the Δasp1 mutant was similar to the wild-type was unexpected because the mutant does not produce aspinolide C and it does not exhibit increased levels of HA production that might compensate for the absence of aspinolide C. Without further experimentation, we can only speculate why the Δasp1 mutant exhibited wild-type antifungal activity. One possibility is that the 8-hydroxy aspinolide A produced by the Δasp1 mutant has antifungal activity that compensates for the absence of aspinolide C. This possibility could be addressed by determining whether 8-hydroxy aspinolide A is antifungal. A second possibility is that the antifungal activities of aspinolide C and HA are not additive or synergistic when produced at wild-type levels. An improved understanding of why loss of aspinolide production increases HA production, and vice versa, could help address this possibility. A third possibility is that T. arundinaceum produces another antifungal metabolite(s) that compensates for the absence of aspinolide C production in the Δasp1 mutant. As noted above, we are currently addressing this possibility by generating double asp2-tri5 mutant that should not produce aspinolides or trichothecenes.
Formation of 3,11-diepiisotrichotriol
To our knowledge, production of 3,11-diepiisotrichotriol by the Δasp1 and Δasp2 mutants in the current study is the first report of this metabolite. Production of 3,11-diepiisotrichotriol by T. arundinaceum is intriguing because, although isotrichotriol is an intermediate in the biosynthesis of the 3-oxygenated trichothecenes produced by Fusarium species, it is not an intermediate in biosynthesis of trichothecene analogs produced by T. arundinaceum (Proctor et al. 2020). Furthermore, the epimer of the Fusarium isotrichotriol has a different stereochemistry than the isotrichotriol epimer produced by the Δasp1 and Δasp2 mutants of T. arundinaceum. This report of 3,11-diepiisotrichotriol by the Δasp1 and Δasp2 mutants will facilitate the determination of whether other strains of T. arundinaceum and other Trichoderma species can produce the metabolite.
Isotrichodiol is the trichothecene biosynthetic intermediate formed by T. arundinaceum that is most similar in structure to isotrichotriol. These two metabolites differ in structure only by the presence (isotrichotriol) and absence (isotrichodiol) of a hydroxyl at C-3. Previous studies indicate that this structural difference at C-3 results from differences in activities of TRI4 homologs in Trichoderma and Fusarium species. Trichoderma TRI4 catalyzes oxygenation of the terpene trichodiene at three positions to form isotrichodiol (Cardoza et al. 2011), while the Fusarium TRI4 catalyzes oxygenation of trichodiene at four positions to form isotrichotriol (McCormick et al. 2006; Tokai et al. 2007). It is not clear whether the formation of the 3-hydroxyl of 3,11-diepiisotrichotriol is catalyzed by a heretofore unrecognized activity of the T. arundinaceum TRI4 homolog or some other enzyme. The low levels of 3,11-diepiisotrichotriol produced by the Δasp1 and Δasp2 mutants of T. arundinaceum suggests that whichever enzyme is responsible it has low levels of trichodiene 3-hydroxylase activity.
Distribution and phylogenetic relationships of aspinolide biosynthetic genes
The 35 Trichoderma genome sequences included in the genome survey in the current study represented a wide breadth of the phylogenetic diversity that exists within the genus Trichoderma. Thus, the detection of putative aspinolide biosynthetic gene cluster only in members of the Brevicompactum clade suggests that the cluster has a limited distribution within the genus. The occurrence of cluster homologs with all 12 putative aspinolide biosynthetic genes in some species of the Brevicompactum clade and homologs with only some of the genes in other species of the clade further suggests that an ancestral species of the Brevicompactum clade had an intact aspinolide gene cluster and that as the ancestor diverged into multiple species, an intact cluster was retained in T. arundinaceum, T. protrudens, and T. turrialbense but partially or completely degenerated in other species.
The presence of closely related homologs of genes in distantly related fungal species combined with absence of the genes in taxa closely related to either species can be a sign of horizontal gene transfer (Campbell et al. 2012; Slot and Rokas 2011). Thus, horizontal gene transfer provides a possible explanation for the presence of the aspinolide cluster in A. ochraceus and the Brevicompactum clade of Trichoderma. However, phylogenetic analysis of ASP1 and ASP2 homologs from other fungi suggest that the putative horizontal transfer might not have involved both Aspergillus and Trichoderma species (Figure S10). In the ASP1 and ASP2 trees, the Aspergillus sequences were nested within a well-supported clade that otherwise consisted only of sequences from the Sordariomycetes. This suggests that the direction of transfer was from a species of the Sordariomycetes to Aspergillus, presumably a common ancestor of A. melleus and A. ochraceus. The poor resolution of the relationship of the Aspergillus clade and other clades in the ASP1 tree precluded hypotheses about which fungus served as a donor in the putative transfer to Aspergillus. In the ASP2 tree, by contrast, there was high bootstrap support to indicate that the Aspergillus sequences are most closely related to a Monosporascus sp. (order Xylariales) sequence and a clade consisting of Chaetomium sp. and Madurella mycetomatis (order Sordariales) sequences. This suggests two potential donors: Monosporascus sp. or a common ancestor of Chaetomium sp. and M. mycetomatis. Identification of aspinolide biosynthetic gene cluster homologs in additional fungi and phylogenetic analyses that more rigorously assess potential horizontal transfer events (Slot and Rokas 2011; Villani et al. 2019) should provide further insight into the putative horizontal transfer of the aspinolide cluster.
Aspinolide biosynthetic pathway
We have proposed an updated aspinolide biosynthetic pathway (Fig. 8) based on three lines of evidence: (a) the pathway previously proposed by Fuchser and Zeeck (1997); (b) the results of biochemical analyses of the Δasp1 and Δasp2 mutants (current study); and (c) the predicted functions, based on sequence homology, of genes in the putative aspinolide biosynthetic gene clusters in T. arundinaceum and A. ochraceus (current study). The proposed pathway begins with condensation of one acetate and four malonate units followed by cyclization to form the 10-member lactone ring that is the common backbone of all aspinolide analogs. The inability of the Δasp2 mutant to produce aspinolides indicates that the ASP2 PKS catalyzes these reactions. The 10-member lactone ring then undergoes a series of reactions that yield multiple aspinolide analogs. The 5-hydroxy intermediate, aspinolide A, has not been detected in Trichoderma cultures but it has been detected in A. ochraceus cultures (Fuchser and Zeeck 1997). The predicted functions of the proteins encoded by some genes in the putative T. arundinaceum aspinolide gene cluster are consistent with the proposed pathway reactions. For example, the pathway includes three oxygenation reactions (4,5-epoxidation, 8-hydroxylation, and 4-hydroxylation) that could be catalyzed by the three cytochrome P450 monooxygenases (Guengerich 2018) encoded by genes that occur in the putative gene clusters in both T. arundinaceum and A. ochraceus. Similarly, esterification of the butenoyl intermediate to the 8-hydroxyl group could be catalyzed by the acyltransferase encoded by a gene that occurs in both clusters. The ability of the Δasp1 mutant to produce the aspinolide analog that lacks the butenoyl substituent but not analogs that have the substituent indicates that the ASP1 PKS catalyzes formation of the diketide precursor of the butenoyl substituent. Finally, the two dehydrogenation reactions in the proposed pathway could be catalyzed by dehydrogenases/oxidoreductases encoded by two genes that occur in both the T. arundinaceum and A. ochraceus gene clusters. However, it is not clear whether one of these reactions, the reduction of the 4,5-epoxide is catalyzed by an enzyme or occurs spontaneously (Guengerich 2018). Functional analyses of the genes in the putative aspinolide biosynthetic gene cluster combined with biochemical analyses of the resulting mutants should provide information on whether the genes function in aspinolide biosynthesis.

Proposed aspinolide biosynthetic pathway. The scheme proposed by Fuchser and Zeeck (1997) was updated using DNA sequence, gene function, and biochemical data reported in the current study. The proposed catalytic activity for the structural changes depicted are indicated at the right of each arrow. Names of aspinolide analogs are indicated in blue type
In conclusion, results from a combination of comparative genomic, gene deletion, and transcriptomic analyses in the current study provide evidence that asp1 and asp2 are required for aspinolide production and that they are located in a putative aspinolide biosynthetic gene cluster. However, additional functional analyses are required to confirm that other genes in the cluster have roles in aspinolide biosynthesis. The ability of both AspC and HA to inhibit fungal growth combined with the retention of antifungal activity of T. arundinaceum strains that cannot produce one or the other of these metabolites highlight the complex nature of the antifungal activity of T. arundinaceum. This complexity is underscored by the reciprocal increase in aspinolide production in HA-nonproducing mutants and HA production in the aspinolide-nonproducing asp2 mutant. It remains to be determined whether T. arundinaceum strains that are completely blocked in trichothecene and aspinolide production retain any antifungal activity and, if they do, what metabolite(s) is responsible for the activity. Another question raised by the results of the current study is the mechanism(s) that regulates increased HA and aspinolide production in aspinolide-nonproducing and HA-nonproducing T. arundinaceum mutants, respectively.
Multiple Trichoderma species are used in the biological control of crop diseases caused by other fungi. In some cases, the control has been attributed, at least in part, to the production of antifungal secondary metabolites (Hermosa et al. 2014; Khan et al. 2020). The results of the current study as well as investigations into the genetics and biochemistry of HA production have provided valuable insights into the antifungal activity of T. arundinaceum and its potential as a biological control agent.
Data availability
The manuscript has no associated data.
References
Brown DW, Kim HS, McGovern AE, Probyn CE, Proctor RH (2022) Genus-wide analysis of Fusarium polyketide synthases reveals broad chemical potential. Fungal Genet Biol 160:103696. https://doi.org/10.1016/j.fgb.2022.103696
Campbell MA, Rokas A, Slot JC (2012) Horizontal transfer and death of a fungal secondary metabolic gene cluster. Genome Biol Evol 4:289–293. https://doi.org/10.1093/gbe/evs011
Cardoza RE, Hermosa MR, Vizcaíno JA, González F, Llobell A, Monte E, Gutiérrez S (2007) Partial silencing of a hydroxy-methylglutaryl-CoA reductase-encoding gene in Trichoderma harzianum CECT 2413 results in a lower level of resistance to lovastatin and lower antifungal activity. Fungal Genet Biol 44:269–283. https://doi.org/10.1016/j.fgb.2006.11.013
Cardoza RE, Malmierca MG, Hermosa MR, Alexander NJ, McCormick SP, Proctor RH, Tijerino AM, Rumbero A, Monte E, Gutiérrez S (2011) Identification of loci and functional characterization of trichothecene biosynthesis genes in filamentous fungi of the genus Trichoderma. Appl Environ Microbiol 77:4867–4877. https://doi.org/10.1128/AEM.00595-11
Cardoza RE, McCormick SP, Malmierca MG, Olivera ER, Alexander NJ, Monte E, Gutiérrez S (2015) Effects of trichothecene production on the plant defense response and fungal physiology: overexpression of the Trichoderma arundinaceum tri4 gene in T. harzianum. Appl Environ Microbiol 81:6355–6366. https://doi.org/10.1128/AEM.01626-15
Cardoza RE, Vizcaino JA, Hermosa MR, Monte E, Gutiérrez S (2006) A comparison of the phenotypic and genetic stability of recombinant Trichoderma spp. generated by protoplast- and Agrobacterium-mediated transformation. J Microbiol 44:383–395
Dalmais B, Schumacher J, Moraga J, P LEP, Tudzynski B, Collado IG, Viaud M, (2011) The Botrytis cinerea phytotoxin botcinic acid requires two polyketide synthases for production and has a redundant role in virulence with botrydial. Mol Plant Pathol 12:564–579. https://doi.org/10.1111/j.1364-3703.2010.00692.x
Degenkolb T, Dieckmann R, Nielsen KF, Gräfenhan T, Theis C, Zafari D, Chaverri P, Ismaiel A, Brückner H, von Döhren H, Thrane U, Petrini O, Samuels GJ (2008) The Trichoderma brevicompactum clade: a separate lineage with new species, new peptaibiotics, and mycotoxins. Mycol Prog 7:177–219. https://doi.org/10.1007/s11557-008-0563-3
Frisvad JC, Frank JM, Houbraken J, Kuijpers AFA, Samson RA (2014) New ochratoxin A producing species of Aspergillus section Circumdati. Stud Mycol:23–43
Fuchser J, Zeeck A (1997) Aspinolides and aspinonene/aspyrone co-metabolites, new pentaketides produced by Aspergillus ochraceus. Liebigs Ann 1997:87–95. https://doi.org/10.1002/jlac.199719970114
Gaffoor I, Trail F (2006) Characterization of two distinct polyketide synthase genes involved in zearalenone biosynthesis in Gibberella zeae. Appl Environ Microbiol 72:1793–1799. https://doi.org/10.1128/AEM.72.3.1793-1799.2006
Ghimire GP, Lee HC, Sohng JK (2009) Improved squalene production via modulation of the methylerythritol 4-phosphate pathway and heterologous expression of genes from Streptomyces peucetius ATCC 27952 in Escherichia coli. Appl Environ Microbiol 75:7291–7293. https://doi.org/10.1128/aem.01402-09
Guengerich FP (2018) Mechanisms of cytochrome P450-catalyzed oxidations. ACS Catal 8:10964–10976. https://doi.org/10.1021/acscatal.8b03401
Gutiérrez S, McCormick SP, Cardoza RE, Kim H-S, Yugueros LL, Vaughan MM, Carro-Huerga G, Busman M, Sáenz de Miera LE, Jaklitsch WM, Zhuang W-Y, Wang C, Casquero PA, Proctor RH (2021) Distribution, function, and evolution of a gene essential for trichothecene toxin biosynthesis in Trichoderma. Front Microbiol 12:3638. https://doi.org/10.3389/fmicb.2021.791641
Gutiérrez S, McCormick SP, Cardoza RE, Lindo L, Alexander NJ, Proctor RH (2020) Trichoderma trichothecenes: beyond their toxic effect. In: Gupta VK, Zeilinger S, Singh HB, Druzhinina I (eds) New and Future Developments in Microbial Biotechnology and Bioengineering. Elsevier, pp 281–301
Hermosa R, Cardoza RE, Rubio MB, Gutiérrez S, Monte E (2014) Secondary metabolism and antimicrobial metabolites of Trichoderma. In: Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy MG (eds) Biotechnology and biology of Trichoderma. Elsevier, Amsterdam, pp 125–137
Izquierdo-Bueno I, Moraga J, Cardoza RE, Lindo L, Hanson JR, Gutiérrez S, Collado IG (2018) Relevance of the deletion of the Tatri4 gene in the secondary metabolome of Trichoderma arundinaceum. Org Biomol Chem 16:2955–2965. https://doi.org/10.1039/c8ob00338f
Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR (1999) Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284:1368–1372. https://doi.org/10.1126/science.284.5418.1368
Khan RAA, Najeeb S, Hussain S, Xie B, Li Y (2020) Bioactive secondary metabolites from Trichoderma spp. against phytopathogenic fungi. Microorganisms 8 https://doi.org/10.3390/microorganisms8060817
Kubicek CP, Steindorff AS, Chenthamara K, Manganiello G, Henrissat B, Zhang J, Cai F, Kopchinskiy AG, Kubicek EM, Kuo A, Baroncelli R, Sarrocco S, Noronha EF, Vannacci G, Shen Q, Grigoriev IV, Druzhinina IS (2019) Evolution and comparative genomics of the most common Trichoderma species. BMC Genomics 20:485. https://doi.org/10.1186/s12864-019-5680-7
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN (2010) RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26:493–500. https://doi.org/10.1093/bioinformatics/btp692
Lindo L, McCormick SP, Cardoza RE, Brown DW, Kim HS, Alexander NJ, Proctor RH, Gutierrez S (2018) Effect of deletion of a trichothecene toxin regulatory gene on the secondary metabolism transcriptome of the saprotrophic fungus Trichoderma arundinaceum. Fungal Genet Biol 119:29–46. https://doi.org/10.1016/j.fgb.2018.08.002
Lindo L, McCormick SP, Cardoza RE, Busman M, Alexander NJ, Proctor RH, Gutiérrez S (2019) Requirement of two acyltransferases for 4-O-acylation during biosynthesis of harzianum A, an antifungal trichothecene produced by Trichoderma arundinaceum. J Agric Food Chem 67:723–734. https://doi.org/10.1021/acs.jafc.8b05564
Lindo L, McCormick SP, Cardoza RE, Kim HS, Brown DW, Alexander NJ, Proctor RH, Gutiérrez S (2019) Role of Trichoderma arundinaceum tri10 in regulation of terpene biosynthetic genes and in control of metabolic flux. Fungal Genet Biol 122:31–46. https://doi.org/10.1016/j.fgb.2018.11.001
Malmierca MG, Barua J, McCormick SP, Izquierdo-Bueno I, Cardoza RE, Alexander NJ, Hermosa R, Collado IG, Monte E, Gutiérrez S (2015) Novel aspinolide production by Trichoderma arundinaceum with a potential role in Botrytis cinerea antagonistic activity and plant defence priming. Environ Microbiol 17:1103–1118. https://doi.org/10.1111/1462-2920.12514
Malmierca MG, Cardoza RE, Alexander NJ, McCormick SP, Collado IG, Hermosa R, Monte E, Gutiérrez S (2013) Relevance of trichothecenes in fungal physiology: disruption of tri5 in Trichoderma arundinaceum. Fungal Genet Biol 53:22–33. https://doi.org/10.1016/j.fgb.2013.02.001
Malmierca MG, Cardoza RE, Alexander NJ, McCormick SP, Hermosa R, Monte E, Gutiérrez S (2012) Involvement of Trichoderma trichothecenes in the biocontrol activity and induction of plant defense-related genes. Appl Environ Microbiol 78:4856–4868. https://doi.org/10.1128/aem.00385-12
McCormick SP, Taylor SL, Plattner RD, Beremand MN (1989) New modified trichothecenes accumulated in solid culture mutant strains of Fusarium sporotrichioides. Appl Environ Microbiol 1989:2195–2199. https://doi.org/10.1128/aem.55.9.2195-2199.1989
McCormick SP, Alexander NA, Proctor RH (2006) Fusarium Tri4 encodes a multifunctional oxygenase required for trichothecene biosynthesis. Can J Microbiol 52:636–642
Minh BQ, Nguyen MA, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30:1188–1195. https://doi.org/10.1093/molbev/mst024
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. https://doi.org/10.1038/nmeth.1226
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Nielsen KF, Gräfenhan T, Zafari D, Thrane U (2005) Trichothecene production by Trichoderma brevicompactum. J Agric Food Chem 53:8190–8196. https://doi.org/10.1021/jf051279b
Proctor RH, Desjardins AE, Plattner RD, Hohn TM (1999) A polyketide synthase gene required for biosynthesis of fumonisin mycotoxins in Gibberella fujikuroi mating population A. Fungal Genet Biol 27:100–112. https://doi.org/10.1006/fgbi.1999.1141
Proctor RH, McCormick SP, Gutiérrez S (2020) Genetic bases for variation in structure and biological activity of trichothecene toxins produced by diverse fungi. Appl Microbiol Biotechnol 104:5185–5199. https://doi.org/10.1007/s00253-020-10612-0
Punt PJ, Oliver RP, Dingemanse MA, Pouwels PH, van den Hondel CAMJJ (1987) Transformation of Aspergillus based on the hygromycin B resistance marker from Escherichia coli. Gene 56:117–124. https://doi.org/10.1016/0378-1119(87)90164-8
Royse DJ, Ries SM (1978) Influence of fungi isolated from peach twigs on the pathogenicity of Cytospora cincta. Phytopathology 68:603–607. https://doi.org/10.1094/Phyto-68-603
Sivasithamparam K, Ghisalberti EL (1998) Secondary metabolism in Trichoderma and Gliocladium. In: Harman GE, Hubicek CP (eds) Trichoderma and Glicocladium, vol 1. Taylor & Francis. London, UK, pp 139–191
Slot JC, Rokas A (2011) Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Curr Biol 21:134–139. https://doi.org/10.1016/j.cub.2010.12.020
Tokai T, Koshino H, Takahashi-Ando N, Sato M, Fujimura M, Kimura M (2007) Fusarium Tri4 encodes a key multifunctional cytochrome P450 monooxygenase for four consecutive oxygenation steps in trichothecene biosynthesis. Biochem Biophys Res Commun 353:412–417. https://doi.org/10.1016/j.bbrc.2006.12.033
Villani A, Proctor RH, Kim HS, Brown DW, Logrieco AF, Amatulli MT, Moretti A, Susca A (2019) Variation in secondary metabolite production potential in the Fusarium incarnatum-equiseti species complex revealed by comparative analysis of 13 genomes. BMC Genomics 20:314. https://doi.org/10.1186/s12864-019-5567-7
Acknowledgements
We acknowledge José Álvarez, Paula Vales, and Inés Popat from the University of León, and Crystal Probyn, Amy McGovern, and Christine Poppe from the United States Department of Agriculture for their excellent technical assistance.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work is a part of the Spanish I + D + i Grants RTI2018-099600-B-I00 and PID2021-123874OB-I00 financed by the MCIN/ AEI/10.13039/501100011033. This work was supported in part by the U.S. Department of Agriculture, Agricultural Research Service.
Author information
Authors and Affiliations
Contributions
R. H. P., S. P. M., and S. G. M. conceived and designed research. R. E. C. performed all genetic experiments and quantified harzianum A production. S. P. M. performed the GC–MS analyses. I. I. B. and I. G. C. carried out the NMR analyses to identify and quantify of metabolites produced by the different strains. D. W. B. conducted the phylogenetic analysis and contributed, together with R. H. P. and S. G. M., to the description of the updated aspinolide biosynthetic pathway. N. M. R. and L. L. contributed to the comparative genomic analysis. R. H. P. and S. G. M. wrote the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethical approval
This article does not contain any studies with human or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Disclaimer
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cardoza, R.E., McCormick, S.P., Izquierdo-Bueno, I. et al. Identification of polyketide synthase genes required for aspinolide biosynthesis in Trichoderma arundinaceum. Appl Microbiol Biotechnol 106, 7153–7171 (2022). https://doi.org/10.1007/s00253-022-12182-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-022-12182-9