ES2739672T3 - Procedimientos de reprogramación de células y usos de los mismos - Google Patents
Procedimientos de reprogramación de células y usos de los mismos Download PDFInfo
- Publication number
- ES2739672T3 ES2739672T3 ES10825907T ES10825907T ES2739672T3 ES 2739672 T3 ES2739672 T3 ES 2739672T3 ES 10825907 T ES10825907 T ES 10825907T ES 10825907 T ES10825907 T ES 10825907T ES 2739672 T3 ES2739672 T3 ES 2739672T3
- Authority
- ES
- Spain
- Prior art keywords
- cells
- cell
- nslc
- expression
- type
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/30—Nerves; Brain; Eyes; Corneal cells; Cerebrospinal fluid; Neuronal stem cells; Neuronal precursor cells; Glial cells; Oligodendrocytes; Schwann cells; Astroglia; Astrocytes; Choroid plexus; Spinal cord tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0619—Neurons
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0623—Stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0647—Haematopoietic stem cells; Uncommitted or multipotent progenitors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0656—Adult fibroblasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0657—Cardiomyocytes; Heart cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0667—Adipose-derived stem cells [ADSC]; Adipose stromal stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0668—Mesenchymal stem cells from other natural sources
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/48—Reproductive organs
- A61K35/54—Ovaries; Ova; Ovules; Embryos; Foetal cells; Germ cells
- A61K35/545—Embryonic stem cells; Pluripotent stem cells; Induced pluripotent stem cells; Uncharacterised stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/24—Iron; Fe chelators; Transferrin
- C12N2500/25—Insulin-transferrin; Insulin-transferrin-selenium
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/06—Anti-neoplasic drugs, anti-retroviral drugs, e.g. azacytidine, cyclophosphamide
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/065—Modulators of histone acetylation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/105—Insulin-like growth factors [IGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/11—Epidermal growth factor [EGF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/13—Nerve growth factor [NGF]; Brain-derived neurotrophic factor [BDNF]; Cilliary neurotrophic factor [CNTF]; Glial-derived neurotrophic factor [GDNF]; Neurotrophins [NT]; Neuregulins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/155—Bone morphogenic proteins [BMP]; Osteogenins; Osteogenic factor; Bone inducing factor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/16—Activin; Inhibin; Mullerian inhibiting substance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/30—Hormones
- C12N2501/38—Hormones with nuclear receptors
- C12N2501/395—Thyroid hormones
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/602—Sox-2
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/60—Transcription factors
- C12N2501/604—Klf-4
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases (EC 2.)
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/998—Proteins not provided for elsewhere
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/09—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from epidermal cells, from skin cells, from oral mucosa cells
- C12N2506/094—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from epidermal cells, from skin cells, from oral mucosa cells from keratinocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/11—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from blood or immune system cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1307—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from adult fibroblasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/13—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells
- C12N2506/1346—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells
- C12N2506/1384—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from connective tissue cells, from mesenchymal cells from mesenchymal stem cells from adipose-derived stem cells [ADSC], from adipose stromal stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2513/00—3D culture
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0676—Pancreatic cells
Abstract
Un procedimiento de obtención de una célula madre de tipo neural (NSLC), que comprende: 1) poner en contacto el ADN y/o la cromatina de una célula proporcionada de un primer tipo que no es una NSLC con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador de ADN, y/o un inhibidor de la metilación del ADN; 2) aumentar los niveles intracelulares en la célula de un primer tipo de un polipéptido Musashi1 (Msi1) y/o un polipéptido Neurogenina 2 (Ngn2); y 3) colocar la célula de la etapa (2) en condiciones de cultivo que apoyan la transformación de la célula de un primer tipo en una NSLC durante un período de tiempo suficiente para permitir una expresión estable de una pluralidad de genes cuya expresión es característica de las propiedades fenotípicas y/o funcionales de una NSLC; por lo que al final de dicho período de tiempo se obtiene una NSLC y en el que dicha NSLC obtenida se caracteriza además por una represión estable de una pluralidad de genes expresados en la célula de un primer tipo.
Description
d e s c r ip c ió n
Procedimientos de reprogramación de células y usos de Ios mismos
Campo de la invención
La presente invención se refiere al campo de la reprogramación de células eucariotas, y particularmente a la desdiferenciación celular. La invención también se refiere a Ios procedimientos para generar células neurales de tipo madre (NSLC) estables a partir de células somáticas humanas (y otras células) y al uso de las células así generadas en terapia humana.
Antecedentes de la invención
Reprogramadón celular
Hay un deseo en Ios campos médico, científico y de diagnóstico para reprogramar una célula fácilmente obtenible en una célula que generalmente es más difícil de obtener, o reprogramar una célula para que tenga funcionalidades nuevas o diferentes, sin fusionar ni intercambiar material con un ovocito u otra célula madre.
Según un primer mecanismo, una célula madre puede dividirse o diferenciarse naturalmente en otra célula madre, progenitor, precursor o célula somática. Según un segundo mecanismo, la célula somática a veces puede cambiar transitoriamente su fenotipo o expresar ciertos marcadores cuando se coloca en ciertas condiciones, y, a continuación, revertir cuando se coloca nuevamente en las condiciones originales. Según un segundo mecanismo, el fenotipo de muchas células se puede cambiar mediante la expresión forzada de ciertos genes (por ejemplo, la transfección estable del gen c-myc en fibroblastos Ios convierte en células inmortales con características de neuroprogenitor), sin embargo, una vez que se elimina esta expresión génica forzada, las células vuelven lentamente a su estado original. Por lo tanto, ninguno de Ios tres mecanismos anteriores debe considerarse una verdadera reprogramación: la primera se considera diferenciación natural que es parte de un programa celular que ya está en marcha (pasando de un estado más indiferenciado a un estado más diferenciado), el segundo es un cambio fenotípico transitorio y el tercero es un tipo de célula constantemente forzado. Una verdadera célula madre: (i) se autorrenueva casi "indefinidamente" (durante mucho más tiempo que una célula somática), (ii) no es una célula cancerosa, (iii) no se mantiene artificialmente por la expresión génica forzada o por medios similares (también debe poder mantenerse en medios de células madre estándar), (ív) puede diferenciarse a progenitor, precursor, somática u otro tipo de célula más diferenciada (del mismo linaje), y (v) tiene todas las características de una célula madre y no solo ciertos marcadores o expresión de genes o aspecto morfológico.
A pesar de las numerosas publicaciones científicas y de patentes que afirman la exitosa reprogramación o desdiferenciación, generalmente en una célula madre, casi ninguna de estas publicaciones desvela una verdadera reprogramación porque están comprendidas en uno de Ios mecanismos mencionados anteriormente. Por ejemplo, Bhasin (documento W02010/088735), Cifarelli y coI. (documento US2010/0003223), Kremer y coI. (documento US2004/o009595) y Winnier y coI. (documento US2010/0047908) todos se refieren a la reprogramación, desdiferenciación y/o células madre (o progenitoras) obtenidas como cambios celulares fenotípicos basados únicamente en un cambio en Ios marcadores de la superficie celular después del cultivo en diferentes medios con suplementos, sin evidencia de una verdadera reprogramación o una célula madre real (autorrenovación no cancerosa con marcadores de células madre y sin marcadores de diferenciación). Lo mismo es cierto para Benneti (documento W02009/079007) que usó una mayor expresión de Oct4 y Sox2. Otros, tales como Akarnatsu y coI. (documento W02010/052904) y You y coI. (documento W02007/097494, documento US2009/0246870) se refieren a la fabricación de células madre, pero esto se produjo a través de la inducción génica artificial constante producida por retrovirus (similar a cMyc) sin evidencia de células madre verdaderas que no sean inmortales/tumorigénicas y estables en lugar de transitorias. Otros, tales como Chen y coI. (documento US2005/0176707) y You y coI. (documento US2009/0227023), han hecho "células multipotenciales", pero no células madre. Además, estas células multipotenciales citadas no eran estables (en el caso de You y coI., las células ni siquiera podían proliferar) y ambas utilizaban suplementos y condiciones de medios constantes para forzar el cambio fenotípico. Sin embargo otros, tales como Oliveri y coI. (documento W02009/018832) y Zahner y coI. (documento US2002/0136709) han reivindicado la fabricación de células pluripotenciales, totipotenciales, multipotenciales y/o unipotenciales automáticamente a través de la desmetilación del ADN en todo el genoma y la acetilación de histonas, pero sin evidencia de un verdadera linea celular estable no cancerosa.
La verdadera reprogramación parece haberse logrado con células madre pluripotenciales inducidas (células iPS) creadas independientemente por el grupo de Yamanaka (Takahashi y coI., 2007) y el grupo de Thomson (Yu y coI., 2007), y potencialmente por otros antes que ellos, y aunque mas tarde se descubrió que muchas de estas células eran cancerosas, algunas de ellas no lo eran. Estas células pueden inducirse mediante una reprogramación verdadera, ya que más tarde se demostró que también pueden inducirse mediante transfección transitoria no integradora de genes (Soldner y coI., 2009; Woltjen y coI., 20o9; Yu y coI., 2009), así como por ARN (Warren y coI., 2010) o proteína (Kim y coI., 2009; Zhou y coI., 2009) de forma individual o por moléculas pequeñas (Lyssiotis y coI., 2009), y por procedimientos similares. Sin embargo, estas células son esencialmente idénticas a las células madre embrionarias y tienen Ios mismos problemas de crecimiento descontrolado, formación de teratoma y potencial
formación de tumores.
Una opción más deseable es tener células madre multipotenciales o células similares a pluripotenciales cuyo linaje y potencial de diferenciación sea más restringido para que no formen fácilmente teratomas y un crecimiento descontrolado. Por lo tanto, hay una necesidad de procedimientos para crear células madre multipotenciales, células de tipo madre multipotenciales y células de tipo madre y procedimiento de reprogramación o transformación de células fácilmente obtenibles en células madre multipotenciales altamente deseables, células multipotenciales de tipo madre y células de tipo madre.
Células de tipo madre neurales (NSLC)
Reparar el sistema nervioso central (SNC) es una de las fronteras que la ciencia médica aún tiene que conquistar. Las afecciones tales como la enfermedad de Alzheimer, la enfermedad de Parkinson y los accidentes cerebrovasculares pueden tener consecuencias devastadoras para los afectados. Una esperanza central para estas afecciones es desarrollar poblaciones celulares que puedan reconstituir la red neuronal y volver a alinear las funciones del sistema nervioso. Por este motivo, hay un gran interés en evolución en las células madre neurales y las células progenitoras. Hasta la actualidad, en general, se pensó que las células progenitoras neurales multipotenciales se comprometen temprano en la ruta de diferenciación a las células restringidas neurales o a las células restringidas de la glía.
Las células madre neurales son prometedores para la regeneración de tejidos a partir de enfermedades o lesiones; sin embargo, tales terapias requerirán un control preciso sobre la función celular para crear los tipos de células necesarios. Todavía no se comprende completamente los mecanismos que regulan la proliferación y diferenciación celular y, por tanto, es difícil explorar completamente la plasticidad de la población de células madre neurales derivada de cualquier región del cerebro o del feto en desarrollo.
El SNC, que tradicionalmente se cree que tiene capacidades regenerativas limitadas, retiene un número limitado de células madre neurales en la edad adulta, particularmente en e giro dentado del hipocampo y la zona subventricular que repone las neuronas del bulbo olfatorio (Singec I y col., 2007; Zielton R, 2008). La disponibilidad de las células precursoras es un requisito previo clave para una reparación basada en trasplantes de defectos en el sistema nervioso maduro. Por tanto, las células donantes para trasplantes neurales se derivan en gran parte del cerebro fetal. Esto crea enormes problemas éticos, además del rechazo inmunitario, y es cuestionable si tal enfoque puede usarse para el tratamiento de un gran número de pacientes, ya que las células madre neurales pueden perder algo de su potencia con cada división celular.
Las células madre neurales ofrecen un potencial terapéutico prometedor para las terapias de reemplazo celular en enfermedades neurodegenerativas (Mimeault y col., 2007). Hasta la fecha, se han realizado numerosos trasplantes terapéuticos que explotan diversos tipos de tejido fetal humano como fuente de material donante. Sin embargo, las consideraciones éticas y prácticas y su inaccesibilidad limitan la disponibilidad como fuente celular para las terapias de trasplante (Ninomiy M y col., 2006).
Para superar barreras y limitaciones a la derivación de células específicas del paciente, un enfoque ha sido utilizar células de la piel e inducir la transdiferenciación a células madre neurales y/o a neuronas (Levesque y col., 2000). La transdiferenciación ha estado recibiendo una atención creciente durante los últimos años y la transdiferenciación de células de mamíferos se ha logrado en el cocultivo o mediante la manipulación de las condiciones del cultivo celular. La alteración del destino celular se puede inducir artificialmente in vitro mediante el tratamiento de cultivos celulares con inhibidores de los microfilamentos (Shea y col., 1990), hormonas (Yeomans y col., 1976) e ionóforos de calcio (Shea, 1990; Sato y col., 1991). Se puede inducir que las células epiteliales de los mamíferos adquieran una forma y función similares a los músculos (Paterson y Rudland, 1985), las células del conducto pancreático exocrino pueden adquirir un fenotipo endocrino secretor de insulina (Bouwens, 1998a, b) y las células madre de la médula ósea pueden diferenciarse en células hepáticas (Theise y col., 2000) y en células neuronales (Woodbury y col., 2000). Otros como Page y col. (documento US 2OO3/0o59939) han transdiferenciado células somáticas a células neuronales mediante el cultivo de células somáticas en presencia de inhibidores del citoesqueleto, la acetilación y la metilación, pero después de la retirada del agente de cebado, la morfología de las neuronas y las sinapsis establecidas no duran mucho más que unas pocas semanas in vitro y nunca se ha demostrado la conversión completa a un tipo de neurona completamente funcional y estable. Estos son, por lo tanto, fenotipos celulares transitorios. La conversión completa a un tipo totalmente funcional y estable de neuroprogenitor o de células madre neurales tampoco se ha demostrado nunca. La adquisición de un fenotipo estable después de la transdiferenciación ha sido uno de los principales desafíos que enfrenta el campo.
Así, hay una necesidad en el campo biomédico de células madre neurales estables, potentes y preferentemente autólogas, células progenitoras neurales, así como neuronas y células gliales para su uso en el tratamiento de diversos trastornos y enfermedades neurológicas. Lo mismo es cierto para muchos otros tipos de células. Recientemente, se ha obtenido evidencia de que los genes de la clase básica Hélice-Bucle-Hélice (bHLH) son reguladores importantes de varias etapas en el desarrollo del linaje neural, y la sobreexpresión de varios factores neurogénicos de bHLH da como resultado la conversión de ectodermo no determinado en tejido neuronal. Las proteínas proneurales de bHLH controlan la diferenciación en células progenitoras y su progresión a través del
programa neurogénico en todo el sistema nervioso (Bertrand y coI., 2002). MASH1, NeuroD, NeuroD2, MATH1-3 y Neurogenina 1-3 son factores de transcripción de bHLH expresados durante la determinación y diferenciación neuronal de mamíferos (Johnson y coI., 1990; Takebyashi y coI., 1997; McCormick y coI., 1996; Akazawa y coI., 1995). Las interrupciones dirigidas de MASH1, Ngnl, Ngn2 o NeuroD en ratones conducen a la pérdida de subpoblaciones específicas de neuronas (Guillemot y coI., 1993; Fode y coI., 1998; Miyata y coI., 1999).
La patente de Estados Unidos N.o6.087.168 (Levesque y coI.,) describe un procedimiento para convertir o transdiferenciar las células basales epidérmicas en neuronas viables. En un ejemplo, este procedimiento comprende la transfección de las células epidérmicas con uno o más vectores de expresión que contienen al menos un ADNc que codifica un factor de transcripción neurogénico responsable de la diferenciación neural. Los ADNc adecuados incluyen: activadores de hélice-bucle-hélice básicos, tal como NeuroDI, NeuroD2, ASH1 y activadores tipo dedos de cinc, tal como Zíc3 y MyTI. A la etapa de transfección le siguió la adición de al menos un oligonucleótido antisentido que se sabe que suprime la diferenciación neuronal al medio de crecimiento, tal como el gen MSX1 humano y/o el gen HES1 humano (u equivalentes homólogos no humanos). Por último, las células transfectadas se cultivaron en presencia de un retinoide y al menos una neurotrofina o citocina, tal como el factor neurotrófico derivado del cerebro (BDNF), factor de crecimiento neural ( NGF), neurotrofina 3 (NT-3) o neurotrofina 4 (NT-4). Esta tecnología produce el 26 % de las células neuronales; sin embargo, no se estableció la funcionalidad ni la estabilidad de estas células. Además, las células madre neurales o las células neuroprogenitoras no se producen de acuerdo con este procedimiento.
Un proceso posterior (Levesque y coI., 2005; patente de Estados Unidos n.o 6949380) menciona la conversión de las células basales epidérmicas en una célula progenitora neural, neuronal o de la glía exponiendo la célula basal epidérmica a un antagonista de la proteína morfogenética ósea (PMO) y cultivando la célula en presencia de al menos un oligonucleótido antisentido que comprende un segmento de un gen MSX 1 y/o gen HES1. No obstante, no hay evidencias ni ejemplos de que se produjeran células progenitoras neurales o gliales de acuerdo con este procedimiento, sin mencionar detalles o evidencia de lograr las características morfológicas, fisiológicas o inmunológicas de las células neuronales. Además, dado que no hay información sobre la funcionalidad, estabilidad, expansión y rendimiento sobre las células que pueden o no haberse producido, es posible que estas células son realmente células precursoras derivadas de la piel (Fernandes y coI., 2004) que se han diferenciado en células neuronales.
En vista de lo anterior, hay, por lo tanto, una necesidad de células madre neurales potentes, estables y, preferentemente, autólogas, células progenitoras neurales, neuronas y células gliales, así como otros tipos de células, células madre y células progenitoras. También existe la necesidad de procedimientos que puedan resultar en una verdadera desdiferenciación de las células y una reprogramación de las células.
La presente invención aborda estas necesidades y proporciona procedimientos que pueden resultar en una verdadera desdiferenciación celular y una reprogramación celular.
Los rasgos adicionales de la invención serán evidentes a partir de la revisión de la divulgación, figuras y descripción de la invención en el presente documento.
Sumario de la invención
La invención es como se define en las reivindicaciones. Los términos "realización" y "aspecto" como se usan en el presente documento se refieren a la divulgación en su totalidad y pueden o no pueden referirse a la invención reivindicada.
Un aspecto particular se relaciona con el desarrollo de una tecnología para reprogramar una célula somática o célula no neuronal a una célula que tiene una o más características morfológicas fisiológicas y/o inmunológicas de una célula madre neural y que posee la capacidad de diferenciarse a lo largo de linajes neuronas y gliales. De acuerdo con algunas realizaciones, la divulgación está más particularmente relacionada con Ios procedimientos para generar células neurales similares a las células madre neurales (NSLC) a partir de células somáticas humanas, células progenitoras humanas y/o de células madre humanas, así como células, líneas celulares y tejidos obtenidos mediante el uso de dichos procedimientos.
La divulgación se refiere además a composiciones y procedimientos para inducir la desdiferenciación de células somáticas humanas en células de tipo madre neurales que expresan marcadores específicos de células madre neurales. De acuerdo con la presente divulgación, es posible efectuar la conversión de células a varios tipos de células neuronales diferenciadas que pueden crearse a partir de un solo tipo de célula tomado de un donante individual y, a continuación, reprogramarse y transplantarse en el mismo individuo. Tras la inducción, las células de acuerdo con la divulgación expresan marcadores específicos de células madre neurales y se convierten en células de tipo madre neurales.
Según un aspecto particular, la divulgación se refiere a un procedimiento para transformar una célula de un primer tipo en una célula deseada de un tipo diferente. El comprende i) obtener una célula de un primer tipo; ii) aumentar de forma transitorio en la célula de un primer tipo Ios niveles intracelulares de al menos un agente de reprogramación, por lo que el aumento transitorio induce la expresión endógena directa o indirecta de al menos un regulador génico;
i¡¡) colocar la célula en condiciones para soportar el crecimiento y/o la transformación de la célula deseada y mantener los niveles intracelulares de al menos un agente de reprogramación durante un período de tiempo suficiente para permitir la expresión estable del al menos un regulador génico en ausencia del agente de reprogramación; y ¡v) mantener la célula en condiciones de cultivo que apoyan el crecimiento y/o la transformación de la célula deseada. Tales condiciones se mantienen durante un período de tiempo suficiente para permitir una expresión estable de una pluralidad de genes secundarios. Según la divulgación, la expresión de uno o más de los genes secundarios es característica de las propiedades fenotípicas y funcionales de la célula deseada, mientras que no es característica de las propiedades fenotípicas y funcionales de una célula madre embrionaria. Por lo tanto, al final del periodo de tiempo, se obtiene la célula deseada de un tipo diferente.
Según otro aspecto particular, la divulgación se refiere a un procedimiento para transformar una célula de un primer tipo en una célula de un segundo tipo diferente. El procedimiento comprende poner en contacto la célula de un primer tipo con uno o más agentes capaces de aumentar dentro de dichos niveles celulares de al menos un agente de reprogramación y remodelar directa o indirectamente la cromatina y/o el ADN de la célula. El al menos un agente de reprogramación se selecciona para inducir directa o indirectamente la expresión de características morfológicas y funcionales de una célula deseada de un tipo diferente o un linaje celular diferente.
De acuerdo con otro aspecto, la divulgación se refiere a un procedimiento para transformar una célula de un primer tipo en una célula de un segundo tipo diferente. El procedimiento comprende poner en contacto la cromatina y/o el ADN de una célula de un primer tipo con un agente capaz de remodelar la cromatina y/o el ADN de dicha célula; y aumentar los niveles intracelulares de al menos un agente de reprogramación. El al menos un agente de reprogramación se selecciona para inducir directa o indirectamente la expresión de características morfológicas y funcionales de una célula deseada de un tipo o linaje celular diferente.
Un aspecto adicional de la divulgación se refiere a un procedimiento para transformar una célula de un primer tipo en una célula de una célula deseada de un tipo diferente, que comprende el aumento de los niveles intracelulares de al menos un agente de reprogramación, en el que el al menos un agente de reprogramación se selecciona para inducir directa o indirectamente la expresión de características morfológicas y funcionales de un segundo tipo de célula deseado; y mantener la célula de un primer tipo en condiciones de cultivo para soportar la transformación de la célula deseada durante un período de tiempo suficiente para permitir la expresión estable de una pluralidad de genes secundarios cuya expresión es característica de las propiedades fenotípicas y funcionales de la célula deseada, en el que al menos uno de los genes secundarios no es característico de las propiedades fenotípicas y funcionales de una célula madre embrionaria. Al final del período de tiempo, se obtiene la célula deseada de un tipo diferente y la célula obtenida se caracteriza además por una represión estable de una pluralidad de genes expresados en el primer tipo de célula.
Un aspecto adicional de la divulgación se refiere a un proceso en el que una célula de un primer tipo se reprograma a una célula deseada de un tipo diferente, comprendiendo el proceso:
- un aumento transitorio de los niveles intracelulares de al menos un agente de reprogramación, en el que el al menos un agente de reprogramación induce una expresión endógena directa o indirecta de al menos un regulador génico, y en el que la expresión endógena de dicho al menos un regulador génico es necesaria para la existencia de la célula deseada de un tipo diferente;
- una expresión estable de dicho al menos un regulador génico;
- expresión estable de una pluralidad de genes secundarios, en el que la expresión estable de los genes secundarios es el resultado de la expresión estable de al menos un regulador génico, y en el que: (i) la expresión estable de la pluralidad de genes secundarios es característica de las propiedades fenotípicas y/o funcionales de la célula deseada, (ii) la expresión estable de al menos uno de dichos genes secundarios no es característica de las propiedades fenotípicas y funcionales de una célula madre embrionaria, y en el que (i) y (ii) son indicativos de una reprogramación exitosa de la célula del primer tipo al célula deseada del tipo diferente.
En realizaciones particulares, el al menos un agente de reprogramación en el proceso es un polipéptido Ms¡1 o un polipéptido Ngn2 junto con un polipéptido MDB2. En realizaciones particulares, el al menos un regulador génico es Sox2 Ms¡1, o ambos. En realizaciones adicionales, el al menos un regulador génico puede ser uno o más de los genes enumerados en la Tabla A para células de tipo madre neurales.
De acuerdo con otro aspecto, la divulgación se refiere a un procedimiento para obtener una célula similar a célula madre (SLC), que comprende:
i) proporcionar una célula de un primer tipo;
¡i) aumentar de forma transitorio los niveles intracelulares de al menos un agente de reprogramación, por lo que el aumento transitorio induce la expresión endógena directa o indirecta de al menos un regulador génico;
¡ii) colocar la célula en condiciones para soportar la transformación en la célula similar a célula madre y mantener los niveles intracelulares de al menos un agente de reprogramación durante un período de tiempo suficiente para permitir la expresión estable del al menos un regulador génico en ausencia del agente de reprogramación;
¡v) mantener la célula en condiciones de cultivo para apoyar la transformación en la célula similar a célula madre durante un período de tiempo suficiente para permitir la expresión estable de una pluralidad de genes
secundarios cuya expresión es característica de las propiedades fenotípicas y/o funcionales de la célula similar a célula madre, en el que al menos uno de los genes secundarios no es característico de las propiedades fenotípicas y funcionales de una célula madre embrionaria. Al final de dicho período de tiempo, se obtiene una célula similar a célula madre.
De acuerdo con otro aspecto, la divulgación se refiere a un procedimiento para obtener una célula similar a célula madre. El procedimiento comprende aumentar los niveles intracelulares de al menos un polipéptido específico para el tipo de célula madre deseado que es capaz de conducir directa o indirectamente la transformación de la célula del primer tipo en la célula similar a célula madre. Para aumentar el rendimiento o tipo de células de tipo madre, el procedimiento puede comprender además poner en contacto la cromatina y/o el ADN de una célula de un primer tipo con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador de ADN, y/o un inhibidor de la metilación del ADN; y/o aumentar los niveles intracelulares de al menos otro polipéptido específico para el tipo de célula madre deseado que es capaz de conducir directa o indirectamente la transformación de la célula del primer tipo en la célula similar a célula madre.
De acuerdo con otro aspecto, la divulgación se refiere a un procedimiento para obtener una célula similar a una célula madre (NSLC). El procedimiento comprende aumentar los niveles intracelulares de al menos un polipéptido específico de células madre neurales que es capaz de conducir directa o indirectamente la transformación de la célula del primer tipo en una NSLC. Para aumentar el rendimiento o tipo de NSLC, el procedimiento comprende además la cromatina de contacto y/o el ADN de una célula de un primer tipo con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador de ADN, y/o un inhibidor de la metilación del ADN; y/o aumentar los niveles intracelulares de al menos un polipéptido específico de células madre neurales que es capaz de conducir directa o indirectamente la transformación de la célula del primer tipo en una NSLC.
Otro aspecto de la divulgación se refiere a un procedimiento para obtener una célula similar a una célula madre (NSLC). En una realización, el procedimiento comprende transfectar una célula de la piel con un polinucleótido que codifica Musashil, Musashil y neurogenina 2, Musashil y la proteína 2 del dominio de unión a metil-CpG (MBD2), o la neurogenina 2 y la proteína 2 del dominio de unión a metil-CpG, reprogramando así la célula de la piel en una NSLC. En otra realización, el procedimiento comprende exponer una célula de la piel a: (i) un inhibidor de la desacetilación de histonas, (ii) un inhibidor de la metilación del ADN, (iii) un acetilador de histonas, y/o (ív) un desmetilador de ADN tal como un polipéptido MBD2 y/o transfectando con un polinucleótido que codifica un polipéptido MBD2; y transfectar aún más la célula (ya sea simultáneamente, antes, o después) con un polinucleótido que codifica MUSASHI1 y/o con un polinucleótido que codifica NGN2, reprogramando así la célula de la piel en una NSLC. Algunas otras células, tales como queratinocitos y células CD34+, también se pueden utilizar y reprogramar. En una realización particular, el procedimiento para obtener una célula similar a célula madre neural (NSLC), comprende:
- proporcionar una célula de un primer tipo;
- introducir en la célula uno o más polinucleótidos capaces de la expresión transitoria de uno o más de los siguientes polipéptidos: Musashil (Msi 1); un Musashil (Msi 1) y una Neurogenina 2 (Ngn2); un Musashil (Msí1) y una proteína 2 de dominio de unión a metil-CpG (MBD2); y neurogenina 2 (Ngn2) y proteína 2 del dominio de unión a metil-CpG (MBD2); y
- colocar la célula en condiciones de cultivo que apoyan la transformación en una NSLC durante un período de tiempo suficiente para permitir una expresión estable de una pluralidad de genes cuya expresión es característica de las propiedades fenotípicas y funcionales de una NSLC.
Al final del período de tiempo, se obtiene un NSLC y el NSLC obtenido se caracteriza además por una represión estable de una pluralidad de genes expresados en el primer tipo de célula.
De acuerdo con otra realización, el procedimiento para obtener una célula similar a célula madre neural (NSLC), comprende:
- proporcionar una célula de un primer tipo que no es NSLC;
- aumentar los niveles intracelulares de al menos un polipéptido específico de células madre neurales, en el que el polipéptido es capaz de conducir directa o indirectamente la transformación de la célula del primer tipo en una NSLC; y
- poner en contacto la cromatina y/o el ADN de la célula de un primer tipo con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador y/o un inhibidor químico de la metilación del ADN. De acuerdo con otra realización, el procedimiento para obtener una célula similar a célula madre neural (NSLC), comprende:
- obtener una no NSLC;
- cotransfectar la no NSLC con un primer polinucleótido que codifica un polipéptido MBD2 y con al menos un segundo polinucleótido que codifica un polipéptido MUSASHI1 y/o que codifica un polipéptido NGN2;
- colocar la célula cotransfectada en condiciones de cultivo para soportar la transformación de NSLC hasta que se obtenga dicha NSLC.
Ciertos aspectos de la invención se refieren a células aisladas, líneas celulares, composiciones, ensamblaje 3D de células y tejidos que comprenden células obtenidas usando los procedimientos descritos en el presente documento. Aspectos adicionales se refiere al uso de tales células aisladas, líneas celulares, composiciones, ensamblaje 3D de células y tejidos de tratamiento médico y procedimientos de regeneración de tejidos u órganos de mamíferos.
No obstante, un aspecto adicional se refiere a un procedimiento para reparar o regenerar un tejido en un sujeto. En una realización, el procedimiento comprende la administración de una célula reprogramada como se define en el presente documento a un sujeto que lo necesite, en el que la administración proporciona una dosis de células reprogramadas suficiente para aumentar o apoyar una función biológica de un tejido u órgano dado, mejorando así la afección del sujeto.
Los beneficios de la presente invención son significativos e incluyen un menor coste de la terapia celular eliminando la necesidad de agentes inmunosupresores, no hay necesidad de embriones ni tejido fetal, eliminando así las limitaciones éticas y de tiempo, menor coste de producción y ningún riesgo para la salud debido a la posible transmisión de virus u otras enfermedades. Además, ya que las células se crean frescas, tienden a ser más potentes que las células que se han pasado varias veces.
Aspectos adicionales, ventajas y características de la presente invención se harán más evidentes al leer la siguiente descripción no restrictiva de realizaciones preferidas que son de ejemplo y no deben interpretarse como limitantes del alcance de la invención.
Breve descripción de las figuras
La Figura 1 es un panel de micrografía óptica (10X) que presenta cambios en la morfología celular de células no transfectadas y transfectadas con Msil y MBD2 en varios puntos de tiempo.
La Figura 2 es un panel de fotomicrografías obtenidas con Cellomics™ (10x) y que revelan células positivas a NCAM en células transfectadas con Msil o Ngn2 en presencia de MBD2. Los HFF se trataron previamente con citocalasina B (10 pg/ml) y se transfectaron con pcMv 6-XL5-Msí1 y pCMV6-XL5-MBD2 o pcMv6-XL4-Ngn2 y pCMV6-XL5-MBD2. Tras 24 horas después de la transfección, el medio se cambió y las células se cultivaron en medio de proliferación (NPBM, Lonza) suplementado con EGF (20 ng/ml) y bFGF (20 ng/ml) durante una semana. La diferenciación se indujo cambiando el medio a NbActive (BrainBits™) suplementado con NGF (20 ng/ml), bFGF (20 ng/ml), ATRA (5 pM) y forscolina (10 |j M). Las células se incubaron a 370C, 5 % de CO2, 5 % de O2 durante 20 días.
La Figura 3 es un panel de fotomicrografías obtenidas con Cellomics™ (10x) y que revelan células positivas para MAP2b en células transfectadas con Msil o Ngn2 en presencia de MBD2. Las células positivas para MAP2b fueron indetectables en células no transfectadas y células transfectadas con Pax6/MBD2. Los HFF se trataron previamente con citocalasina B (10 pg/ml) y se transfectaron con pCMV6-XL5-Msí1, pCMV6-XL4-Ngn2 o pCMV6-XL5-Pax6 y pCMV6-XL5-MBD2. Tras 24 horas después de la transfección, el medio se cambió y las células se cultivaron en medio de proliferación (NPBM, Lonza) suplementado con EGF (20 ng/ml. Peprotech) y bFGF (20ng/ml, Peprotech) durante una semana. La diferenciación se indujo cambiando el medio a NbActive (BrainBits™) suplementado con NT-3 (20 ng/ml), bFGF (20 ng/ml), ATRA (5 pM) y forscolina (10 j M). Las células se incubaron a 37 oc, 5 % de CO2, 5 % de O2 durante 2 semanas.
La Figura 4A es un panel de fotografías que muestra que las neuroesferas formadas por NSLC del Ejemplo V se disociaron completamente en suspensiones de células individuales utilizando Accutase y se controló una sola célula a lo largo del tiempo para revelar la capacidad de formación de neuroesferas (A, observación con microscopio óptico). Las neuroesferas fueron positivas para la tinción como Sox2.
La Figura 4B es un panel de fotografías de los resultados de inmunohistoquímica obtenidos con Cellomics™. Se realizó inmunohistoquímica, el día 20, para detectar fabricantes de neuroesferas y en comparación con los niveles de expresión en las neuroesferas formadas por células neuroprogenitoras humanas normales (hNPC, Lonza). Además de Sox2, células teñidas positivas para los marcadores de células madre neurales Musashi, CD133, nestina y GFAP. Las células también se tiñeron de forma positiva para plll-tubulina (un marcador para las neuronas), 04 (un marcador para oligodendrocitos) y GFAP (un marcador para astrocitos), lo que indica el potencial de diferenciación tripotencial de ambos conjuntos de células (NSLC y hNPC), y negativo para NGFrec y NeuN (marcadores para neuronas diferenciadas) que indica que las células no se diferenciaron terminalmente. La Figura 5 es un panel de fotomicrografías de los resultados de inmunohistoquímica obtenidos con Cellomics™. La inmunohistoquímica se realizó en HFF, NSLC y hNPC para detectar la expresión de marcadores para fibroblastos así como células madre neurales (Sox2, nestina, GFAP) en cultivos adherentes (que impidieron que las células flotaran y formaran neuroesferas). Los núcleos se tiñeron con Hoechst (imágenes de nivel superior). Los HFF expresaron marcadores de fibroblastos, mientras que las NSLC creadas a partir de estos HFF no lo hicieron. En comparación, las NSLC expresaron marcadores de células madre neurales de manera similar a las hNPC, mientras que los HFF no expresaron ninguno de estos marcadores.
La Figura 6 es un panel de fotomicrografías que muestra NSLC humanas. Se indujo la diferenciación de NSLC
humanas en linajes neuronales en presencia de medio de diferenciación NS-A (StemCell Technologies) en presencia de BDNF (20 ng/ml, Peprotech) y bFGF (40 ng/ml, Peprotech) durante tres semanas. En diferentes puntos de tiempo de diferenciación, la inmunotinción con Cellomics™ (10x) reveló una diferenciación de las células como lo demuestra la disminución de las células positivas para Sox2 y el aumento en el número y la intensidad de la tinción de células positivas para p75, pIN-tubulina y GABA, así como la morfología diferenciada, mientras que el número total de células aumentó como se muestra mediante tinción de Hoechst.
La Figura 7 es otro panel de fotomicrografías. Los HFF, queratinocitos y CD34+ se transfectaron con pCMV6-Msi1-Ngn2 y pCMV6-XL5 - MBD2. Tras 24 horas después de la transfección, el medio se cambió a medio de proliferación (StemCell Technologies) suplementado con EGF (20 ng/ml. Peprotech) y bFGF (20ng/ml, Peprotech) durante dos semanas y después se analizaron. Las fotomicrografías que utilizan Cellomics™ (10x) muestran que las NSLC creados a partir de los tres tipos de células son positivos para nestina, Sox2 y GFAP (marcadores para células madre neurales), mientras que los HFF originales no lo son.
La Figura 8 es un panel de fotomicrografías que muestran el efecto del medio CDM en la transdiferenciación de HFF hacia neuronas. Los HFF se trataron previamente con citocalasina B (10 pg/ml) e inhibidor de desacetilación de histonas (VPA, 4 mM) e inhibidor de metilación de ADN (5-Aza, 5 pM y se cultivaron en medio CDM que contiene una relación 3:1 de medio Eagle modificado por Dulbecco (DMEM, alto contenido de glucosa (4,5 g/l) con L-glutamina y piruvato de sodio) y medio F-12 de Ham complementado con los siguientes componentes: EGF (4,2 x lo-10 M), bFGF (2,8 x 10'10M), ITS (8,6 x 10's M), dexametasona (1,0 x 10-7M), fosfato de ácido L-ascórbico, sal de magnesio n-hidrato (3,2 x 10 ^ ) , L-3,3',5-triyodotironina (2,0 x 10‘10 M), etanolamina (10 ^ ) , GlutaMAX™ (4 x 10-3 M), glutatión (3,3 x 10‘6M). Después de 24 horas, el medio de cultivo se reemplazó con 75 % de medio CDM y 25 % de medio de proliferación neuronal (Lonza, N.0 de catálogo CC-3210); durante los siguientes 3 días, la relación de los medios se cambió a 50 %:50 %, 25 %:75 % y, a continuación, 100 % de medio de proliferación neuronal al tercer día. Se tomaron fotomicrografías con Cellomics™ (10X) después de la inmunotinción de las células con pIN-tubulina (marcador neuronal) y Hoechst (para teñir los núcleos) en diferentes puntos temporales. Las células empezaron a transdiferenciarse en unos pocos días y las células transdiferenciadas fueron positivas para pIN-tubulina; sin embargo, después de una semana se observó una inversión espontánea a la forma fibroblástica y pérdida de la expresión de pIM-tubulina.
La Figura 9 es un panel de fotomicrografías que muestran la caracterización de células reprogramadas dentro de CDM en diferentes puntos de tiempo después de la transfección con Msi1 y Ngn2. Las células transfectadas se trataron con citocalasina B (10 pg/ml), El VpA (4 mM) y 5-AZA (5 pM) que dio como resultado una interrupción de los microfilamentos y el redondeo de las células y laxitud de la cromatina. La inmunohistoquímica en el CDM tridimensional se realizó después de una y dos semanas utilizando Cellomics™ (10X). Las células fueron positivas para el marcador neuronal maduro, tal como MAP2b, pero estuvieron ausentes en el CDM de control no transfectado.
La Figura 10 es otro panel de fotomicrografías. Las células el día 4 de CDM se lipotransfectaron con los dos vectores pCMV6-XL5-Msí1 y pCMV6-XL4-Ngn2 individualmente o en combinación con pCMV-XL5-MBD2 durante un período de 6 horas. En paralelo, la transfección se realizó en HFF nuevos después de las 6 horas usando nucleofección y estos HFF nuevos se colocaron en la parte superior del CDM al mismo tiempo que los medios de lipofectamina se cambiaron a medio de CDM nuevo después de 6 horas. Después de 24 horas, el medio se cambió a medio de proliferación neural (NPBM, Lonza) con la presencia de noggina (50 ng/ml, Peprotech), hFGF recombinante (20 ng/ml, Peprotech) y hEGF recombinante (20 ng/ml, Peprotech) durante una semana. El día 7 se indujo diferenciación, añadiendo medio de diferenciación NS-A (StemCell Technologies) durante 24 días. La inmunohistoquímica se realizó en varios puntos de tiempo utilizando Cellomics™ (10X). El CDM se tiñó con un anticuerpo específico contra nestina (un marcador para las células madre neurales), y las células dentro del CDM expresaron nestina en todos los puntos de tiempo analizados (días 8, 15 y 21) después de la transfección. Las células dentro del CDM de control no transfectado no expresaron ninguna nestina.
La Figura 11 es un panel que muestra una imagen de una electroforesis en gel de poliacrilamida. Las NSLC cultivadas como cultivos adherentes o cultivos en suspensión (como neuroesferas) expresan la telomerasa (que se expresa en todas las células madre, pero no en células somáticas diferenciadas normales). Tanto las NSLC de pase temprano (p5) como tardío (p27) expresan telomerasa. (Los HFF originales a partir de los cuales se crearon las NSLC no expresaron telomerasa). Las muestras (NSLC) se centrifugaron y la concentración de proteína del sobrenadante se determinó mediante el ensayo BCA. Se añadieron 900 ng de proteína de cada extracto celular directamente a la mezcla de reacción TRAp que contenía el tampón de reacción TRAP, dNTP, cebador del sustrato molde (TS), mezcla de cebadores TRAp y Taq polimerasa. Las mezclas de reacción se incubaron a 30 °C durante 30 minutos para la síntesis del molde, seguido de un procedimiento de PCR (95 °C/15 min para la desnaturalización inicial, 94 oc/30 s, 59 °C/30 s, 72 °C/1 min durante 32 ciclos) para la amplificación de los productos de telomerasa extendida. Para detectar la actividad telomerasa, la electroforesis en gel de poliacrilamida (PAGE) se realizó para los productos de reacción en un gel TBE no desnaturalizante al 10 %. Después de la electroforesis, el gel se tiñó con SYBR® Green I Nucleic Acid Gel Stain durante 30 minutos, seguido de la captura de imágenes mediante el sistema de documentación en gel (Alpha Innotech). Las 4 muestras fueron positivas para telomerasa (como lo indica la escala de productos TRAP).
La Figura 12 es un panel que muestra una imagen que muestra el análisis de transferencia Southern de dos muestras de NSLC diferentes analizadas para la integración de los genes MsI1 y Ngn2 dos semanas después de la transfección transitoria. La sonda de PCR marcada con Dig reveló señales distintas en las muestras de control positivo en las que el ADN plasmídico Msi1/Ngn2 se insertó en el ADN genómico de HFF para la equivalencia de 1, 10 o 100 integraciones por genoma. Hubo algunas bandas débiles e idénticas que aparecieron en el ADN genómico digerido con enzimas de restricción de las muestras de HFF y NSLC no transfectadas n.01 y n.02, lo que sugiere que no había integración de ADN plasmídico en el ADN genómico de NSLC. Estas bandas débiles pueden representar el gen Ngn2 endógeno ya que la sonda de PCR marcada con Dig de 1,2 kb contiene una pequeña parte del gen Ngn2. Hubo señales positivas en el carril de la escalera de ADN kb ya que las bandas pertenecen a varios plásmidos digeridos hasta el final con enzimas de restricción apropiadas (NEB). Este dato muestra que ninguna NSLC, o solo un pequeño número de ellas, tenía integración de plásmidos en el genoma del huésped después de la transfección transitoria y que los genes transfectados transitoriamente solo estaban presentes en las células durante un corto período de tiempo (menos de dos semanas).
La Figura 13 es un panel con un gráfico de líneas y un gráfico de barras que muestra mejoras y puntuaciones clínicas significativamente mejores en ratones EAE tratados con NSLC. Se inmunizaron ratones hembra C57BL/6 de 8 semanas de edad con MOG35-55 (Sheldon Biotechnology Center McGill University) en CFA que contenía 5 mg/ml de Mycobacterium tuberculosis H37Ra (Difeo, inc) desecada (muerta y secada) en dos sitios en la parte posterior, y se inyectaron 200 ng de toxina pertussis (List Biologieal Laboratories, Inc) en PBS por vía intraperitoneal los días 0 y 2. Una vez que los ratones comenzaron a mostrar síntomas de EAE (el día 13 después de la Inmunización), se inyectaron por vía intravenosa 200 |jl de NSLC (1 millón de células), hNPC (1 millón de células), solución salina o solución salina con ciclosporina. Todos los ratones, excepto el grupo de control con solución salina, recibieron inyecciones diarias de ciclosporina. Se puntuó la enfermedad clínica en los ratones diariamente; los datos representan puntuaciones promedio diarias. Los ratones que recibieron una inyección única de NSLC tuvieron una gravedad de la enfermedad significativamente menor que los ratones que recibieron hNPC o ciclosporina solo.
La Figura 14 es un gráfico de líneas que muestra los resultados de las evaluaciones rotarod de acuerdo con el Ejemplo XVII, parte 2. Se entrenó a las ratas en el rotarod antes del comienzo del experimento. Se colocó a las ratas en un rotarod estacionario y giratorio (girando a 20 rpm) y se monitorizó la cantidad de tiempo que pasaban las ratas caminando sobre el rotarod antes de caerse. Las mediciones se tomaron antes de la ablación y el tratamiento quirúrgico del hemisferio izquierdo del cerebro (antes de la cirugía) y después (después de la cirugía). Los puntos de datos representan el número medio de caídas de cada animal durante cada sesión de prueba de 60 segundos realizada a una velocidad constante de 20 rpm. Cada grupo constaba de ocho ratas.
La Figura 15 es un gráfico de líneas que muestra los resultados de las evaluaciones de la viga para caminar según el Ejemplo XVII, parte 2. Las ratas se midieron según su capacidad para cruzar una viga de 100 em de largo después de la ablación y el tratamiento quirúrgico del hemisferio izquierdo del cerebro. Dos días después de la cirugía, ningún grupo pasó la prueba y los animales no pudieron mantenerse en equilibrio en la viga. Una semana después de la cirugía, todos los animales muestran una mejora en su capacidad para caminar, pero no se observó diferencia significativa entre los diferentes grupos tratados. Desde la semana 4 hasta la semana 26, los animales tratados con NSLC muestran una mejora significativa en su capacidad para caminar en comparación con los otros grupos.
La Figura 16 es un panel que muestra fotografías de ADSC transfectadas transitoriamente con varios vectores pluripoteneiales usando nueleofeetor como se describe en el Ejemplo XIX. Después de la transfección, las células se cultivaron en placas de 6 pocilios en suspensión con una mezcla 50:50 de medio completo ADSC (StemPro™ -43) y medio de células madre embrionarias (mTeSRI™, StemCell Technologies). Después de dos días de cultivo, las células se volvieron a transfeetar con los mismos plásmidos y se sembraron en placas de 96 pocilios recubiertas con Matrigel™ (BD Biosclenees) en presencia de medio completo mTeSRI™ suplementado con tiazovivina (0,5 j M), un inhibidor de ALK-5 (s B341542, Stemgent, 2 j M), y un inhibidor de MEK (PD0325901, Stemgent, 0,5 j M). El medio se cambió todos los días y las células se cultivaron durante 22 días a 37 oc, 5 % de CO2, 5 % de O2, seguido de tinción AP e inmunohlstoquímlea para analizar la expresión de marcadores de plurlpotenela. Las células formaron colonias y se descubrió que expresaban ambos marcadores de plurlpotenela Oet4 y AP después de transfeetar células con pEF-Rex1-EF-0et4-2A-Klf4-2A-RFP.
La Figura 17 es un panel que muestra fotografías de ADSC transfectadas transitoriamente con pCMV6-XL5-Rex1/pCMV6-XL5-Klf4 y pCMV6-XL5-Rex1/pCMV6-XL4-Oet4. Después de la segunda transfección, las ADSC se cultivaron en placas de 96 pocilios recubiertas con Matrigel™ durante 24 días en presencia de medio mTeSRI™ suplementado con Sb341542 y PD0325901 a 37 oc, 5% de CO2, 5% de O2. Con el fin de caracterizar subpoblaciones de células después de la transfección, se usaron tinción en vivo, inmunohlstoquímlea y tinción AP. 1-5 % del total de células transfectadas con Rex1/Oet4 o Rex1/Klf4 mostraron un fenotipo SSEA-4+ y TRA-1-81+ (marcadores de plurlpotenela temprana). La observación a lo largo del tiempo mostró que el fenotipo de estas colonias se mueve desde un fenotipo SSEA-4+ temprano hasta un fenotipo Oet4 /Sox2/Nanog+ tardío a partir del Día 22, que estaba más cerca del estado reprogramado final y una célula de tipo plurlpotenelal.
La Figura 18 es un panel que muestra fotografías de ADSC transfectadas transitoriamente con varios vectores pluripotenciales. Después de la transfección, las células se sembraron en placas en medio de StemPro™ MSC SFM en Matrigel™ (Bd Biosciences) recubiertas de 24 pocilios y se incubaron a 37 °C, 5 % de CO2, 5 % de O2. El día 1, el medio se cambió a una mezcla de 75 % de StemPro™ MSC y 25 % de medio de células hES; el porcentaje de medio StemPro™ MSC SFM se redujo todos los días durante cuatro días para obtener un medio de células hES al 100 % para el día 4. A continuación, se cambió el medio cada dos días. El medio de células hES consistió en medio Eagle modificado por Dulbecco (DMEM, Invitrogen) suplementado con un 20 % de Knockout™ Serum Replacement (KSR, Invitrogen), GlutaMAX™ 1 mM, aminoácidos no esenciales 100 |j M, pmercaptoetanol 100 j M y 10 ng/ml de Fgf-2. Con el fin de caracterizar subpoblaciones de células después de la transfección, se usaron tinción en vivo, inmunohistoquímica y tinción AP. Las células transfectadas con Oct4/UTF1/MBD2, Oct4/Dppa4/MBD2, FoxD3/Dppa4/MBD2, Oct4/FoxD3/Dppa4 y Sox2/FoxD3/UTF1 dieron positivo para el fenotipo SSEA-4+, TRA1-60 y TRA-1-81+ (marcadores de pluripotencia temprana) el día 14.
La Figura 19 es un panel que muestra fotografías de HFF transfectadas transitoriamente. Los HFF se transfectaron de forma transitoria utilizando el dispositivo Nucleofector® II (Lonza) siguiendo el procedimiento descrito en el Ejemplo II, con la excepción de que se utilizó 1 jg de cada uno de los 3 plásmidos de ADN siguientes: pCMV-Oct4nuc-IRES2-Sox2nuc, pCMV-Klf4nuc-IREs2-Cmyc-nuc y pCMV-Nanognuc-IRES2-Lin28. Las células se trataron previamente con o sin VPA y 5-Aza. Después de la transfección, las células se sembraron en placas en el medio de fibroblastos, suplementado con o sin VPA (2 mM) y 5-AZA (2,5 j M) en placas de 6 pocilios recubiertas con Matrigel™ (BD Biosciences) y se incubaron a 37 °C, 5 % de CO2. Los días 1 y 2, el medio se cambió a 100 % de medio mTeSRI™ (StemCell Technologies) suplementado con o sin VPA y 5-AZA. Los días 3 y 6, las células se volvieron a transfectar como antes y se sembraron en placas recubiertas con Matrigel ™ en medio mTeSRI™ suplementado con o sin VPA y 5-Az a . Los medios se cambiaron diariamente como anteriormente. El medio se suplementó con Y27632 (Stemgent, 10 j M) desde el día 7 hasta el día 14 para estimular la viabilidad y la expansión clonal de posibles células reprogramadas. Las células se analizaron el día 20 utilizando el kit de detección de fosfatasa alcalina (Millipore) y mediante análisis de inmunohistoquímica. Algunas células se tiñeron positivamente para los marcadores de pluripotencia AP, SSEA-4 y TRA-1-81 (similar a la línea de células madre embrionarias humanas Mel2 (control positivo)). Estos clones se obtuvieron solo en la condición que no contenía inhibidores (es decir: VPA y 5-AZA). No se observaron clones para la condición tratada con estos inhibidores.
La Figura 20 es un panel que muestra fotografías de NSLC y BG-01 transfectadas. NSLC y BG-01 NS se transfectaron como se ha descrito previamente en el Ejemplo II mediante dos vectores episomales, pEF-Oct4nuc-IRES2-MBD2 (NC1) o pCMV-FoxD3-2A-Oct4-2A-Klf4 (F72). Después de la transfección, se recogieron las células y se sembraron en placas de Petri sin recubrimiento en presencia de medio de proliferación y medio mTeSRI™ (50:50) en condiciones de proliferación a 37 °C, 5 % de CO2. Después de 48 horas, las células se volvieron a transfectar con el mismo plásmido y se sembraron en placas de 96 pocilios recubiertas con Matrigel ™ y se cultivaron en presencia de medio mTeSRI ™ complementado por las pequeñas moléculas BIX01294 (Stemgent, 2 j M) y BayK8644 (Stemgent, 2 j M) 37 0C, 5 % de O2 durante 22 días, después de lo cual se realizaron tinciones en vivo e inmunohistoquímica para caracterizar subpoblaciones de células para marcadores de pluripotencia. Las células formaron colonias positivas para TRA-1-81 y SSEA-4 indicativas de células de tipo pluripotencial.
La Figura 21 es un panel que muestra imágenes de campo brillante el día 17 de fibroblastos transfectados con Msi1/Ngn2 y pCMV6-XL5-Mb D2 colocados en diferentes condiciones de medios y que muestran diferentes morfologías y grados de diferenciación. (a) Células en medio de proliferación neural desde el día 1 hasta el día 12 y, a continuación, en medio de diferenciación neural con citocinas desde el día 12 hasta el 17. (b) Células en medio de proliferación neural desde el día 1 hasta el día 12 y, a continuación, en medio NbActive4 con citocinas desde el día 12 hasta el 17. (c) Células en medio de diferenciación neural con citocinas más Fgf-2 desde el día 1 hasta el día 12 y, a continuación, en el mismo medio pero sin Fgf-2 desde el día 12 al 17. (d) Células en medio NbActive4 con citocinas más Fgf-2 desde el día 1 hasta el día 12 y en ese mismo medio pero sin Fgf-2 desde el día 12 al 17. (e) Células en medio CDM II con citocinas más Fgf-2 desde el día 1 hasta el día 12 y, a continuación, en ese mismo medio pero sin Fgf-2 desde el día 12 al 17.
La Figura 22 es un panel que muestra imágenes de los resultados de inmunoquímica el día 17 de fibroblastos transfectados con Msi1/Ngn2 y pCMV6-XL5-MBD2 en la Figura 21. Figura 22A y 22B: Las células estaban en medio de proliferación NS-A desde el día 1 hasta el día 12 y, a continuación, en medio de diferenciación NS-A (A) o medio NBActive4 (B) con citocinas desde el día 12 al 17. Había más células en B, pero la diferenciación desde el día 12-17 fue demasiado corta para inducir la expresión de plll-tubulina en ambos casos. Figuras 22C-E: Las células estaban en medio de diferenciación NS-A (C) o medio NbActive4 (D) desde el día 1-17 (con suplementación con FGF-2 desde el día 1-12) o medio CDM ll desde el día 1-12 y, a continuación, medio de diferenciación NS-A desde el día 12-17 (E). Había un gran número de células en C y un número mucho menor de células en D y E. Las células fueron inmunopositivas para GFAP y plll-tubulina en todos los casos y, al colocar las células en medios de diferenciación o no proliferación desde el día 1 en adelante, parece haber inducido una transformación más directa en neuronas y glía, con plll-tubulina más intensa que las células positivas para GFAP en E.
La Figura 23 es un panel que muestra dos mapas de calor que proporcionan una visión global de la comparación de la expresión génica entre NSLC frente a HFF (Conjunto 1) o NSLC frente a hNPC (Conjunto 2). NSLC tiene un perfil de expresión génica distinto en comparación con HFF o hNPC. Basado en la intensidad (a mayor intensidad, mayor cambio relativo en la expresión), NSLC es mucho más similar a hNPC que a HFF.
La Figura 24 es un panel que muestra imágenes de NSLC. Las NSLC se analizaron para determinar si son una población de células precursoras derivadas de la piel (SKP). Las SKP capaces de proliferar en respuesta a EGF y bFGF, expresan nestina y fibronectina, y pueden diferenciarse en progenie neuronal y mesodérmica, incluso en adipocitos. Para este fin se realizó un protocolo estándar para convertir las SKP en adipocitos, en el que se cultivaron células madre derivadas de adipocitos (ADSC) y NSLC en medio de proliferación StemPro™ y se indujo diferenciación hacia adipocitos mediante el cultivo de estas células en medio de diferenciación consistente en DMEM/F12 (50:5o), ITS (1:10o), HEPES (1:10o), GlutaMAX™ (1:10o), T3 (0,2 nM), Rosiglitasona (0,5 pg/ml), IBMX (100 pM) y Dexametasona (1 pM). Tres días después, IBMX y Dexametasona se retiraron del medio. El día 10, las células se fijaron con una solución de formaldehído al 4 % durante 10 minutos y se tiñeron con una solución de tinción Oil Red O (Invitrogen). Las células adiposas aparecieron rojas con gotitas de lípidos (manchas blancas brillantes en la imagen de la izquierda) teñidas específicamente con Oil Red O; sin embargo, las NSLC se tiñeron de forma negativa y no tenían presencia de gotitas de lípidos en las células, pero en cambio adoptó la morfología de las células neuronales. Estos resultados indican que las NSLC no son una población de células precursoras derivadas de la piel (SKP).
Descripción detallada de la invención
La presente invención se refiere a procedimientos para la desdiferenciación celular y la reprogramación celular. Un aspecto significativo de la presente divulgación es que permite el uso de células propias de un paciente para desarrollar diferentes tipos de células que pueden trasplantarse después de las etapas de la desdiferenciación in vitro y la reprogramación in vitro. Así, esta tecnología elimina los problemas asociados con el trasplante de células no hospedadoras, tales como, rechazo inmunológico y el riesgo de transmisión de enfermedades. Además, ya que las células son "de nueva creación", tienen el potencial de ser más potentes que las fuentes alternativas de células naturales que ya se han dividido varias veces.
Definiciones
Como se usa en el presente documento y en las reivindicaciones adjuntas, las formas en singular "un", "una y "el/la", incluyen los referentes en plural a menos que el contexto indique claramente lo contrario. Así, por ejemplo, la referencia a "una célula" incluye una o más de dichas células o una línea celular derivada de dicha célula, la referencia a "un agente" incluye uno o más de dichos agentes y la referencia a "el procedimiento" incluye referencias a etapas equivalentes y procedimientos conocidos por los expertos en la materia que podrían modificarse o sustituirse por los procedimientos descritos en el presente documento.
Como se usa en el presente documento, el término "polinucleótido" se refiere a cualquier secuencia o molécula de ADN o ARN, que comprende secuencias nucleotídicas codificantes. El término pretende abarcar todos los polinucleótidos, sean de origen natural o no natural en una célula, tejido u organismo en particular. Esto incluye ADN y fragmentos del mismos, ARN y fragmentos del mismo, a DNc y fragmentos de mismo, etiquetas de secuencia expresada, secuencias artificiales que incluyen secuencias artificiales aleatorias.
Como se usa en el presente documento, el término "polipéptido" se refiere a cualquier secuencia de aminoácidos que tenga una actividad biológica funcional deseada (por ejemplo, desmetilación del ADN). El término pretende abarcar proteínas completas, fragmentos de las mismas, proteínas de fusión y similares, Incluyendo cadenas o composiciones de carbohidratos o lípidos.
"Transdiferenciación" se refiere a un cambio directo de una célula ya diferenciada a otro tipo de célula diferenciada.
"Desdiferenciación" se refiere a la pérdida de características fenotípicas de una célula diferenciada activando o desactivando genes o vías metabólicas.
"Marcador" se refiere a un gen, polipéptido o función biológica que es característica de un tipo de célula particular o fenotipo celular.
"Secuencia de ADN modificada genéticamente" se refiere a una secuencia de ADN en la que los elementos de la secuencia de ADN se organizan dentro de la secuencia de ADN de una manera que no se encuentra en la naturaleza.
"Secuencia señal" se refiere a una secuencia de ácido nucleico que, cuando se incorpora en una secuencia de ácido nucleico que codifica un polipéptido, dirige la secreción del polipéptido traducido desde las células que expresan dicho polipéptido, o permite que el polipéptido atraviese fácilmente la membrana celular hacia una célula. La secuencia señal se ubica preferentemente en el extremo 5' de la secuencia de ácido nucleico que codifica el polipéptido, de tal manera que la secuencia polipeptídica codificada por la secuencia señal se localice
en el extremo N del pollpéptldo traducido. Por "péptido señal" se entiende la secuencia peptídlca que resulta de la traducción de una secuencia señal.
"Promotor ubicuo" se refiere a un promotor que dirige la expresión de un polipéptido o péptidos codificados por secuencias de ácido nucleico a las que el promotor está unido operativamente. Los promotores ubicuos preferidos incluyen el inmediato temprano del citomegalovirus humano (c MV); el promotor temprano 40 del virus de simio (SV40); el virus del sarcoma de Rous (RSV); o el promotor tardío principal de adenovirus.
"Perfil de expresión génica" significa un ensayo que mide la actividad de múltiples genes a la vez, creando una imagen global de la función celular. Por ejemplo, estos perfiles pueden distinguir entre células madre neurales humanas y células somáticas que se están dividiendo o diferencian activamente.
"Transfección" se refiere a un procedimiento de administración de genes que introduce secuencias de nucleótidos extrañas (por ejemplo, moléculas de ADN) en una célula, preferentemente, por un procedimiento no viral. En realizaciones preferidas de acuerdo con la presente invención, se introduce ADN extraño en una célula mediante transfección transitoria de un vector de expresión que codifica un polipéptido de interés, por lo que el ADN extraño se introduce pero se elimina con el tiempo por la célula y durante la mitosis. Por "transfección transitoria" se entiende un procedimiento en el que los vectores de expresión introducidos y el polipéptido codificado por el vector, no están integrados permanentemente en el genoma de la célula huésped o en cualquier parte de la célula y, por lo tanto, pueden eliminarse de la célula huésped o su progenie con el tiempo. Las proteínas, los polipéptidos u otros compuestos también pueden administrarse en una célula utilizando procedimientos de transfección.
"Célula neuroprogenitora" se refiere a una célula inmadura del sistema nervioso, que puede diferenciarse en neuronas y glía (oligodendrocitos y astrocitos). "Célula madre neural" es una célula madre multipotencial derivada de la capa germinal del ectodermo que tiene, como característica fisiológica, una capacidad para formar células neuroprogenitoras y en condiciones fisiológicas que favorecen la diferenciación para formar neuronas y glía. "Células de tipo madre neurales" o "NSLC" se refiere a cualquier célula madre multipotencial derivada de células que tenga, como característica fisiológica, una capacidad para formar otras células de tipo madre neurales y células de tipo neuroprogenitoras y en condiciones fisiológicas que favorecen la diferenciación para formar células similares a neuronas y células de tipo glía.
"Neuroesfera" se refiere a un agregado celular de células madre neurales y células neuroprogenitoras que forman una esfera flotante formada como resultado de la proliferación de células madre neurales y células neuroprogenitoras en condiciones de proliferación apropiadas. Las NSLC también forman neuroesferas que consisten en agregados de NSLC y células de tipo neuroprogenitor.
"célula reprogramada" se refiere a una célula que ha sufrido una transdiferenciación, desdiferenciación, o transformación estables. Algunas células reprogramadas pueden ser inducidas posteriormente a rediferenciarse. La célula reprogramada expresa de manera estable un marcador específico de célula o un conjunto de marcadores, morfología y/o función biológica que no era característica de la célula original. "Célula somática reprogramada" se refiere a un proceso que altera o invierte el estado de diferenciación de una célula somática, que puede ser una conversión completa o parcial del estado diferenciado a un estado menos diferenciado o un nuevo estado diferenciado.
"Regeneración" se refiere a la capacidad de contribuir a la reparación o construcción de novo de una célula, tejido u órgano.
"Diferenciación" se refiere al proceso de desarrollo del compromiso de linaje de una célula. La diferenciación se puede analizar midiendo un aumento en uno o más marcadores específicos de diferenciación celular en relación con la expresión de los marcadores celulares indiferenciados.
"Linaje" se refiere a una vía de desarrollo celular, en la que una célula más indiferenciada experimenta cambios fisiológicos progresivos para convertirse en un tipo celular más diferenciado que tiene una función característica (por ejemplo, las neuronas y las células de la glía son de linaje neuroprogenitor, que es de un linaje ectodémico que se formó a partir de blastocitos y células madre embrionarias (ES).
"Tejido" se refiere a un conjunto de células (idénticas o no) y una matriz extracelular (ECM) que juntas llevan a cabo una función específica o un conjunto de funciones.
"CDM" significa un tejido vivo equivalente o matriz, un armazón vivo o matriz derivada de células.
Transformación celular
Algunos aspectos de la divulgación se refieren a procedimientos y células para transformar o reprogramar una célula somática dada en una célula pluripotencial, multipotencial y/o unipotencial. Algunos aspectos de la divulgación se refieren a procedimientos para condicionar una célula somática para reprogramarla en una célula pluripotencial, multipotencial o unipotencial.
Los términos "transformar" o "reprogramar" se usan indistintamente para hacer referencia al fenómeno en el que una célula se desdiferencia o transdiferencia para volverse pluripotencial, multipotencial y/o unipotencial. Posteriormente, la célula desdiferenciada podría rediferenciarse en un tipo de célula diferente. Las células se pueden reprogramar o convertir en diversos grados. Por ejemplo, es posible que solo una pequeña parte de las células se convierta o que una célula individual se reprograme para que sea multipotencial pero no necesariamente pluripotencial. Así, Ios términos "transformar" o "reprogramar" procedimientos pueden referirse a procedimientos en Ios que es posible reprogramar una célula de tal manera que la "nueva" célula muestre características morfológicas y funcionales de un linaje celular específico nuevo o diferente (por ejemplo, la transformación de células fibroblastos en células neuronales).
Como se usa en el presente documento, el término "célula somática" se refiere a cualquier célula diferenciada que forma el cuerpo de un organismo, aparte de las células madre, células progenitoras y células de la línea germinal (es decir, ovogonias y espermatogonias) y las células derivadas de las mismas (por ejemplo, ovocitos, espermatozoides). Por ejemplo, Ios órganos internos, la piel, Ios huesos, la sangre y el tejido conjuntivo están todos formados por células somáticas. Las células somáticas de acuerdo con la divulgación pueden ser células diferenciadas aisladas de adultos o pueden ser células somáticas fetales. Las células somáticas se obtienen de animales, preferentemente sujetos humanos y se cultivan de acuerdo con Ios protocolos estándar de cultivo celular disponibles para Ios expertos en la técnica.
En la técnica, "células madre" se refiere a aquellas células que conservan la capacidad de renovarse mediante la división de las células mitóticas y que pueden diferenciarse en una amplia gama de tipos de células especializadas. Incluye tanto células madre embrionarias que se encuentran en Ios blastocistos como células madre adultas que se encuentran en tejidos adultos. "Células totipotenciales" se refiere a las células que tienen la capacidad de convertirse en células derivadas de las tres capas germinales embrionarias (mesodermo, endodermo y ectodermo) y un organismo completo (por ejemplo, un ser humano si se encuentra en el útero de una mujer en el caso de Ios seres humanos). Las células totipotenciales pueden dar lugar a un embrión, las membranas embrionarias adicionales y todos Ios tejidos y órganos postembrionarios. El término "pluripotencial", como se usa en el presente documento, pretende significar la capacidad de una célula para dar lugar a células diferenciadas de las tres capas germinales embrionarias. "Células multipotenciales" se refiere a las células que pueden producir solo células de una familia de células muy relacionadas (por ejemplo, las células madre hematopoyéticas se diferencian en glóbulos rojos, glóbulos blancos, plaquetas, etc.). "Células unipotenciales" se refiere a las células que tienen la capacidad de desarrollarse/diferenciarse en un solo tipo de tejido/tipo de célula (por ejemplo, células de la piel).
La presente divulgación permite la reprogramación de cualquier célula a un tipo diferente de célula. Aunque la presente solicitud se enfoca principalmente en la preparación de células de tipo células madre, especialmente, células neuronales de tipo células madre neurales (NSLc ), la divulgación no está tan restringida porque se pueden generar muchos tipos diferentes de células de acuerdo con Ios principios descritos en el presente documento. De forma similar, aunque la sección de Ejemplos describe realizaciones en las que Ios fibroblastos, queratinocitos, células CD34+, células madre derivadas de tejido adiposo (ADSC), células madre neurales (incluidas las NSLC) y células dentro de una matriz derivada de células (CDM) se reprograman, la divulgación no se limita a tales células. La divulgación puede emplearse para la reprogramación de prácticamente cualquier célula de interés.
En consecuencia, un aspecto general de la divulgación se refiere a un procedimiento para transformar una célula de un primer tipo en una célula de un segundo tipo diferente. Como se usa en el presente documento, ejemplos de células de un primer tipo incluyen, pero no se limitan a las mismas, células germinales, células madre embrionarias y sus derivaciones, células madre adultas y sus derivaciones, células progenitoras y sus derivaciones, células derivadas de mesodermo, endodermo o ectodermo, y una célula de linaje mesodérmico, endodérmico o ectodérmico, tal como una célula madre derivada de tejido adiposo (ADSC), célula madre mesenquimatosa, célula madre hematopoyética (células CD34+), célula precursora derivada de la piel, célula del folículo piloso, fibroblasto, queratinocito, célula epidérmica, célula endotelial, célula epitelial, célula epitelial granulosa, melanocito, adipocito, condrocito, hepatocito, linfocito (linfocitos B y T), granulocito, macrófago, monocito, célula mononuclear, células de Ios islotes pancreáticos, célula de Sertoli, neurona, célula glial, célula muscular cardíaca y otra célula muscular.
Tal como se desvela en el presente documento, las células de un segundo tipo incluyen, pero no se limitan a las mismas, células germinales, células madre embrionarias y sus derivaciones, células madre adultas y sus derivaciones, células progenitoras y sus derivaciones, células derivadas de mesodermo, endodermo o ectodermo, y una célula de linaje mesodérmico, endodérmico o ectodérmico, tal como una célula madre derivada de tejido adiposo, célula madre mesenquimatosa, célula madre hematopoyética, célula precursora derivada de la piel, célula del folículo piloso, fibroblasto, queratinocito, célula epidérmica, célula endotelial, célula epitelial, célula epitelial granulosa, melanocito, adipocito, condrocito, hepatocito, linfocito (linfocitos B y T), granulocito, macrófago, monocito, célula mononuclear, células de Ios islotes pancreáticos, célula de Sertoli, neurona, célula glial, célula muscular cardíaca y otra célula muscular. Además, cada una de las células "similares o de tipo" (una célula que tiene características similares pero no completamente idénticas del tipo natural conocido de la célula) también se incluye en Ios ejemplos de células de un segundo tipo.
Según un aspecto particular, el procedimiento de transformar una célula de un primer tipo en una célula de un segundo tipo diferente comprende las etapas de:
i) proporcionar una célula de un primer tipo;
II) aumentar de forma transitorio en la célula de un primer tipo los niveles intracelulares de al menos un agente de reprogramadón, por lo que el aumento transitorio induce la expresión endógena directa o indirecta de al menos un regulador génico;
iii) colocar la célula en condiciones para soportar la transformación de la célula deseada y mantener los niveles intracelulares de al menos un agente de reprogramación durante un período de tiempo suficiente para permitir la expresión estable del al menos un regulador génico en ausencia del agente de reprogramación; y
iv) mantener la célula en condiciones de cultivo que apoyan la transformación de la célula deseada durante un período de tiempo suficiente para permitir una expresión estable de una pluralidad de genes secundarios cuya expresión es característica de las propiedades fenotípicas y funcionales de la célula deseada. Al menos uno de los genes secundarios expresados de manera estable no es característico de las propiedades fenotípicas y funcionales de una célula madre embrionaria. Al final de dicho período de tiempo, la célula del primer tipo se ha transformado en la célula deseada de un tipo diferente. Preferentemente, la célula de un tipo diferente obtenida después de la transformación se caracteriza además por una represión estable de una pluralidad de genes expresados en el primer tipo de célula.
De acuerdo con diversas realizaciones, la etapa iii) puede llevarse a cabo consecutivamente a la etapa ii), simultáneamente con la etapa ii), o antes de la etapa ii).
Según un aspecto relacionado, la divulgación se refiere a un proceso en el que una célula de un primer tipo se reprograma a una célula deseada de un tipo diferente, comprendiendo el proceso:
- un aumento transitorio de los niveles intracelulares de al menos un agente de reprogramación, en el que el al menos un agente de reprogramación induce una expresión endógena directa o indirecta de al menos un regulador génico, en el que la expresión endógena de al menos un regulador génico es necesaria para la existencia de la célula deseada de un tipo diferente;
- una expresión estable de dicho al menos un regulador génico;
- expresión estable de una pluralidad de genes secundarios, en el que la expresión estable de la pluralidad de genes secundarios es el resultado de la expresión estable de al menos un regulador génico, y en el que: (i) la expresión estable de la pluralidad de genes secundarios es característica de las propiedades fenotípicas y/o funcionales de la célula deseada, (ii) la expresión estable de al menos uno de los genes secundarios no es característica de las propiedades fenotípicas y funcionales de una célula madre embrionaria, y en el que (i) y (ii) son indicativos de una reprogramación exitosa de la célula del primer tipo al célula deseada del tipo diferente.
Como se usa en el presente documento, "aumento transitorio" se refiere a un aumento que no es necesariamente permanente y, por lo tanto, que puede disminuir o desaparecer con el tiempo. Por ejemplo, cuando se refiere al aumento transitorio de los niveles intracelulares de al menos un agente de reprogramación en una célula, significa que el aumento en el presente durante un período de tiempo suficiente para causar que ocurran eventos celulares particulares (por ejemplo, induciendo la expresión endógena estable de un regulador génico). Normalmente, un aumento transitorio no es permanente y no se asocia, por ejemplo, a la integración del genoma de un vector de expresión.
Como se usa en el presente documento, la expresión "agente de reprogramación" se refiere a un compuesto que es capaz de inducir directa o indirectamente la expresión de características morfológicas y/o funcionales de la célula deseada de un tipo diferente. Los compuestos preferidos incluyen aquellos capaces de conducir directa o indirectamente la transformación de la célula del primer tipo en la célula deseada de un tipo diferente. En la realización preferida, el agente de reprogramación se selecciona para inducir una expresión endógena directa o indirecta de al menos un regulador génico como se define en el presente documento. Hay muchos compuestos que pueden ser útiles para reprogramar una célula de acuerdo con la invención y estos compuestos pueden usarse solos o en combinaciones. En diversas realizaciones, el agente de reprogramación es un polinucleótido o polipéptido seleccionado de acuerdo con la TABLA A:
t a b l a a :

(continuación)

(continuación)
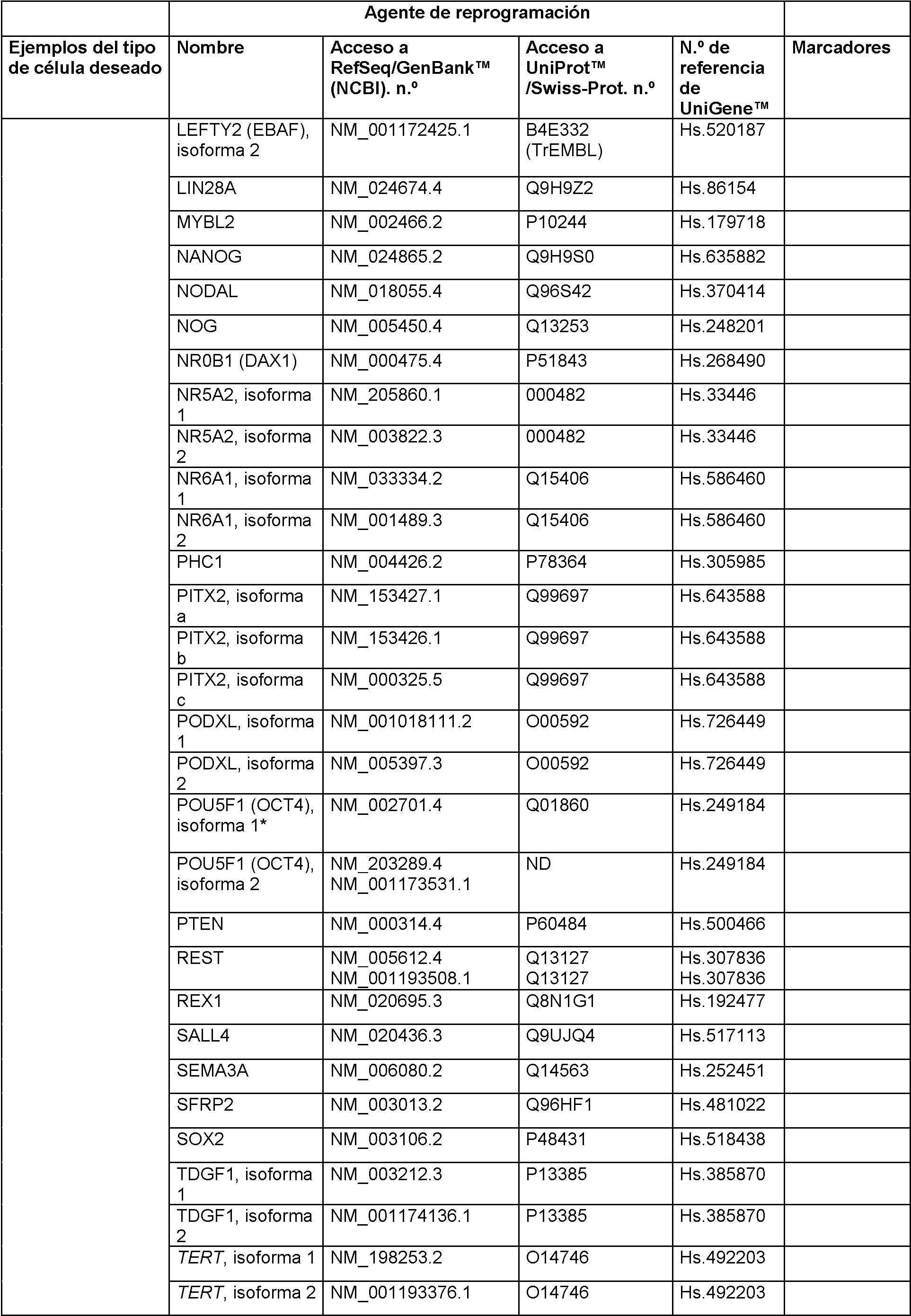
(continuación)

(continuación)

(continuación)

(continuación)
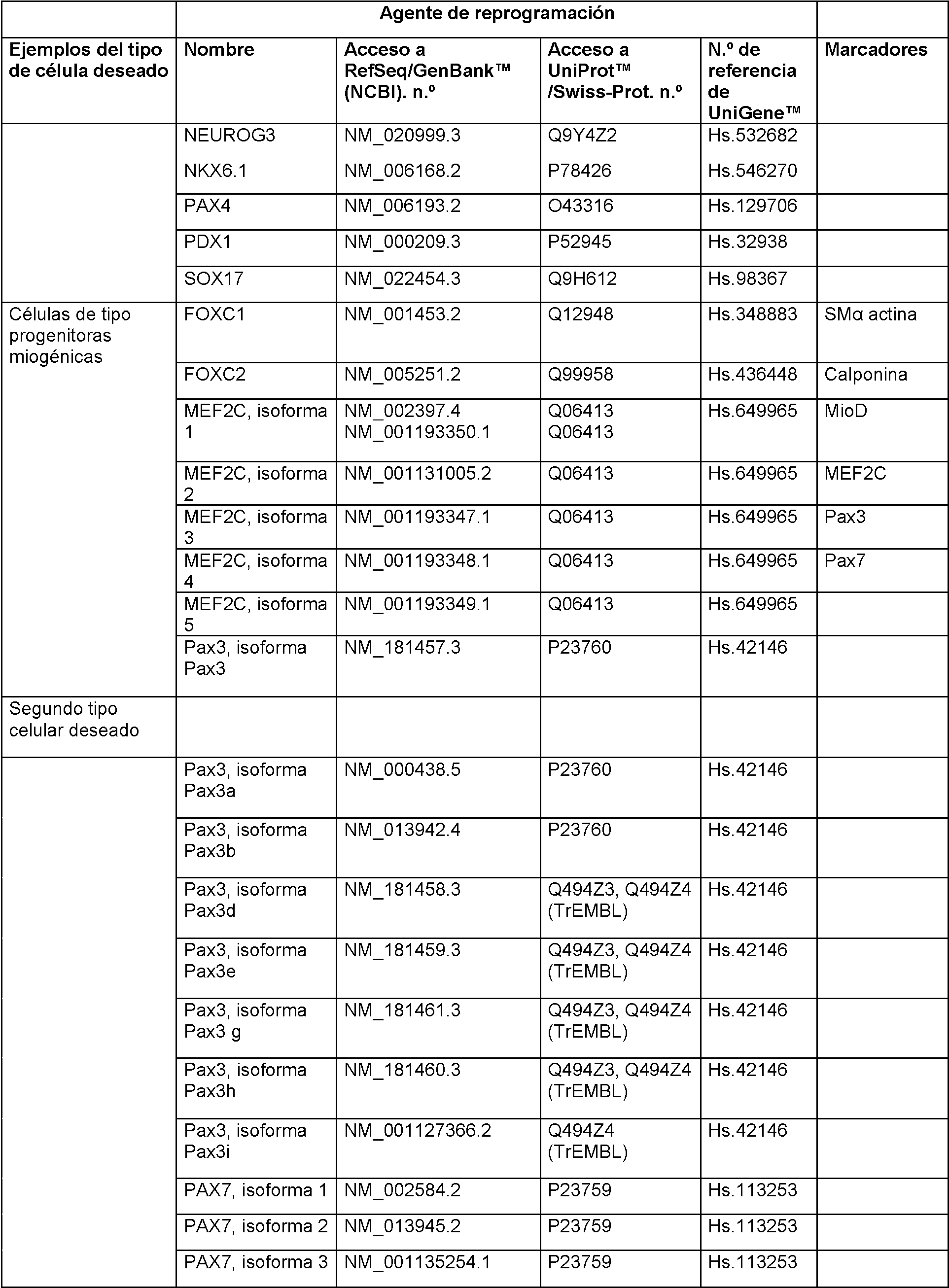
En algunas realizaciones, el agente de reprogramación es un polipéptido que comparte al menos el 75 %, 80 %, 85 %, 90 %, 95 %, 97 %, 99 % o más de la funcionalidad o identidad de secuencia de cualquiera de los agentes de reprogramación en la tabla anterior.
Identificar el "período de tiempo suficiente" para permitir la expresión estable del al menos un regulador génico en ausencia del agente de reprogramación y el "período de tiempo suficiente" en el que la célula debe mantenerse en condiciones de cultivo que apoyan la transformación de la célula deseada está dentro del alcance de los expertos en la técnica. El período de tiempo suficiente o apropiado variará de acuerdo con varios factores, incluyendo, pero sin limitación, el tipo particular y el estado epigenético de las células (por ejemplo, la célula del primer tipo y la célula deseada), la cantidad de material de partida (por ejemplo, el número de células a transformar), la cantidad y el tipo de agentes de reprogramación, el (los) regulador (es) génico(s), las condiciones de cultivo, la presencia de compuestos que aceleran la reprogramación (por ejemplo, compuestos que aumentan la rotación del ciclo celular, modifican el estado epigenético, y/o mejoran la viabilidad celular), etc. En diversas realizaciones, el período de tiempo suficiente para permitir una expresión estable de al menos un regulador génico en ausencia del agente de reprogramación es de aproximadamente 1 día, aproximadamente 2-4 días, aproximadamente 4-7 días, aproximadamente 1-2 semanas, aproximadamente 2-3 semanas o aproximadamente 3-4 semanas. En diversas realizaciones, el período de tiempo suficiente en el que las células deben mantenerse en condiciones de cultivo que apoyan la transformación de la célula deseada y permiten una expresión estable de una pluralidad de genes secundarios es de aproximadamente 1 día, aproximadamente 2-4 días, aproximadamente 4-7 días o aproximadamente 1-2 semanas, aproximadamente 2-3 semanas, aproximadamente 3-4 semanas, aproximadamente 4-6 semanas o aproximadamente 6-8 semanas. En realizaciones preferidas, al final del periodo de transformación, el número de células deseadas transformadas es sustancialmente equivalente o incluso más alto que una cantidad de células, un primer tipo proporcionado al principio.
La presente divulgación abarca varios tipos de compuestos que son adecuados para aumentar en una célula de un primer tipo los niveles intracelulares de al menos un agente de reprogramación. Preferentemente, el compuesto también debe poder remodelar directa o indirectamente la cromatina y/o el ADN de la célula, dando como resultado directa o indirectamente la expresión de características morfológicas y funcionales de la célula deseada de un tipo diferente. Los compuestos preferidos son agentes de reprogramación como se definen en el presente documento o cualquier otro compuesto que tenga una actividad similar y que tenga la capacidad de activar o mejorar la expresión de la versión endógena de los genes enumerados en la tabla de agentes de reprogramación expuestos anteriormente en el presente documento y que sean capaces de impulsar la transformación directa o indirectamente. de la célula del primer tipo en la célula deseada de un tipo diferente.
Como se explicará más adelante, el aumento de los niveles intracelulares de al menos un agente de reprogramación puede lograrse por diferentes medios. En realizaciones preferidas, el agente de reprogramación es un polipéptido y los niveles intracelulares crecientes de dicho polipéptido incluyen la transfección (o cotransfección) de un vector de expresión que tiene un polinucleótido (por ejemplo, ADN o ARN) que codifica el polipéptido(s) o por un suministro intracelular de polipéptido(s). De acuerdo con la invención, la expresión transitoria es generalmente preferible. Los compuestos adecuados adicionales pueden incluir compuestos capaces de aumentar la expresión de la versión endógena de los genes enumerados en la tabla de agentes de reprogramación y reguladores génicos, incluidos, pero sin limitación, factores de reprogramación enumerados en la Tabla B.
t a b l a B:
un

De acuerdo con los principios de la divulgación, el aumento de los niveles intracelulares de al menos un agente de
reprogramación debe inducir una expresión endógena directa o indirecta de al menos un regulador génico. Como se usa en el presente documento, "regulador génico" se refiere a un polinucleótido o polipéptido cuya expresión está asociada con una serie de eventos intracelulares que conducen a la transformación de una célula dada de un primer tipo en una célula pluripotencial, multipotencial y/o unipotencial. Normalmente, la expresión de un regulador génico activa directa o indirectamente los genes necesarios para las características fenotípicas y funcionales de las células pluripotenciales, multipotenciales y/o unipotenciales, mientras se reprimen los genes de la célula de un primer tipo. El regulador génico puede ser el mismo o ser diferente al agente de reprogramación. Los ejemplos de reguladores génicos de acuerdo con la divulgación incluyen, aunque no de forma limitativa, los polinucleótidos y polipéptidos enumerados en el presente documento anteriormente en la TABLA a .
En algunas realizaciones, el regulador génico es un polipéptido que comparte al menos el 75 %, 80 %, 85 %, 90 %, 95 %, 97 %, 99 % o más de la funcionalidad o identidad de secuencia de cualquiera de los reguladores génicos proporcionados en la Tabla A anterior.
Como se usa en el presente documento, "condiciones que soportan el crecimiento" o "condiciones que apoyan la transformación" cuando se refiere a una célula deseada se refiere a varias condiciones de cultivo adecuadas (temperatura, pH, tensión de O2, medios celulares, factores, compuestos, sustrato de crecimiento (ejemplo, laminina, colágeno, fibronectina, Matrigel™, superficie de baja adherencia, superficie nanoestructurada o cargada, etc.), entorno 3D, etc.) que favorece el crecimiento del tipo celular deseado y/o favorece la transformación hacia dicho tipo celular deseado. Los expertos en la materia saben que el crecimiento o la transformación de tipos celulares particulares se estimula en condiciones específicas, mientras que otros los inhiben y está dentro de su habilidad para seleccionar condiciones adecuadas (por ejemplo, condiciones de cultivo) que favorecen el crecimiento o la transformación de los tipos de células deseados.
Los términos "propiedades fenotípicas y funcionales", cuando se refiere a una célula deseada o a una célula madre embrionaria, significa las características biológicas, bioquímicas, fisiológicas y visuales de una célula, incluyendo la expresión de ciertos genes y marcadores de superficie celular, que pueden medirse o evaluarse para confirmar su identidad o función(es).
Un ejemplo de un agente de reprogramación adecuado de acuerdo con realizaciones preferidas de la divulgación es MUSASHI1. En algunas realizaciones, este polipéptido es preferido para conducir una primera célula, tal como un fibroblasto, en una célula similar a célula madre neural (NSLC). En otras realizaciones, el al menos un agente de reprogramación que aumenta dichos niveles intracelulares es (son) Musashil (Msí1) solo; Musashil (Msí1) y Neurogenina 2 (Ngn2); Musashil (Msí1) y proteína 2 del dominio de unión a metil-CpG (MBD2); o neurogenina 2 (Ngn2) y proteína 2 del dominio de unión a metil-CpG (MBD2). Se prefieren niveles intracelulares adecuados de estos polipéptidos ya que tienden a expresarse a lo largo de un linaje celular completo, desde tan pronto como células madre embrionarias (o incluso antes) a células presomáticas (o incluso más tarde).
MBD2 es un miembro de una familia de proteínas de unión a metil-CpG que se ha indicado que es un represor transcripcional y una ADN desmetilasa (dMTasa). Como se usa en el presente documento, el término "MBD2" generalmente se refiere a la proteína 2 del dominio de unión a metil-CpG humana. El número de referencia de GeneBank™ (NCBI) del MBd2 humano es NM_003927.3/AF072242, el número de referencia de UniProt™ es NP-003918/Q9UBB5 y el número de referencia de UniGene™ es Hs.25674.
Como se usa en el presente documento, el término "Msil" generalmente se refiere al homólogo 1 de musashi humano. El número de referencia de GeneBank™ (NCBI) de Msil humano es NM_002442.2/AB012851, el número de referencia de UniProt™ es NP-002433/O43347 y el número de referencia de UniGene™ es Hs.158311.
Como se usa en el presente documento, el término "Ngn2" generalmente se refiere a la neurogenina 2 humana. El número de referencia de GeneBank™ (NCBI) del Ngn2 humana es NM_024019.2/BC036847, el número de referencia de UniProt™ es NP-076924/Q9H2A3 y el número de referencia de UniGene™ es Hs.567563.
Según aspectos adicionales, el procedimiento para transformar una célula de un primer tipo en una célula deseada de un tipo diferente comprende las etapas de:
1) poner en contacto la célula de un primer tipo con uno o más compuestos capaces de aumentar los niveles intracelulares de al menos un agente de reprogramación dentro de la célula y remodelar directa o indirectamente la cromatina y/o el ADN de la célula; o
2) poner en contacto la cromatina y/o el ADN de una célula de un primer tipo con un agente capaz de remodelar la cromatina y/o el ADN de la célula; y aumentar los niveles intracelulares de al menos un agente de reprogramación.
Según diversas realizaciones, la etapa 2) puede realizarse consecutivamente a la etapa 1), simultáneamente con la etapa 1), o antes de la etapa 1).
Según un aspecto particular, la divulgación se refiere a un procedimiento para obtener una célula de tipo célula madre (NSLC), que comprende:
- proporcionar una célula de un primer tipo que no es NSLC;
- aumentar los niveles intracelulares de al menos un polipéptido específico de células madre neurales, en el que el polipéptido es capaz de conducir directa o indirectamente la transformación de la célula del primer tipo en una NSLC; y
- poner en contacto la cromatina y/o el ADN de una célula de un primer tipo con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador y/o un inhibidor químico de la metilación del ADN. Con respecto a la segunda etapa, la expresión "remodelar la cromatina y/o el ADN" se refiere a cambios estructurales dinámicos en la cromatina. Estos cambios pueden variar desde cambios locales necesarios para la regulación transcripcional, a cambios globales necesarios para abrir la estructura de la cromatina o la segregación del cromosoma para permitir la transcripción del nuevo conjunto de genes característicos de la célula deseada de un tipo diferente, hasta el cierre de la estructura de la cromatina o la segregación de cromosomas para evitar la transcripción de ciertos genes que no son característicos de la célula deseada de un tipo diferente. En algunas realizaciones, la apertura de la estructura de la cromatina se refiere más específicamente a la acetilación de las histonas y la desmetilación del ADN, mientras que el cierre de la estructura de la cromatina se refiere más específicamente a la desacetilación de las histonas y la metilación del ADN.
Como se usa en el presente documento, "compuesto" se refiere a un compuesto capaz de realizar una función biológica deseada. El término incluye, pero sin limitación, ADN, ARN, proteína, polipéptidos, y otros compuestos, incluyendo factores de crecimiento, citocinas, hormonas o moléculas pequeñas. Como se usa en el presente documento, los compuestos capaces de remodelar la cromatina y/o el ADN incluyen, aunque no de forma limitativa, acetiladores de histonas, inhibidores de la desacetilación de histonas, desmetiladores del ADN, inhibidores de la metilación del ADN y combinación de los mismos.
"Inhibidor de la metilación del ADN" se refiere a un agente que puede inhibir la metilación del ADN. Los inhibidores de la metilación del ADN han demostrado la capacidad de restaurar la expresión génica suprimida. Los agentes adecuados para inhibir la metilación del ADN incluyen, pero no se limitan a, 5-azacitidina, 5-aza-2'-desoxicitidina, 1-p-D-arabinofuranosil-5-azacitosina y dihidro-5-azacitidina y zebularina (ZEB), BIX (inhibidor de la histona lisina metiltransferasa) y RG108.
"Inhibidor de la desacetilación de histonas" se refiere a un agente que evita la eliminación de los grupos acetilo de los restos de lisina de las histonas que, de otro modo, conducirían a la formación de una cromatina condensada y transcripcionalmente silenciada. Los inhibidores de la histona desacetilasa se clasifican en varios grupos, incluyendo: (1) ácidos hidroxámicos, tales como tricostatina (A), (2) tetrapéptidos cíclicos, (3) benzamidas, (4) cetonas electrófilas y (5) grupo de compuestos de ácido alifático, tales como fenilbutirato y ácido valproico. Los agentes adecuados para inhibir la desacetilación de histonas incluyen, aunque no de forma limitativa, ácido valproico (VPA), fenilbutirato tricostatina A (TSA), butirato de sodio y benzamidas. El VPA promueve el destino neuronal e inhibe el destino glial simultáneamente a través de la inducción de factores de transcripción neurogénicos, incluyendo NeuroD.
"Acetilador de histonas" se refiere a un agente que inserta grupos acetilo en los restos de lisina de las histonas que abren la cromatina y la convierten en un estado transcripcionalmente activo. Los agentes de acetilación de histonas adecuados incluyen, aunque no de forma limitativa, poliamina, CREB (proteína de unión al elemento cAMP) y BniP3. "Desmetilador de ADN" se refiere a un agente que elimina los grupos metilo del ADN y posee la capacidad de inhibir la hipermetilación y restaurar la expresión génica suprimida. Se espera que una desmetilasa active los genes eliminando los residuos metílicos represivos. Los desmetiladores de ADN adecuados incluyen, aunque no de forma limitativa, MBD2 y Gadd45b.
En algunas realizaciones, el agente de reprogramación tiene una o más de las siguientes funciones: disminuye la expresión de uno o más marcadores de células del primer tipo (por ejemplo, consulte la Tabla C) y/o aumenta la expresión de uno o más marcadores de la célula deseada del tipo diferente (por ejemplo, consulte la Tabla A). Las células que exhiben un marcador seleccionable para la célula deseada de un tipo diferente se seleccionan después y evalúan para las características de la célula deseada de un tipo diferente.
De acuerdo con la divulgación, la transformación en la célula deseada da como resultado la expresión estable de una pluralidad de genes secundarios cuya expresión es característica de las propiedades fenotípicas y/o funcionales de la célula deseada. Los genes cuya expresión es característica de las propiedades fenotípicas y/o funcionales de la célula deseada incluyen, pero sin limitación, los que figuran en la Tabla A.
En algunas realizaciones, La expresión de genes secundarios cuya expresión es característica de las propiedades fenotípicas y funcionales de los resultados celulares deseados en la expresión de marcadores definidos de acuerdo con la siguiente tabla

En algunas realizaciones, la transformación de una célula de un primer tipo en la célula deseada da como resultado una represión estable de una pluralidad de genes expresados típicamente en la célula del primer tipo. Ejemplos de tales genes suprimidos incluyen, aunque no de forma limitativa, los definidos en la Tabla C:
Tabla C: Ejemplos de genes suprimidos

continuación

En realizaciones preferidas, la represión estable de uno cualquiera o más de los genes enumerados en la Tabla C que se expresan en el primer tipo de célula también se caracteriza por la desaparición de los marcadores correspondientes (consulte la Tabla C).
Los expertos en la materia entenderán que existen muchas etapas alternativas para facilitar la reprogramación celular. Estos incluyen desestabilizar la estructura del citoesqueleto de la célula (por ejemplo, exponiendo la célula a citocalasina B), aflojar la estructura de la cromatina de la célula (por ejemplo, mediante el uso de agentes tales como 5 -azacitidina (5-Aza) y ácido valproico (VPA) o agentes desmetiladores del ADN, tal como MBD2), transfectar la célula con uno o más vectores de expresión que contienen al menos un ADNc que codifica un factor de transcripción neurogénico (por ejemplo, Msil o Ngn2), que utiliza un medio apropiado para la célula deseada de un tipo diferente y un medio de diferenciación apropiado para inducir el compromiso de diferenciación de la célula deseada de un tipo diferente, inhibir las vías represivas que afectan negativamente a la inducción al compromiso de la célula deseada de un tipo diferente, cultivar las células en un sustrato apropiado para la célula deseada de un tipo diferente (por ejemplo, laminina para NSLC o una superficie de baja unión para el cultivo de neuroesferas flotantes) y sembrar las células en un entorno al que la célula deseada de un tipo diferente ( o "similar a" una célula) normalmente estaría expuesta in vivo tal como la temperatura adecuada, el pH y el medio ambiente con poco oxígeno (por ejemplo, aproximadamente 2-5 % de O2). En diversas realizaciones, la divulgación abarca estos y otros procedimientos y técnicas relacionados para facilitar la reprogramación de células.
En consecuencia, el procedimiento de transformar una célula de un primer tipo en una célula de un segundo tipo diferente puede comprender etapas facultativas adicionales. En una realización, el procedimiento de transformación de una célula comprende además la etapa de pretratar la célula de un primer tipo con un disruptor del citoesqueleto. Tal como se usan en el presente documento, "citoesqueleto" se refiere a la red filamentosa de la F-actina, las cadenas ligera y pesada de la miosina, los microtúbulos y los filamentos intermedios (IF) compuestos por una de las tres subunidades químicamente distintas, actina, tubulina o una de varias clases de la proteína de los IF. En consecuencia, el término "disruptor del citoesqueleto" se refiere a cualquier molécula que pueda inhibir el citoesqueleto celular para desestabilizar la célula y, en consecuencia, eliminar los mecanismos de retroalimentación entre la forma de la célula y la función celular y nuclear. Los disruptores del citoesqueleto adecuados incluyen,
aunque no de forma limitativa, la familia de citocalasina de Ios inhibidores del citoesqueleto de actina, tal como citocalasina B o D, y Ios inhibidores de la miosina, tal como 2,3-butanediona monoxima. Tal tratamiento previo puede impulsar la reprogramación. En una realización preferida, la célula se cultiva en presencia de al menos un inhibidor del citoesqueleto un día antes, durante o después de la introducción de uno o varios factores de transcripción neurogénicos.
La colocación de la célula en condiciones para soportar la transformación de la célula deseada y/o mantener la célula en condiciones de cultivo que soportan la transformación de la célula deseada puede comprender cultivar la célula en un medio que comprende uno o más factores apropiados para inducir la expresión de las características morfológicas y funcionales de la célula deseada de un tipo diferente. En algunas realizaciones, uno o más factores son factores de reprogramación útiles para reprogramar una célula y estos factores de reprogramación se pueden usar soIos o en combinaciones.
En otras realizaciones, la etapa de cultivar la célula en un medio que comprende uno o más factores apropiados para inducir la expresión de las características morfológicas y funcionales de la célula deseada de un tipo diferente se lleva a cabo después o simultáneamente a las etapas iii) o ív), o después o simultáneamente a las etapas 1) o 2), como se ha definido anteriormente en el presente documento.
Los expertos en la técnica conocen muchos tipos diferentes de medios y muchos factores de reprogramación que pueden ser útiles para reprogramar una célula y estos factores de reprogramación se pueden usar soIos o en combinaciones. En diversas realizaciones, el factor de reprogramación se selecciona de acuerdo con la TABLA B. En algunas realizaciones, Ios factores de reprogramación tienen una o más de las siguientes funciones: disminuir la expresión de uno o más marcadores del primer tipo de célula y/o aumentar la expresión de uno o más marcadores de la célula deseada. Las células que exhiben un marcador seleccionable para la célula deseada se seleccionan y se evalúan para determinar su unipotencia, multipotencia, pluripotencia, o características similares (según corresponda).
En realizaciones particulares, las células se cultivan en medio sin suero antes, durante o después de cualquiera de las etapas i) a iv como se ha definido anteriormente en el presente documento, o durante o después de las etapas 1) o 2), como se ha definido anteriormente en el presente documento.
Obtención de células de tipo madre neurales (NSLC)
De acuerdo con las realizaciones preferidas para crear células de tipo madre neurales (NSLC), Ios procedimientos de la invención se llevan a cabo de manera que las células se traten con agentes seleccionados, compuestos y factores para promover la reprogramación y/o desdiferenciación hacia células de tipo madre (SLC). Dichas células somáticas reprogramadas pueden tratarse posteriormente con agentes y/o cultivarse en condiciones adecuadas para promover la reprogramación hacia las células de tipo madre neurales (NSLC) y la expansión de las NSLC a largo plazo. Las NSLC tienen el potencial de diferenciarse en células de tipo neuronal y/o glial, así como células neuronales y/o gliales, para el tratamiento potencial de enfermedades y lesiones neurológicas, tales como la enfermedad de Parkinson y la lesión de la médula espinal. Los procedimientos descritos en el presente documento también son útiles para producir células histocompatibles para terapia celular.
En consecuencia, algunos aspectos de la presente divulgación se refieren a la generación de neuronas a partir de un paciente individual, haciendo así posibles Ios trasplantes autólogos como modalidad de tratamiento para muchas afecciones neurológicas, incluyendo neurotraumatismo, ictus, enfermedades neurodegenerativas, tales como esclerosis múltiple, enfermedad de Parkinson, enfermedad de Huntington, enfermedad de Alzheimer. Así, la divulgación proporciona terapias neurológicas para tratar la enfermedad o el traumatismo de interés.
Por lo tanto, otro aspecto de la divulgación se refiere a un procedimiento para obtener una célula de tipo madre neural (NSLC), que comprende bien:
1) poner en contacto la célula de un primer tipo con uno o más polipéptidos reguladores de células madre neurales capaces de aumentar Ios niveles intracelulares de polipéptidos específicos de células madre neurales dentro de dicha célula y remodelar directa o indirectamente la cromatina y/o el ADN de la célula y conducir directa o indirectamente la transformación de la célula del primer tipo en una NSLC; o
2) poner en contacto la cromatina y/o el ADN de una célula de un primer tipo con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador de ADN, y/o un inhibidor de la metilación del ADN; y aumentar Ios niveles intracelulares de al menos un polipéptido específico de células madre neurales que es capaz de dirigir directa o indirectamente la transformación de la célula del primer tipo en una NSLC.
En realizaciones preferidas, la etapa 1) comprende aumentar Ios niveles intracelulares de un polipéptido MUSASHI1. Como se explicará más adelante, esto se puede lograr por diferentes medios, incluyendo, pero sin limitación, la expresión transitoria del polipéptido MUSASHI1, preferentemente, transfectando un vector de expresión que codifica el polipéptido.
En realizaciones preferidas, la etapa 2) comprende aumentar Ios niveles intracelulares de un polipéptido MBD2 o
tratar las células con VPA y 5-AZA. Como se explicará más adelante, esto se puede lograr por diferentes medios, incluyendo, pero sin limitación, expresión transitoria del polipéptido MBD2, preferentemente, transfectando un vector de expresión que codifica el (Ios) polipéptido (s) y/o tratando previamente y/o tratando las células con VPA y 5-AZA.
En una realización particular, la reprogramación de una célula de un primer tipo a otro tipo de célula que muestre al menos dos marcadores seleccionadles para células madre neurales requiere la transfección de la célula de un primer tipo con un vector que contiene un ADNc que codifica un factor de transcripción neurogénico y un desmetilador de ADN. Para mejorar la desdiferenciación, las células se exponen o se exponen previamente a uno o más agentes que inhiben la metilación del ADN, inhiben la desacetilación de histonas y/o interrumpen el citoesqueleto celular. Por ejemplo, la desdiferenciación se puede mejorar mediante el tratamiento previo de las células con un agente que interrumpe el citoesqueleto celular y, a continuación, la transfectación de las células con uno o más vectores que contienen dos factores de transcripción neurogénicos en presencia de un desmetilador de ADN y/o un inhibidor de la metilación del ADN y la desacetilación de histonas. El desacetilador de histonas, el inhibidor de la desacetilación de histonas, el desmetilador de ADN y/o un inhibidor de la metilación del ADN son como se ha definido previamente.
Como se ha definido anteriormente, el procedimiento puede comprender además una etapa preliminar de pre tratamiento de la célula de un primer tipo con un interruptor del citoesqueleto, como se ha definido anteriormente y/o cultivar la célula en un medio que comprende uno o más factores de reprogramación apropiados para el aspecto y el mantenimiento de las características morfológicas y funcionales de las NSLC como se ha definido anteriormente (por ejemplo, un compuesto retinoide, un factor neurotrófico, bFGF, EGF, SHH, Wnt 3a, neuropéptido Y, estrógeno). En alguna realización, el procedimiento comprende además inhibir las rutas de señalización de BMP celulares (por ejemplo, por NOGGINA, fetuína o folistatina).
En realizaciones preferidas, la generación de una NSLC a partir de una primera célula comprende el uso de uno o más agentes de reprogramación. Los agentes adecuados incluyen, aunque no de forma limitativa, Musashi-1 (MsI1) y Neurogenina 2 (Ngn2). Otros agentes potenciales se enumeran en la Tabla A y B.
La presente divulgación también se refiere al uso de vectores de expresión de ADN que codifican una proteína o transcrito que regula por aumento la expresión de neurogénesis. La secuencia de ADN modificada genéticamente, codificar un agente de reprogramación definido, tal como MsI1 y Ngn2, se puede introducir en las células utilizando vectores monocistrónicos, bicistrónicos o policistrónicos. La expresión de un gen de multipotencia endógeno indica que el ADNc codifica una proteína cuya expresión en la célula resulta directa o indirectamente en la desdiferenciación de la célula. Las células de mamífero recién desdiferenciadas son capaces de volver a diferenciarse en linajes neuronales para regenerar dichas células de mamífero, tejidos y órganos.
La presente divulgación se refiere además a un procedimiento para generar NSLC mediante la Introducción de una secuencia de ADN modificada genéticamente en células somáticas humanas mediante transfección transitoria. Dado que el ADN introducido en el proceso de transfección no se inserta en el genoma nuclear, el ADN extraño disminuye con el tiempo y cuando las células sufren mitosis. Los vectores no virales permanecen en forma no replicativa, tienen baja inmunogenicidad y son fáciles y seguros de preparar y usar. Adicionalmente, Ios plásmidos pueden alojar fragmentos grandes de ADN.
En una realización particular, el procedimiento comienza con la obtención de células del individuo y la reprogramación de células in vitro para generar NSLC. El aspecto significativo de la presente invención es la reprogramación estable de una célula no neuronal en una NSLC que puede dar lugar a diferentes tipos de células neuronales o gliales (Incluidas células de tipo neuronal o de tipo glial). Estos se pueden volver a implantar en el mismo paciente del que se obtuvieron las células, haciendo así una modalidad de tratamiento autólogo para muchas afecciones neurológicas, incluyendo neurotraumatismo, ictus y posible enfermedad neurodegenerativa. Estos también pueden implantarse en un individuo diferente del que se obtuvieron las células. En consecuencia, Ios procedimientos de la presente invención pueden ser útiles para tratar, prevenir o estabilizar una enfermedad neurológica, tal como enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis múltiple o lesión de la médula espinal. Esta tecnología proporciona una amplia fuente de células madre neurales, células neuroprogenitoras, neuronas y glia para tratamiento clínico, que se pueden realizar mediante la implantación de NSLc in vivo o induciendo la diferenciación in vitro y la implantación de células neuroprogenitoras o neuronas específicas o glía in vivo.
En otra realización, el procedimiento comprende aislar células somáticas o no neuronales y exponer las células a uno o más agentes que alteran la morfología celular y la estructura de la cromatina, y transfectar las células con uno o más genes que contienen al menos un ADNc que codifica un factor de transcripción neurogénico. La etapa de transfección de genes puede reemplazarse con agentes alternativos que inducen la expresión del (de Ios) factor(es) de transcripción neurogénico(s) en la célula. La Inducción de modificaciones epigenéticas en el ADN y las histonas (especialmente la desmetilación del ADN y una estructura cromática abierta) facilitan la verdadera reprogramación de las células. En otra realización, las células se incuban en un ambiente de poco contenido en oxígeno, por ejemplo, 5 % de O2, ayudando de esta manera a la reprogramación de las células.
Esta metodología permite la reprogramación de una célula en una NSLc . El curso posterior del desarrollo y la
expansión de la célula reprogramada dependen de las señales del entorno in situ a las que está expuesta. Las realizaciones de la divulgación incluyen además cultivar la célula reprogramada en un medio de proliferación apropiado para expandir la NSLC generada, por ejemplo, medio de proliferación de progenitores neurales (StemCell Technologies) con presencia de factor de crecimiento epidérmico (EGF) y factor de crecimiento de fibroblastos básico (bFGF), para promover la proliferación de células madre neurales.
Las NSLC obtenidas de acuerdo con la invención se pueden diferenciar en linajes neuronales, astrocitos, y/u oligodendrocitos en medio de diferenciación apropiado, por ejemplo, medio de diferenciación NS-A (StemCell, Technologies) o medio NbActive (BrainBits ™) que incluye un compuesto retinoide, tal como el ácido todo-transretinoico o vitamina A, y BDNF, para inducir la diferenciación de NSLC hacia células neuronales y/o gliales. Las células neuronales incluyen células que muestran una o más características morfológicas, fisiológicas, funcionales y/o inmunológicas específicas neurales asociadas a un tipo de célula neuronal. Los criterios útiles incluyen: características morfológicas (por ejemplo, procesos largos o neuritas), características fisiológicas y/o inmunológicas, tal como la expresión de un conjunto de marcadores o antígenos específicos de neuronas, síntesis de uno o más neurotransmisores, tales como dopamina o ácido gamma aminobutírico (GABA), y características funcionales, tales como canales iónicos o Ios potenciales de acción característicos de las neuronas.
De acuerdo con el procedimiento, las células reprogramadas pueden seleccionarse en función de las propiedades de adherencia diferencial en comparación con las células no transfectadas; por ejemplo, las células reprogramadas pueden formar neuroesferas flotantes o crecer bien en laminina, mientras que Ios fibroblastos no transfectados se adhieren y crecen bien en placas normales tratadas con cultivos celulares. Las células reprogramadas incluyen células que muestran uno o más marcadores y morfología específicos de las células madre y la pérdida de algunos o todos Ios marcadores específicos relacionados con las células originales. Adicionalmente, parte de la funcionalidad de las células de tipo neural (NLC) se puede evaluar en diferentes puntos de tiempo mediante, por ejemplo, pinzamiento zonal, inmunotinción para sinaptofisina y MAP2b, y por medios inmunoquímicos, tal como mediante un ensayo inmunoabsorbente ligado a enzimas (ELISA).
En determinadas realizaciones, la presente invención proporciona NSLC que pueden iniciar y dirigir la regeneración del sistema nervioso central en un sitio de daño tisular y pueden personalizarse para pacientes individuales utilizando sus propias células como el donante o la célula de partida. La presente invención se puede usar para generar células de un paciente individual, haciendo así posibles Ios trasplantes autólogos como modalidad de tratamiento para muchas afecciones neurológicas. Así, esta tecnología elimina Ios problemas asociados con Ios trasplantes de células no huésped, tales como, rechazo inmunológico y el riesgo de enfermedad transmitida. La gran ventaja de la presente invención es que proporciona un suministro esencialmente ilimitado para injertos autólogos adecuados para trasplante. Por lo tanto, evitará algunos problemas significativos asociados con la fuente actual de materiales y procedimientos de trasplante.
Administración de polinucleótidos
En determinadas realizaciones, la invención se refiere al uso de polinucleótidos, por ejemplo, un polinucleótido que codifica un polipéptido MBD2, un polipéptido MUSASHI1 y/o un polipéptido Ngn2. Los medios para introducir polinucleótidos en una célula son bien conocidos en la técnica. Se pueden usar procedimientos de transfección de una célula, tal como nucleofección y/o lipofección, u otros tipos de procedimientos de transfección. Por ejemplo, un polinucleótido que codifica un polipéptido deseado puede clonarse en vectores intermedios para la transfección en células eucarióticas para la replicación y/o expresión. Los vectores intermedios para el almacenamiento o la manipulación del ácido nucleico o la producción de proteínas pueden ser vectores procariotas, (por ejemplo, plásmidos), vectores lanzadera, vectores de insectos o vectores virales, por ejemplo. Un polipéptido deseado también puede codificarse por un ácido nucleico de fusión.
Para obtener la expresión de un ácido nucleico clonado, por lo general, se subclona en un vector de expresión que contiene un promotor para dirigir la transcripción. Los promotores bacterianos y eucariotas adecuados son bien conocidos en la técnica y se describen, por ejemplo, en Sarnbrook y Russell (Molecular Cloning: a laboratory manual, Cold Spring Harbor Laboratory Press). El promotor utilizado para dirigir la expresión de un ácido nucleico de elección depende de la aplicación particular. Por ejemplo, típicamente se utiliza un promotor constitutivo fuerte para la expresión y purificación. En cambio, cuando una proteína o compuesto de desdiferenciación se va a utilizar in vivo, se utiliza un promotor o compuesto constitutivo o inducible, dependiendo del uso particular de la proteína. Además, se puede usar un promotor débil, tal como HSV TK o un promotor que tiene una actividad similar. El promotor normalmente también puede incluir elementos que responden a la transactivación, por ejemplo, elementos de respuesta a la hipoxia, elementos de respuesta a Ga14, elemento de respuesta del represor lac y sistemas de control de moléculas pequeñas, tales como Ios sistemas regulados por tet y el sistema RU-486.
Además de un promotor, un vector de expresión típicamente contiene una unidad de transcripción o casete de expresión que contiene elementos adicionales requeridos para la expresión del ácido nucleico en células huésped, ya sea procariota o eucariota. Un casete de expresión típico, por lo tanto, contiene un promotor unido operativamente, por ejemplo, a la secuencia de ácido nucleico, y señales requeridas, por ejemplo, para la poliadenilación eficiente del transcrito, la terminación transcripcional, la unión al ribosoma y/o la terminación de la traducción. Los elementos adicionales del casete pueden incluir, por ejemplo, potenciadores y señales intrónicas de
corte y empalme heterólogas.
Los vectores de expresión que contienen elementos reguladores de virus eucariotas se utilizan a menudo en vectores de expresión eucariotas, por ejemplo, vectores SV40, vectores del virus del papiloma y vectores derivados del virus de Epstein-Barr. Otros ejemplos de vectores eucariotas incluyen pMSG, pAV009/A+, pMTO10/A+, pMAMneo-5, baculovirus pDSVE y cualquier otro vector que permita la expresión de proteínas bajo la dirección del promotor temprano de SV40, promotor tardío de SV40, promotor de la metalotioneína, promotor del virus del tumor mamario murino, promotor del virus del sarcoma de Rous, promotor de polihedrina u otros promotores que se muestran efectivos para la expresión en células eucariotas.
Los procedimientos de transfección estándar se pueden utilizar para producir líneas celulares de bacterias, mamífero, levadura, insecto, u otras que expresan grandes cantidades de proteínas de desdiferenciación, que se pueden purificar, si se desea, utilizando técnicas estándar. La transformación de células eucariotas y procariotas se realiza de acuerdo con técnicas estándar.
Se puede usar cualquier procedimiento para introducir secuencias de nucleótidos extrañas en células huésped. Estos incluyen, aunque no de forma limitativa, el uso de la transfección con fosfato de calcio, transfección mediada por DEAE-dextrano, polibreno, fusión de protoplastos, electroporación, administración mediada por lípidos (por ejemplo, liposomas), microinyección, bombardeo de partículas, introducción de ADN desnudo, vectores plasmídicos, vectores virales (tanto episomales como integradores) y cualquiera de los otros procedimientos bien conocidos para introducir ADN genómico clonado, ADNc, ADN sintético u otro material genético extraño en una célula huésped (véase, por ejemplo, Sarnbrook y col., citado anteriormente. Solo es necesario que el procedimiento de ingeniería genérica concreta usado pueda introducir con éxito al menos un gen en la célula huésped capaz de expresar la proteína de elección.
Se pueden usar procedimientos de transferencia génica basados en virus y no virales convencionales para introducir ácidos nucleicos en células o tejidos diana de mamífero. Tales procedimientos pueden usarse para administrar ácidos nucleicos que codifican polipéptidos de reprogramación a células in vitro. Preferentemente, los ácidos nucleicos se administran para usos de terapia génica in vivo o ex vivo. Los sistemas de administración de vectores no virales incluyen plásmidos de ADN, ácido nucleico desnudo y ácido nucleico en complejos con un vehículo de administración tal como un liposoma. Los sistemas de administración de vectores virales incluyen virus de ADN y ARN, que tienen genomas episomales o integrados después de la administración a la célula.
Los procedimientos de administración no viral de ácidos nucleicos incluyen lipofección, microinyección, balística, virosomas, liposomas, inmunoliposomas, policatión o conjugados de lípido-ácido nucleico, ADN desnudo, viriones artificiales, y captación de ADN potenciada por agentes. Los reactivos de lipofección se venden comercialmente (por ejemplo, Transfectam™ y Lipofectin™). Se conocen lípidos catiónicos y neutros adecuados para la lipofección eficaz de polinucleótidos de reconocimiento de receptores. El ácido nucleico se puede administrar a las células (administración ex vivo) o a los tejidos diana (administración in vivo). La preparación de complejos de lípidos:ácidos nucleicos, incluyendo liposomas dirigidos tales como complejos inmunolipídicos, es de sobra conocida por los expertos en la materia.
El uso de sistemas basados en virus de ARN o ADN para la administración de ácidos nucleicos aprovecha los procesos altamente evolucionados para dirigir un virus a células específicas del cuerpo y el tráfico de la carga viral al núcleo. Los vectores virales pueden administrarse directamente a los pacientes (in vivo) o pueden usarse para tratar células in vitro, en el que las células modificadas se administran a pacientes (ex vivo). Los sistemas basados en virus convencionales para la administración incluyen vectores retrovirales, lentivirales, poxvirales, adenovirales, virus adenoasociados, virus de la estomatitis vesicular y herpesvirales, aunque la integración en el genoma del huésped es posible con ciertos vectores virales, incluyendo procedimientos de transferencia génica de retrovirus, lentivirus, virus adenoasociados, lo que a menudo da como resultado la expresión a largo plazo del transgén insertado. Adicionalmente, se han observado altas eficiencias de transducción en diferentes tipos de células y tejidos diana. pLASN y MFG-S son ejemplos de vectores retrovirales que se han utilizado en ensayos clínicos. En aplicaciones para las que se prefiere la expresión transitoria, los sistemas basados en adenovirus son útiles. Los vectores basados en adenovirus son capaces de una eficiencia de transducción muy alta en muchos tipos de células y son capaces de infectar y, por lo tanto, administrar ácido nucleico a células tanto en división como células que no están en división. Con tales vectores, se han obtenido títulos y niveles de expresión altos. Los vectores de adenovirus se pueden producir en grandes cantidades en un sistema relativamente simple.
Los vectores de terapia génica pueden administrarse in vivo mediante la administración a un paciente individual, típicamente por administración sistémica (por ejemplo, intravenosa, intraperitoneal, intramuscular, infusión subdérmica o intracraneal) o aplicación tópica. Como alternativa, los vectores pueden administrarse a las células ex vivo, tales como las células explantadas de un paciente individual (por ejemplo, linfocitos, aspirados de médula ósea, biopsia de tejido) o células madre hematopoyéticas de donantes universales, seguido de la reimplantación de las células en un paciente, por lo general, después de la selección de las células que han sido reprogramadas.
La transfección celular ex vivo para diagnóstico, investigación o para terapia génica (por ejemplo, a través de la
reinfusión de las células transfectadas en el organismo huésped) es bien conocida por Ios expertos en la técnica. En una realización preferida, las células están aisladas del organismo sujeto, se transfectan con un ácido nucleico (gen o ADNc) y se vuelven a infundir en el organismo sujeto (por ejemplo, paciente). Los expertos en la técnica conocen bien diversos tipos de células adecuados para la transfección ex vivo.
Los vectores (por ejemplo, retrovirus, adenovirus, liposomas, etc.) que contienen ácidos nucleicos terapéuticos también se pueden administrar directamente al organismo para la transfección de células in vivo. Como alternativa, se puede administrar ADN desnudo. La administración se realiza por cualquiera de las rutas que normalmente se utilizan para introducir una molécula en contacto final con células de la sangre o tejidos. Los procedimientos adecuados para administrar tales ácidos nucleicos están disponibles y son bien conocidos por Ios expertos en la técnica, y, aunque se puede usar más de una ruta para administrar una composición particular, una ruta particular a menudo puede proporcionar una reacción más inmediata y más efectiva que otra ruta.
Los vehículos farmacéuticamente aceptables se determinan en parte por la composición particular que se administra, así como por el procedimiento particular utilizado para administrar la composición. En consecuencia, existe una amplia variedad de formulaciones adecuadas de composiciones farmacéuticas.
Administración de polipéptidos
En la mayoría, si no todos Ios procedimientos descritos en el presente documento, una posibilidad alternativa consiste en evitar el uso de un polinucleótido y poner en contacto una célula de un primer tipo de célula directamente con un compuesto (por ejemplo, un polipéptido) para el cual se desea un nivel intracelular aumentado. En otras realizaciones, por ejemplo en ciertas situaciones in vitro, las células se cultivan en un medio que contiene uno o más polipéptidos funcionales.
Un factor importante en la administración de polipéptidos es asegurar que el polipéptido tenga la capacidad de atravesar la membrana plasmática de una célula o la membrana de un compartimento intracelular como el núcleo. Las membranas celulares están compuestas por bicapas lipídicas-proteicas que son libremente permeables a pequeños compuestos lipófilos no iónicos y son inherentemente impermeables a Ios compuestos polares, macromoléculas y agentes terapéuticos o diagnósticos. Sin embargo, se han descrito proteínas, lípidos y otros compuestos, que tienen la capacidad de translocar polipéptidos a través de una membrana celular. Por ejemplo, Ios "polipéptidos de translocación de membrana" tienen subsecuencias de aminoácidos anfifílicos o hidrófobos que tienen la capacidad de actuar como vehículos de translocación de membrana. Los polipéptidos para Ios que se desea un aumento del nivel intracelular según la invención pueden unirse a secuencias peptídicas adecuadas para facilitar su captación en las células. Otros restos químicos adecuados que proporcionan una captación celular mejorada también pueden unirse, tanto de forma covalente como de forma no covalente, a Ios polipéptidos. También se pueden usar otros vehículos adecuados que tienen la capacidad de transportar polipéptidos a través de las membranas celulares.
Un polipéptido deseado también se puede introducir en una célula animal, preferentemente una célula de mamífero, a través de liposomas y derivados de liposomas tales como inmunoliposomas. El término "liposoma" se refiere a vesículas que comprenden una o más bicapas lipídicas ordenadas concéntricamente, que encapsulan una fase acuosa. La fase acuosa contiene típicamente el compuesto que se administrará a la célula. En determinadas realizaciones, puede ser deseable apuntar a un liposoma usando restos de direccionamiento que son específicos para un tipo de célula particular, tejido, y similares. El direccionamiento de Ios liposomas utilizando diversos restos de orientación (por ejemplo, ligandos, receptores y anticuerpos monoclonales) se ha descrito previamente.
Células v líneas celulares
La divulgación abarca las células, líneas celulares, células madre y preparaciones de células purificadas derivadas de cualquiera de Ios procedimientos descritos en el presente documento. En algunas realizaciones, las células, líneas de células, células madre y preparaciones de células purificadas de la divulgación son de origen mamífero, incluyendo, pero sin limitaciones, seres humanos, primates, roedor, perro, gatos, caballo, vaca u oveja. En realizaciones preferidas, proceden de un ser humano.
En consecuencia, otro aspecto de la divulgación se refiere a células modificadas, líneas celulares, células pluripotenciales, multipotenciales o unipotenciales y preparaciones de células purificadas, en las que cualquiera de estas células comprende un polinucleótido exógeno que codifica Musashil (Msí1); Msí 1 y Ngn2; Msil y MBD2; y Ngn2 y MBD2; Msil, Ngn2 y MBD2; Msí 1, Ngn2, Nestina y MBD2; y otras combinaciones potenciales de la Tabla A que incluyen, preferentemente, Msí 1 y Ngn2 y MBD2. En realizaciones preferidas, la célula de acuerdo con la divulgación es una célula de tipo madre, más preferentemente una célula de tipo célula madre neural (NSLC), poseyendo la célula una o más de las siguientes características:
- expresión de uno o más marcadores de células madre neurales seleccionados del grupo que consiste en Sox2, nestina, GFAP, Msil y Ngn2;
- disminución de la expresión de uno o más genes específicos de la célula de la que se obtuvo la NSLC (por ejemplo, consulte la Tabla C);
- forma neuroesferas en el ensayo de formación de colonias de neuroesferas;
- que se puede cultivar en suspensión o como un cultivo adherente;
- capaz de proliferar sin la presencia de un agente de reprogramación exógeno durante más de 1 mes, preferentemente durante 2 meses, durante 3 meses, durante 5 meses e incluso durante más de un año;
- capaz de dividirse cada 36 horas a un pase bajo;
- positiva para la actividad telomerasa;
- capaz de diferenciarse en una célula de tipo neuronal, célula de tipo astrocito, célula de tipo oligodendrocito y combinaciones de las mismas;
- disminución de la expresión de la telomerasa y uno o más marcadores de células madre neurales después de la diferenciación;
- tienen uno o más procesos morfológicos de tipo neurita (axones y/o dendritas) de más de un diámetro de célula de longitud después de la diferenciación en una célula de tipo neuronal;
- expresión de al menos un antígeno neural específico seleccionado del grupo que consiste en tubulina neural específica, proteína 2 asociada a los microtúbulos, NCAM, y marcador para un neurotransmisor después de la diferenciación en una célula de tipo neuronal;
- expresión de uno o más marcadores neurales funcionales (por ejemplo, sinapsina) después de la diferenciación en una célula de tipo neuronal;
- capaz de liberar uno o más factores neurotróficos (por ejemplo, BDNF) después de la diferenciación en una célula de tipo neuronal;
- negativo en un ensayo de formación de colonias de tumores;
- negativo para el crecimiento de tumores en ratones SCID;
- negativo para el crecimiento de teratoma en ratones SCID;
- capaz de mejorar significativamente una o más medidas funcionales después de la colocación de un número adecuado de NSLC en el hueco en un modelo de ablación cerebral;
- capaz de mejorar significativamente o mantener una o más medidas funcionales después de inyectar un número adecuado de NSLC en un modelo de EAE; y
- capaz de mejorar una o más medidas funcionales más significativamente que las hNPC en lesiones del SNC o modelos neurodegenerativos.
Ejemplos de todos los puntos anteriores se pueden encontrar en la sección Eemplos de esta solicitud.
En realizaciones preferidas, una NSLC posee todas las características siguientes:
- capacidad de autorrenovarse durante un tiempo significativamente mayor que una célula somática;
- no es una célula cancerosa;
- es estable y no se mantiene artificialmente por la expresión génica forzada o por medios similares y se puede mantener en medios de células madre neuronales estándar;
- puede diferenciarse en una célula progenitora, precursora, somática o en otro tipo de célula más diferenciada del mismo linaje;
- tiene las características de una célula madre y no solo ciertos marcadores o expresión génica o aspecto morfológico; y
- no exhibe crecimiento descontrolado, formación de teratoma y formación de tumores in vivo.
En una realización particular, las células reprogramadas (NSLC) son capaces de proliferar durante varios meses sin perder sus marcadores de células madre neurales y su capacidad para diferenciarse en células de tipo neurona, de tipo astrocito y de tipo oligodendrocito. La generación de los linajes neurales se caracteriza por su morfología, cambios fenotípicos y funcionalidad.
En algunas realizaciones, las células pueden tener una o más de las siguientes características y propiedades: autorrenovación, diferenciación multilinaje in vitro e in vivo, clonogenicidad, un cariotipo normal, extensa proliferación in vitro en condiciones de cultivo bien definidas y la capacidad de congelarse y descongelarse, así como cualquiera de las propiedades o características conocidas y/o deseadas habitualmente típicas de las células madre. Las células pueden expresar además marcadores moleculares de células multipotenciales o pluripotenciales (es decir, marcadores génicos y de superficie tal como se ha definido anteriormente).
Otro aspecto de la divulgación se refiere a la producción de células madre y/o progenitoras autólogas (propias) específicas del tejido. Estas células madre y/o progenitoras pueden usarse en aplicaciones de terapia celular para tratar enfermedades de degeneración celular. Las enfermedades de degeneración celular incluyen, por ejemplo, enfermedades neurodegenerativas, tales como ictus, enfermedad de Alzheimer, enfermedad de Parkinson, esclerosis múltiple, esclerosis lateral amiotrófica, degeneración macular, enfermedades osteolíticas, tales como osteoporosis, osteoartritis, fracturas óseas, roturas de huesos, diabetes, lesión hepática, enfermedades degenerativas, infarto de miocardio, quemaduras y cáncer. Se prevé que las células de acuerdo con la divulgación se puedan implantar o trasplantar en un huésped. Una ventaja de la invención es que se pueden producir grandes cantidades de células madre autólogas para la implantación sin el riesgo de un rechazo mediado por el sistema inmunitario. Esas células pueden conducir a la producción de tejido adecuado para el trasplante en el individuo. Dado que el tejido deriva del receptor de trasplante, no debe estimular una respuesta inmunitaria, como lo haría el tejido de un donante no relacionado. Dichos trasplantes pueden constituir tejidos (por ejemplo, vena, arteria, piel, músculo), trasplantes de órganos sólidos (por ejemplo, corazón, riñón, hígado), trasplantes de células neuronales o
trasplantes de médula ósea, tales como Ios que se utilizan en el tratamiento de diversas neoplaslas malignas, tales como, por ejemplo, leucemias y linfomas. También se pueden usar trasplantes de células madre neurales, neuroprogenitoras o neuronales (así como las NSLC y sus derivados) en el tratamiento de, por ejemplo, trastornos neurológicos, ictus, lesión de la médula espinal, enfermedad de Parkinson y similares, así como potencialmente algunos trastornos no neurológicos, tales como un infarto cardíaco.
Otro aspecto de la divulgación se refiere a un procedimiento para producir tejidos modificados por ingeniería ex vivo para su posterior implantación o trasplante en un huésped, en el que Ios componentes celulares de esos tejidos modificados por ingeniería comprenden células de acuerdo con la divulgación o células derivadas de Ios mismos. Por ejemplo, Ios cultivos expandidos de las células de la divulgación pueden diferenciarse mediante tratamiento in vitro con factores de crecimiento y/o morfógenos. A continuación, se implantan poblaciones de células diferenciadas en el huésped receptor cerca del sitio de la lesión o el daño, o se cultivan in vitro para generar tejidos modificados mediante ingeniería genética, como se ha descrito.
Los procedimientos y las células de la divulgación descritos en el presente documento se pueden usar para inmortalizar células, por ejemplo para generar una línea celular. Usando Ios procedimientos desvelados en el presente documento, puede transformarse una célula somática en una que posee un fenotipo desdiferenciado, facilitando así la generación de líneas celulares a partir de diversos tejidos. Por lo tanto, la divulgación abarca tales células inmortalizadas.
Además, Ios procedimientos de derivación de las células pueden ser útiles en aplicaciones científicas y terapéuticas que incluyen, pero sin limitación, (a) descubrimiento científico e investigación que involucre el desarrollo celular y la investigación genética (por ejemplo, Ios usos en lugar de las células madre humanas como una línea celular modelo para estudiar la diferenciación, desdiferenciación, o reprogramación de células humanas), (b) desarrollo y descubrimiento de medicamentos (por ejemplo, detección de la eficacia y toxicidad de ciertos medicamentos candidatos y productos químicos, detección de posibles fármacos o agentes que median en la diferenciación, desdiferenciación, o reprogramación de células), (c) terapia génica (por ejemplo, como un dispositivo de administración para terapia génica), y (d) tratamiento de lesiones, traumatismos, enfermedades y trastornos, que incluyen, pero sin limitación, enfermedad de Parkinson, enfermedad de Alzheimer, lesión de la médula espinal e ictus.
Aspectos adicionales se refieren a procedimientos terapéuticos, procedimientos de tratamiento y procedimientos de regeneración de un tejido u órgano en un mamífero (por ejemplo, un sujeto humano). Un procedimiento particular se refiere a un procedimiento para regenerar un tejido u órgano de mamífero que comprende poner en contacto el tejido u órgano a regenerar con una SLC, NSLC u otra célula deseada o construcción de tejido artificial tal como se define en el presente documento. Las SLC, NSLC, célula deseada o la construcción de tejido artificial pueden colocarse cerca del tejido u órgano que se va a regenerar administrando al sujeto usando cualquier vía adecuada (por ejemplo, inyectando la célula por vía intratecal, directamente en el tejido u órgano, o en el torrente sanguíneo).
Otro procedimiento para reparar o regenerar un tejido u órgano en un sujeto que lo necesite comprende administrar al sujeto un compuesto que induce una expresión endógena directa o indirecta de al menos un regulador génico en células del tejido u órgano y/o un compuesto que induce una expresión endógena directa o indirecta de al menos un regulador génico en células capaces de transformación o desdiferenciación in vivo en el sujeto. En consecuencia, la expresión de al menos un regulador génico reprograma las células en células deseadas de un tipo diferente (por ejemplo, células de tipo madre neurales), y estas células de un tipo diferente son efectivas para reparar o regenerar dicho tejido u órgano.
Otro procedimiento comprende obtener células o tejido de un paciente (por ejemplo, células madre hematopoyéticas, fibroblastos o queratinocitos), reprogramar una pluralidad de tales células o el tejido, y reintroducir las células reprogramadas o el tejido en el paciente. Un aspecto relacionado se refiere a composiciones farmacéuticas que comprenden una pluralidad de una célula deseada, SLC y/o células de tipo madre neurales (NSLC) o tejido reprogramado como se define en el presente documento.
Los procedimientos terapéuticos de la divulgación pueden ser aplicables a la regeneración o reparación de diversos tejidos y órganos, incluidos, pero sin limitación, el cerebro, la médula espinal y la retina.
Un aspecto relacionado se refiere a composiciones farmacéuticas que comprenden una pluralidad de células deseadas, SLC y/o células de tipo madre neurales (NSLC) como se define en el presente documento.
Tejidos
Otro aspecto de la divulgación se refiere a un tejido que contiene células reprogramadas como se define en el presente documento que puede implantarse en un sujeto que lo necesite.
En algunas realizaciones, la presente divulgación proporciona la reprogramación de células dentro de un tejido, por ejemplo, una construcción de tejido 3D producida in vitro que comprende células y matriz extracelular producida por estas células. Además, las células transfectadas se pueden sembrar sobre estas construcciones de tejido 3D que se pueden hacer completamente de forma autóloga, evitando así el rechazo de huésped, haciéndolo completamente
inmunocompatible y como vehículo para transplantar células reprogramadas in vivo. Ventajosamente, estas células recién creadas se pueden usar en su estado indiferenciado y/o diferenciado dentro de estos tejidos para fines de diagnóstico in vitro o se pueden trasplantar a un paciente que necesite tal construcción en enfoques de terapia celular/reemplazo de tejido.
La divulgación abarca además tejido multicapa 3D de tipo neuronal. Las células dentro de CDM reprogramadas en células de tipo madre neurales de acuerdo con la divulgación se diferencian fácilmente en células de tipo neuronales, células de tipo astrocito y células de tipo oligodendrocito dentro de la CDM. Por lo tanto, es posible usar CDM y los procedimientos de reprogramación de la invención para reprogramar las células dentro de la CDM para formar tejido multicapa 3D de tipo neuronal (hasta> 30 capas de células). Dicho tejido 3D comprende neuronas (o específicamente, células de tipo neuronas), astrocitos (o específicamente, células de tipo astrocito), y oligodendrocitos (o específicamente, células de tipo oligodendrocitos) y puede hacerse completamente de forma autóloga, pueden manejarse manualmente e implantarse con relativa facilidad, o pueden usarse como un modelo in vitro de tejido del SNC.
Un aspecto particular se refiere a una construcción de tejido artificial que comprende un ensamblaje 3D de células cultivadas in vitro y matriz extracelular producida por estas células. Las células pueden ser células deseadas, SLC y/o una pluralidad de células de tipo madre neurales (NSLC) obtenidas utilizando cualquiera de los procedimientos descritos en el presente documento.
Procedimientos de detección selectiva
Otro aspecto de la divulgación se refiere a procedimientos para identificar nuevos compuestos (por ejemplo, moléculas pequeñas, medicamentos, etc.) capaces de transformar una célula de un primer tipo en una célula deseada de un tipo diferente. Estos nuevos compuestos pueden ser útiles para fines de investigación o como medicamentos para uso en la reparación o regeneración de tejidos en un sujeto.
La sección de ejemplos proporciona principios, procedimientos y técnicas útiles para seleccionar e identificar tales compuestos activos deseables. Por ejemplo, los expertos en la materia entenderán que es posible detectar compuestos que induzcan la transformación de una célula de un primer tipo a una NSLC al reemplazar la "inducción" o la "actividad biológica" proporcionada por el aumento transitorio de Musashil, NGN2 o MBD2 en la célula por un compuesto candidato a analizar (por ejemplo, una biblioteca de pequeñas moléculas o compuestos) y la medición de la actividad o eficacia del compuesto candidato para generar la NSLC. Los individuos o la mezcla de compuestos activos se seleccionarán si tienen la misma actividad y/o si pueden proporcionar los mismos o similares efectos que estos polipéptidos (por ejemplo, la transformación celular y/o la aparición de cualquier marcador deseable o características deseables como se ha definido anteriormente en el presente documento). Por ejemplo, un compuesto o mezcla de compuestos capaces de transformar un fibroblasto en una NSLC podría identificarse mediante:
(i) La configuración, cultivo y transformación de los fibroblastos en NSLC como en el Ejemplo 1;
(ii) la selección de una biblioteca de compuestos reemplazando MsI1, Ngn2 y/o MBD2 con cada compuesto candidato en un pocilio diferente;
(ill) la identificación de un compuesto "de éxito" cuando el compuesto candidato sea capaz de transformar los fibroblastos en NSLC aproximadamente tan bien como en MsI1, Ngn2 y/o MBD2 reemplazados;
(Iv) Si el compuesto de la parte (ill) no reemplazó a todos los MsI1, Ngn2 y MBD2, y no es capaz de transformar los fibroblastos en NSLC por sí mismos, incluyendo el compuesto de (ill) en cada pocilio, seleccionando una biblioteca de compuestos reemplazando el MsI1, Ngn2 y/o Mb D2 que no se eliminó en la parte (ii) con cada compuesto candidato en un pocilio diferente;
(v) la identificación de un compuesto "de éxito" cuando el compuesto candidato sea capaz de transformarse, junto con el compuesto de la parte (ill), Ios fibroblastos en NSLC aproximadamente tan bien como el MsI1, Ngn2 y/o MBD2 reemplazados;
(vI) si el compuesto de la parte (V) no reemplazó todos Ios MsI1, Ngn2 y/o MBD2, y no es capaz de transformar Ios fibroblastos en NSLC junto con el compuesto de la parte (ill), incluyendo el compuesto de (ill) y (v) en cada pocilio, seleccionando una biblioteca de compuestos reemplazando el MsI1, Ngn2 o MBD2 que no se eliminó en la parte (ii) y (Iv) con cada compuesto candidato en un pocilio diferente;
(vII) la identificación de un compuesto "de éxito" cuando el compuesto candidato sea capaz de transformarse, junto con el compuesto de las partes (ill) y (v), Ios fibroblastos en NSLC aproximadamente tan bien como el MsI1, Ngn2 o MBD2;
(vIII) una combinación de Ios compuestos de la parte (ill), (v) y (vII) podrán transformar Ios fibroblastos en NSLC; se pueden hacer modificaciones a estos compuestos y analizarlos después para identificar versiones más efectivas o seguras de estos compuestos.
Los mismos principios son aplicables para otros tipos deseados de células de tipo madre, incluidas las células de tipo pluripotencial, células tipo mesoendodérmico, células de tipo progenitoras pancreáticas, etc. Las Tablas A y B, y la sección de ejemplos proporcionan, para cada uno de estos tipos de células, una lista de genes y/o compuestos potenciales para considerar en tales procedimientos de selección.
En consecuencia, la presente divulgación abarca estos y cualquier procedimiento de detección equivalente en el que
Ios compuestos candidatos se prueban para determinar su eficacia en la transformación de una célula de un primer tipo en una célula deseada de un tipo diferente cuando se compara con la eficacia del factor de reprogramación y/o el regulador génico tal como se define en el presente documento.
Administración de factores neurotróficos
La administración local de factores neurotróficos se ha sugerido como un procedimiento para tratar varias afecciones neurológicas. Las estrategias que utilizan moléculas neurotróficas se centran en prevenir la pérdida progresiva de neuronas, mantener las conexiones y la función neuronales (neuroprotección) e inducir respuestas regenerativas adicionales en las neuronas, tale como el aumento de la rotación de Ios neurotransmisores y/o la brotación axonal (neuroregeneración). A día de hoy, se han explorado varias estrategias terapéuticas para administrar factores neurotróficos en modelos animales, pero hasta ahora las pruebas de Ios efectos de Ios factores de crecimiento en el cerebro y el sistema nervioso se han limitado a la inyección periférica directa de grandes dosis de estos factores, lo que conlleva un riesgo significativo de efectos secundarios. En consecuencia, un aspecto relacionado de la divulgación se refiere a la superación de estos problemas mediante el uso de células NSLC y líneas celulares de acuerdo con la divulgación que puede expresar y secretar de manera estable factores de crecimiento de interés potencial después del trasplante.
En resumen, la presente divulgación proporciona una fuente abundante de células de tipo madre neurales, células de tipo neurona, células de tipo astrocito o células de tipo oligodendrocito para tratamientos clínicos potenciales que requieren el trasplante de células madre neurales, neuronas, astrocitos u oligodendrocitos 1) para compensar la pérdida de células huésped (por ejemplo, neuronas) o 2) como vehículos para administrar medicamentos de base genética. Además, la invención proporciona una herramienta neurológica novedosa para su uso en investigación básica y detección de fármacos.
Ejemplos
Los ejemplos que se detallan a continuación proporcionan procedimientos de ejemplo para obtener células reprogramadas y desdiferenciadas, incluidas las células de tipo madre neurales (NSLC). También se proporcionan protocolos de ejemplo, herramientas moleculares, sondas, cebadores y técnicas.
Ejemplo I
Preparación de células fibroblastos humanos
Se adquirieron células fibroblastos de prepucio humano (HFF) de la Colección Americana de Cultivos Tipo (ATCC, Manassas, Va) y se expandió en matraces de cultivo celular con medio de Eagle modificado por Dulbecco (DMEM, Invitrogen), suplementado con 10 % de suero de ternera fetal inactivado con calor (FCS, Hyclone Laboratories), aminoácidos no esenciales 0,1 mM y piruvato de sodio 1,0 mM (Invitrogen) a 370C, 5 % de CO2. El medio se cambió dos veces a la semana. Las células se digirieron con tripsina utilizando tripsina al 0,25 % durante 4 minutos a 37 oc, seguido de la adición de solución inhibidora de tripsina, sedimentando las células por centrifugación, lavando las células una vez con PBS y sembrando las células en placas en una proporción de 1:2 en matraces de cultivo de tejidos hasta que se alcanzó un número adecuado de células.
A continuación, las células se digirieron con tripsina y se sembraron en placas (8 x 104 células/pocillo) en placas de cultivo celular recubiertas previamente con laminina (10 pg/ml, Sigma) en dos composiciones diferentes de medio CDM: medio CDM I que consiste en una proporción 3:1 de medio Eagle modificado por Dulbecco (DMEM, alto contenido de glucosa (4,5 g/l) con L-glutamina y piruvato de sodio) y medio F-12 de Ham suplementado con Ios siguientes componentes: EGF (4,2 x 10'10 M), bFGF (2,8 x 10'10M), ITS (8,6x10-5 M), dexametasona (1,0x10'7M), L-3,3',5-triyodotironina (2,0 x 10'10 M), etanolamina (10'4M), GlutaMAx™ (4x10'3M) y glutatión (3,3x10'6M), pero sin la presencia de ácido L-ascórbico.
Medio CDM II que consiste en una relación 3:1 de medio Eagle modificado por Dulbecco (DMEM, alto contenido de glucosa (4,5 g/l) con L-glutamina y piruvato de sodio) y medio F-12 de Ham suplementado con Ios siguientes componentes: EGF (2,5 ng/ml), bFGF (10ng/ml), etanolamina (2,03 mg/ml), insulina (10 mg/ml), ácido selenioso (2,5 pg/ml), dexametasona (19,7 pg/ml), L-3,3',5-triyodotironina (675 ng/ml), GlutaMAX™ (4x10'3M) y glutatión (3,3x10' 6M).
Transfección transitoria de HFF mediante lipofectamina utilizando vectores construidos
Después de dos días de cultivo, las células se transfectaron con pCMV6-XL5-MBD2 (2 pg) (un desmetilador de ADN) utilizando reactivo de lipofectamina (Invitrogen) según el protocolo del fabricante. El complejo de ADN-lípido se añadió a las células y se incubó durante 24 horas a 37 oc, 5 % de CO2. Después de 24 horas de transfección con el desmetilador de ADN, el medio se cambió y las células se transfectaron con pCMV6-XL5-Musashi1 (2 pg, Origene) o pCMV6-XL4-Ngn2 (2 pg, Origene) durante 24 horas. Después de 24 horas, el medio se cambió a medio basal progenitor neural (NPBm , Lonza) suplementado con Noggina (20 ng/ml, Peprotech), EGF (20 ng/ml, Peprotech) y bFGF (20 ng/ml, Peprotech) y se cultivó en este medio de proliferación. Las células se retransfectaron después de tres días y se incubaron a 37 oc, 5 % de CO2 y 5 % de O2. Después de 7 días en condiciones de proliferación, el 50
% del medio de proliferación se cambió a medio de diferenciación (NbActive, BrainBits™) suplementado con forskolina (10 pM, Tocris), ácido a // -trans-retinoico (ATRA, 5 pM, Spectrum), bFGF (20ng/ml, Peprotech), NGF (20 ng/ml, Peprotech) y BDNF (20 ng/ml, Peprotech); el medio se cambió todos los días aumentando el porcentaje de medio de diferenciación sobre el medio de proliferación y las células se cultivaron durante 20 días.
La observación visual de las células reprogramadas se realizó mediante observación al microscopio ótico todos los días después de la transfección utilizando un campo brillante con un aumento de 10X. Las muestras se recolectaron en diferentes puntos de tiempo (6 , 12 y 20 días) para analizar la expresión de los genes neuronales y los niveles de proteínas mediante una matriz de genes e inmunohistoquímica. Después de la transfección, la reprogramación de células mostró un cambio rápido en la morfología celular dentro de los 3 días posteriores a la transfección (Figura 1). Las células estaban más redondeadas y el citoplasma de la célula se retrajo hacia el núcleo formando cuerpos celulares contraídos con extensiones citoplásmicas extendidas y exhibiendo una apariencia neuronal pericaaria los días 6 y 12, que se mantuvo hasta el día 20. Sin embargo, esta morfología no se observó en las células no transfectadas los días 6 y 12.
Análisis de matriz de genes
La caracterización de las nuevas células de ingeniería después de la transfección se realizó utilizando una matriz de genes neuronales que contiene 48 ADNc parciales que codifican estos genes y controles.
El ARN se aisló de las muestras utilizando QIAshredder™ (Qiagen) y RNeasy™ Plus mini Kit (Qiagen) según las instrucciones del fabricante. El tratamiento con ADNasa I se realizó en la columna RNeasy ™ para eliminar más el ADN plasmídico transfectado utilizando el Conjunto de ADNasa sin ARNasa (Qiagen). El Ar N se eluyó en 35 pl de agua libre de ARNasa. antes de la síntesis de ADNc, todas las muestras de ARN se cuantificaron utilizando el NanoDrop 1000™ (ThermoScientific). El ADNc se preparó utilizando el kit de archivo de ADNc de alta capacidad (Applied Biosystems) según las instrucciones del fabricante. Se usaron 400 ng de ARN en cada 50 pl de reacción rT. Las muestras de ADNc resultante se usaron inmediatamente para el análisis TLDA. Para cada tarjeta de la matriz de baja densidad Taqman™ (TLDA), hay ocho puertos de carga separados que se alimentan en 48 pocilios separados para un total de 384 pocilios por tarjeta. Cada pocilio de 2 pl contiene cebadores y sondas específicos definidos por el usuario, capaces de detectar un solo gen. En este estudio, se configuró un TlDA de Marcadores Neuronales 2 personalizado en ocho conjuntos de 48 genes idénticos, es decir, 1 puerto de carga para cada conjunto de 48 genes. Los genes se eligieron según la literatura. Cada conjunto de 48 genes también contiene tres genes de mantenimiento: ACTINA, GAPDH y PPIA.
Se hizo una mezcla maestra específica de la muestra para cada muestra mezclando ADNc (160 ng para cada puerto de carga), Mezcla maestra de expresión génica 2X Taqman ™ (Applied Biosystems) y agua libre de nucleasas (USB) para un total de 100 pl por puerto de carga. Después de mezclar suavemente y centrifugar, la mezcla se transfirió después a un puerto de carga en una tarjeta TLDA. La matriz se centrifugó dos veces durante 1 minuto cada una a 1200 rpm para distribuir las muestras desde el puerto de carga a cada pocilio. Luego se selló la tarjeta y se realizó la amplificación por PCR utilizando el sistema de PCR en tiempo real Applied Biosystems 7900HT™ Fast. Las condiciones del termociclador fueron las siguientes: 2 minutos a 500C, 10 minutos a 94,5 °C y 30 segundos a 97 °C, 1 minuto a 59,7 °C durante 40 ciclos. Se preparó 1 TLDA para 8 muestras.
Los valores de expresión relativos se calcularon utilizando el procedimiento Ct comparativo. En resumen, esta técnica utiliza la fórmula 2'AAcT para calcular la expresión de genes diana normalizados a un calibrador. El ciclo de umbral (Ct) indica el número de ciclo en el que la cantidad de objetivo amplificado alcanza un umbral fijo. Los valores de Ct oscilan entre 0 y 40 (este último representa el número de ciclo de PCR del límite superior predeterminado que define el fallo para detectar una señal). Los valores de ACt [ACt = Ct (gen diana) - Ct (Promedio de 3 genes de mantenimiento)] se calculara para los HFF Ctrl y después se usaron como el calibrador para las muestras respectivas. A todos los valores de expresión génica se les asignó un valor relativo de 1.00 para el calibrador, que se usa para determinar la expresión génica comparativa de forma que AACt = ACt (Tratados) - ACt (HFF Ctrl). La expresión relativa se calcula usando la fórmula 2'AAcT.
La comparación cuantitativa de la expresión génica de astrocitos, neuronas y oligodendrocitos permitió la identificación de la mayoría de los genes que se expresan diferencialmente en células reprogramadas. Los datos de la Tabla 1 se analizaron mediante el uso de un algoritmo de análisis de significación para identificar los genes reproducibles para enriquecerse en células reprogramadas en comparación con las células no transfectadas. Después de la transfección con Msil o Ngn2 en presencia de MBD2, la expresión de progenitores de oligodendrocitos tales como NKx2.2, olig2 y MAG, y dos marcadores para astrocitos (GFAP y AQP4) aumentaron considerablemente. Asimismo, varios marcadores de células neuronales tempranas se mejoraron después de la transfección de HFF. Los datos de TDLA revelaron un notable incremento en marcadores específicos para interneuronas, tal como somatostatina y calbindinal. La inducción de doblecortina (DCX), que se expresa mediante la migración de células inmaduras durante el desarrollo y el ARNm de acetilcolina (Ac h e ), un marcador temprano de células neuronales, se expresaban altamente en las células reprogramadas (Tabla 1). La transfección aumentó la expresión de 3 de tipo dihidropirimidinasa (DPYSL3), un marcador temprano de neuronas recién formadas, a cinco veces con Msil y siete veces con Ngn2. La expresión de la proteína 2 asociada a los microtúbulos (MAP2), un marcador esencial para el desarrollo y mantenimiento de la morfología neuronal temprana y la molécula de adhesión
de células neuronales se expresaron altamente con MsI1 y Ngn2 (Tabla 1). La expresión de enolasa-2, un marcador de neuronas maduras, aumentó 20 veces con MsI1 y Ngn2. El miembro de la familia NeuroD NeuroDI se expresó altamente después de la transfección con MsI1 a 84 veces y a 34 por Ngn2.
La expresión génica de factores de crecimiento tales como IGF-1, IGF2, NPY y CSF-3 también se mejoró en las células reprogramadas. La expresión de los genes de VEGF y GDNF se reguló por aumento a casi cinco veces y siete veces mediante MsI1 y Ngn2, respectivamente. Sin embargo, la expresión de BDNF, EGF y bFGF no se activó e incluso se reguló por disminución en comparación con las células no transfectadas. La expresión de la proteína asociada al crecimiento (GAP-43), un marcador de neuronas asociado con el crecimiento y la regeneración y la expresión de netrina, implicado en el desarrollo y la orientación neuronal, estaban considerablemente enriquecidas en células reprogramadas. Se incrementó la expresión de receptores para factores de crecimiento y neurotrofina, tales como el receptor de tipo III tirosina quinasa, tirosina quinasa neurotrófica y receptor tirosina quinasa neurotrófico. La vimentina y la fibronectina, marcadores para fibroblastos, se regularon por disminución en las células reprogramadas en comparación con los cultivos de fibroblastos de control no transfectados.
Tabla 1: Matriz de genes de células de fibroblastos humanos transfectados por MsI1/MBD2 y Ngn2/MBD2. La matriz de genes se realizó en muestras después de dos semanas de diferenciación. Los valores de expresión se dan en relación con los fibroblastos no transfectados.

continuación

Análisis inmunohistoquímico
Las células se fijaron con una solución de formaldehído al 4 %/PBS durante 10 minutos a temperatura ambiente y posteriormente se permeabilizaron durante 5 minutos con Triton X-100™ al 0,1 % en formaldehído al 4 %/PBS. Después de dos breves lavados con PBS, la unión inespecífica del anticuerpo se bloqueó mediante una incubación de 30 minutos con un 5 % de suero normal de cabra en PBS. Luego se añadieron anticuerpos primarios en suero de cabra normal/PBS al 5% de la siguiente manera: anti-nestina de ratón (1:100, BD) como un microfilamento
intermedio presente en células madre neurales y anti-NCAM de ratón (1:10o, Neuromics) como molécula de adhesión neuronal. Después de una incubación de 2 horas, las células se lavaron 4 veces durante 5 minutos cada una con 0,1 % de Tween™/PBS. Se usó un anticuerpo secundario apropiado marcado con fluorescencia para la visualización; 546 anti-ratón de cabra (1:20o, invitrogen) prepaado en 5 % de suero normal de cabra/PBS. Después de la incubación durante una hora, las células se lavaron en 0,1 % de Tween™/PBS tres veces durante 5 minutos cada una. La tinción de ADN con Hoechst33342 (Invitrogen) se usó como marcador de núcleos (dilución 1:500o en PBS, incubación durante 10 minutos). Las imágenes de fluorescencia se tomaron con el sistema de microscopía Cellomics™ ArrayScan HCS Reader. Para determinar una estimación del porcentaje de células que adoptan fenotipos neuronales o gliales, se seleccionaron campos aleatorios y para cada campo se determinó el número total de células (según se determinó contando los núcleos teñidos de Hoechst) y el número total de células positivas para marcadores neuronales o gliales.
Para confirmar que estas células exhibían marcadores de linajes neuronales, las células se inmunotiñeron para nestina y NCAM. Este análisis reveló que las células reprogramadas expresaban ambas proteínas. Como se muestra en la Figura 2, la NCAM estuvo presente en las células durante los 6 días posteriores a la transfección y aumentó a los 12 y 20 días después de la diferenciación, mientras que el patrón inverso se observó para la tinción de nestina.
Este estudio demostró la capacidad de reprogramar las células HFF utilizando un factor de transcripción neurogénico con la presencia de un desmetilador de ADN hacia las células que expresan genes neuronales y proteínas específicas de las células madre neurales y células neuronales. Estas células reprogramadas se mantuvieron estables en cultivo durante al menos 2 semanas.
Ejemplo II
Comparación de la eficiencia de la reprogramación de tres genes neurogénicos diferentes
Las células HFF se cultivaron como se describe en el Ejemplo I y se sembaron en placas en medio CDM i. Las células se transfectaron utilizando el dispositivo Amaxa Nucleofector ™™ (Lonza). Los HFF se cosecharon con TrypLE ™ (Gibco), se resuspendieron en medio CDM y se centrifugaron durante 10 min a 90 xg (1 x 106 células/tubo). El sobrenadante se descartó y se resuspendió suavemente en 100 ml de Solución Nucleofector™ Básica (kit Nucleofector™ básico para fibroblastos primarios de mamífero, Lonza). Cada 100 |jl de suspensión celular se combinó con una mezcla diferente de ADN plasmídico (por ejemplo, la muestra 1 se mezcló con 2 mg de pCMV6-XL5-Pax6 y 2 jg de pCMV6-XL5-MBD2). La suspensión celular se transfirió a una cubeta certificada por Amaxa y se transfectó con el programa apropiado (U023). La muestra se transfirió sin ninguna resuspensión adicional a una placa de cultivo recubierta con LAS-Lisina/Alanina (BrainBits™, 50 jg/ml) y las células se incubaron a 37 °0C, 5 % de CO2. Estas etapas se repitieron para cada muestra que se transfectó. Después de 24 horas, el medio se cambió a medio de proliferación. Después de dos días, las células se volvieron a transfectar usando lipofectamina como se describe en el Ejemplo I y se incubaron a 37 oc, 5 % de CO2 y 5 % de O2. Después de 6 días, la diferenciación se indujo con medio de diferenciación que reemplazó gradualmente al medio de proliferación durante varios días. Las células se recogieron el día 14 para RT-PCR y análisis de inmunohistoquímica.
Análisis de la expresión génica
El aislamiento y la cuantificación del ARN se realizaron como se ha descrito anteriormente en el Ejemplo I. Se preparó ADNc utilizando el kit RT de ADN de alta capacidad (Applied Biosystems) según las instrucciones del fabricante con una concentración final de ADNc de 2 ng/jl. Luego se realizó una PCR en tiempo real para cada gen de interés utilizando la mezcla maestra de FAST PCR (Applied Biosystems) y los ensayos de expresión de genes Taqman™ ® (Applied Biosystems) que se enumeran a continuación:

continuación

La reacción FAST de 96 pocilios se realizó con 8 ng de ADNc por pocilio en una reacción de 10 |jI con 40 ciclos. Las condiciones del termociclador fueron las siguientes: 20 segundos a 95 °C y 1 segundo a 95 °C, 20 segundos a 60 °C durante 40 ciclos.
Los valores de expresión relativa se calcularon como se ha descrito anteriormente en el Ejemplo I, excepto el promedio de 2 genes de mantenimiento (GAPDH y PPIA) se usó para la normalización en lugar del promedio de 3 genes de mantenimiento. La identificación de los genes del linaje neuronal se investigó después de la transfección con tres vectores independientes que contienen Msí1, Ngn2 y Pax6.
Tal como se muestra en la Tabla 2, después de 14 días tras la transfección, la expresión relativa de ARNm de linaje neuronal fue indetectable en células no transfectadas (HFF), mientras que las células transfectadas con Msil o Ngn2 en presencia de MBD2 expresaron marcadores de células madre neurales (Nestina y Sox2), sin embargo, la expresión de Sox2 fue mucho más altamente expresada que la nestina después de la transfección con Ngn2 o Msí1. Los genes específicos neuronales y de astrocitos (pIN-tubulina, MAP2b, GfAP y ACHE) aumentaron también. El nivel de ARNm de los genes asociados tripotenciales de pIM-tubulina, MAP2b, acetilcolina y GFAP no fueron detectables en células transfectadas con Pax6 , lo que indica que Pax6 solo no estuvo implicado en el proceso de reprogramación hacia el linaje neuronal.

Análisis inmunohistoquímico
La tinción fluorescente imunohistoquímica se realizó como se ha descrito anteriormente en el Ejemplo I. De acuerdo con los datos de RT-PCR, El análisis inmunohistoquímico de estos cultivos reveló que las células reprogramadas (con Msil o Ngn2) generaban neuronas morfológicamente complejas que eran positivas para MAP2b, lo que indica la diferenciación de NSLC a células de tipo neuronas (NLC) (Figura 3). Sin embargo, la tinción positiva para estos marcadores no fue detectable después de la transfección con Pax6/MBD2. Además, las neuronas recién formadas expresaron los marcadores y desarrollaron neuritas largas con conos de crecimiento en sus extremos, expresaron genes específicos neurales y dejaron de proliferar cuando se expusieron a condiciones de diferenciación.
Ejemplo 111
Transfección de HFF mediante varias combinaciones de vectores y alteración del citoesqueleto celular
Se probaron varias combinaciones de reguladores neurogénicos y citocinas para modificaciones epigenéticas para determinar su efecto sobre la eficiencia de la reprogramación. Comenzando un día antes de la transfección, las células se trataron con o sin citocalasina B (Calbiochem), con la concentración disminuida todos los días durante cinco días durante los cambios en los medios (comenzando con 10 pg/ml de citocalasina B el día 1 a 75 pg/ml, 5 pg/ml, 25 pg/ml y 0 pg/ml durante los siguientes cuatro días) para investigar el efecto de la interrupción del citoesqueleto celular en el proceso de reprogramación. Las células se transfectaron de forma transitoria como se describe en el Ejemplo II con uno o dos vectores que contienen un factor de transcripción neurogénico por nucleofección. Las células se cotransfectaron con uno de dos desmetiladores de ADN, MBD2 o GAdd45B, (por ejemplo, se transfectaron 2x106 células con pCMV6-XL5Msí1 (2 pg) y pCMV6-XL5-MBD2 (2 pg)). Después de 24 horas, el medio se cambió a medio de proliferación neural (kit de proliferación NeuroCult™, StemCell Technologies) que consiste en DMEM/F12 (1:1), glucos (0,6 %), bicarbonato de sodio (0,1 %), glutamina (20 mM), HEPES (5 mM), insulina (230 pg/ml), transferrina (100 pg/ml), progesterona (200 nM), putrescina (90 pg/ml) y selenito de sodio (300 nM) y suplementado con Noggina (2o ng/ml, Peprotech), hFGF recombinante (2o ng/ml, Peprotech) y hEGF recombinante (20 ng/ml, Peprotech) y las células se cultivaron durante dos semanas a 370C, 5 % de CO2 y 5 % de O2. Las células se analizaron para determinar los marcadores de células madre neurales.
Análisis de la expresión génica
El análisis de la expresión génica se realizó para los marcadores específicos de células madre neurales (Sox2, Nestina, GFAP) y un marcador específico de fibroblastos (CoI5A2) mediante RT-PCR como se ha descrito anteriormente en el Ejemplo I. El análisis por RT-PCR mostró que la expresión relativa de Sox2, nestina y GFAP mejoraban después de transfectar las células con Ios factores de transcripción neurogénicos. Como se muestra en la Tabla 3, la transfectación de las células con un factor de transcripción Msil en presencia de Gadd45b se asoció con una regulación por aumento de la expresión relativa de Sox2 (22,3 ± 5,26) y GFAP (10,14 ± 0,15) y la expresión de estos genes fue aumentó considerablemente cuando se transfectaron las células con Ngn2 20 veces y 10 veces respectivamente. La combinación de Ios dos fatores neurogénicos (Msil y Ngn2) con Gadd45b mejoró aún más la expresión de Sox2 y GFAP. Transfectar las células con un factor de transcripción (Msil o Ngn2) en presencia de MBD2 se asoció con una regulación por aumento de la expresión relativa de Sox2, Nestina y GFAP y regulación por disminución de CoI5A2, mientras que la cotransfección con Gadd45b no aumentó la expresión de nestina y la expresión de CoI5A2 no se reguló. La mejora de la expresión relativa de las células madre neurales se observó al transfectar las células con dos genes neurogénicos en combinación con MBD2; se observó un pequeño aumento en la expresión en presencia de citocalasina B en ciertas condiciones. Un aumento en la expresión relativa de Ios marcadores específicos de células madre neurales (Sox2, Nestina, GFAP) y una disminución en el gen específico de fibroblastos (COL5A2) se observó después de la transfección con Msi1/Ngn2/MBD2, Msi1/Ngn2/Gadd45b, Msí1/MBD2 o Ngn2/MBD2 (Tabla 3). Este estudio demostró que MBD2 aumentó más la eficiencia de reprogramación que GDA45b y demostró que la citocalasina B no tuvo ningún efecto en Ios cultivos de control.


Análisis inmunohistoquímico
La tinción fluorescente imunohistoquímica se realizó como se ha descrito anteriormente en el Ejemplo I. La tabla 4 muestra el porcentaje de Nestina y Sox2 en cada condición, con el porcentaje más alto de células positivas a Sox2 (38,18 ± 1,75 %) y nestina (28,18 ± 2,77 %) observadas después de transfectar las células simultáneamente con ambos factores de transcripción neurogénicos y en presencia de un desmetilador de ADN y citocalasina B. Se detectó un ligero aumento de células positivas a Sox2 (10,42 ± 10,27 %) y células positivas a nestina (4,85 ± 1 , 10 %) después de la transfección con un factor de transcripción Msi1 y MBD2. Se observó la misma tendencia de las células positivas para nestina y Sox2 después de la transfección con Ngn2 y MBD2. La interrupción del citoesqueleto celular con citocalasina B mejoró significativamente la reprogramación, pero no tuvo ningún efecto de reprogramación por sí mismo (Tabla 4).
Tabla 4: Porcentaje de células positivas para Sox2 y nestina después de la transfección de células fibroblastos con diferentes vectores de expresión con o sin la presencia de citocalasina B. Después de la transfección, las células se cultivaron en medio de proliferación (StemCell Technologies) suplementado con EGF (20 ng/ml, Peprotech) y FGF (20 ng/ml, Peprotech) durante dos semanas a 37 °C/5 % de CO2/5 % de O2. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± SD (n = 3-5).

Se probaron varios desmetiladores de ADN por su efecto en la eficiencia de la reprogramación. Las células se cotransfectaron con un vector (MSI1/NGN2) que contenía dos factores neurogénicos pCMV6-Msi1-Ngn2 con varios desmetiladores de ADN. Simultáneamente, se probó otro factor neurogénico para determinar su efecto sobre la diferenciación de las células hacia NSC, pCMV-XL-Nestina individualmente o en combinación con pCMV-Msi1-Ngn2, PCMV-XL5-Msí1, o pCMV-XL4-Ngn2 en presencia de MBD2 como se ha descrito anteriormente en el Ejemplo II.
Las células se cotransfectaron con pCMV-Msi1-Ngn2 con diferentes desmetiladores de ADN (MBD1, MBD2, MBD3, MBD4, MeCP2, AICDA). Se realizó otro ensayo para evaluar el efecto de la nestina en la eficiencia de la reprogramación; por lo tanto, las células se transfectaron con nestina individualmente o en combinación con un vector que contiene un factor neurogénico (Msi1 o Ngn2) o ambos factores neurogénicos en presencia de MBD2. Las células se cultivaron después de la transfección en presencia de medio de proliferación suplementado con EGF (20 ng/ml), FGF (20 ng/ml) y Noggina (20 ng/ml) con y sin VPA (1 mM) durante 12 días a 370C, 5 % de CO2 y 5 % de O2.
El análisis de la expresión génica y la inmunohitoquímica se realizaron para analizar la expresión de genes y proteínas específicas neurales (plll-tubulina, GFAP, Sox2, Nestina) como se describe en el Ejemplo II. Transfectar células con Msi1 y Ngn2 en presencia de varios desmetiladores de ADN reveló y confirmó datos previos que muestran que entre los diversos desmetiladores de ADN utilizados en este estudio, m BD2 promueve la expresión de los genes de células madre neurales (Sox2, GFAP, Nestina) como se muestra en la Tabla 5. Adicionalmente, la transfección de células con nestina con y sin la presencia de un factor neurogénico no tuvo ningún efecto sobre la eficiencia de reprogramación en células neurales similares. Sin embargo, la cotransfección con nestina y Msi1/Ngn2/MBD2 mejoró la expresión de los genes de las células madre neurales y este aumento fue más pronunciado en presencia de VPA.
Tabla 5: Análisis por RT-PCR de la expresión relativa de marcadores de células neuronales precursoras tales como nestina, Sox2, plll-tubulina y GFAP después de la transfección de células fibroblastos con varias combinaciones de pCMV-Msi1-Ngn2 (MSI1/NGN2), pCMV-XL5-Msí1, pCMV-XL4-Ngn2, pCMV-XL-Nestina con diferentes combinaciones de desmetiladores de Ad N, con y sin cotratamiento con VPA.

continuación

El análisis inmunohistoquímico realizado en paralelo con los datos de RT-PCR indicó que las células Sox2 positivas no eran detectables al transfectar las células con Msi1/Ngn2 en presencia de MBD1, MBD3, MBD4, MeCP1 o AICADA (Tabla 6 ) y que, entre los diferentes tipos de genes de desmetilador de ADN probados, solo MBD2 desempeña un papel positivo significativo en la eficiencia de reprogramación de HFF hacia Ns LC cuando se usan los genes neurogénicos anteriores. El análisis inmunohistoquímico reveló un pequeño aumento de células Sox2 inmunopositivas (89,49 ± 3,18) después de cotransfectar las células con nestina y Msi1/Ngn2 en presencia de MBD2 (Tabla 6).
Tabla 6: Porcentaje de células positivas para Sox2 después de la transfección de células fibroblastos con diferentes vectores de expresión con o sin la presencia de varios desmetiladores de ADN. Después de la transfección, las células se cultivaron en medio de proliferación (StemCell Technologies) suplementado con EGF (20 ng/ml, Peprotech) y FGF (20 ng/ml, Peprotech) durante dos semanas a 37 °C/5 % de CO2/5 % de O2. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± SD (n = 3-5).
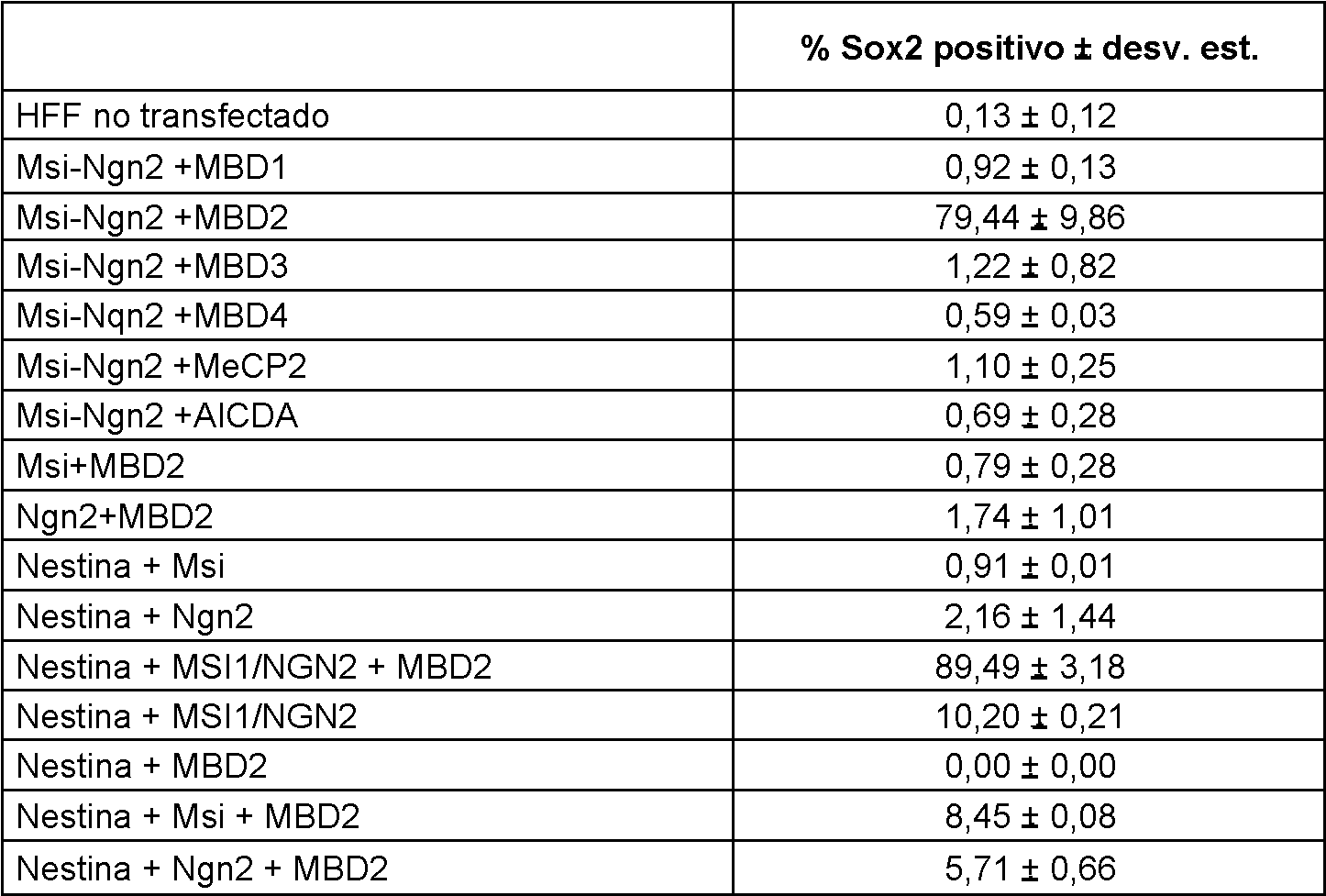
Se diseñó otro estudio para probar el efecto de varios genes neurogénicos en la eficiencia de la reprogramación hacia células de tipo madre neurales. Las células HFF se cultivaron como se describe en el Ejemplo I y se transfectaron utilizando el dispositivo Nucleofector™® de 96 pocilios Shuttle® (Lonza) siguiendo el procedimiento descrito en el Ejemplo IV, excepto para el control de HFF no tratado y el control de HFF no transfectado (para determinar el efecto de los medios completos y los tratamientos de compuestos en las células). Las células que habían sido tratadas previamente con VPA y 5-Aza y las células no tratadas se transfectaron con las mezclas de ADN como se describe en la Tabla 7. Las células se sembraron en placas recubiertas con laminina y se incubaron a
37 0C, 5 % de CO2. Los medios se cambiaron diariamente de acuerdo con la Tabla 7. Las células se analizaron Ios días 3, 7, 12 mediante análisis inmunohistoquímico y el día 9 mediante una matriz de genes para la expresión génica multipotencial y pluripotencial.
Análisis de matriz de genes
Un lote adicional de células tratadas de acuerdo con Oa y la en la Tabla 7 se analizó el día 9, Junto con HFF, hNPC y NSLC de pase 5 (congeladas de experimentos previos del Ejemplo 111) por la matriz de genes de pluripotencia (a BI) (Tablas 8a y b) y un conjunto de genes (Tabla 8c) para determinar el perfil de expresión génica de Ios genes del linaje seleccionado de pluripotencia, ectodérmico, endodérmico, mesodérmico y neural en las NSLC de pase 1 y pase 5 en comparación con Ios HFF (a partir de Ios cuales se crearon) y las células neuroprogenitoras humanas normales (hNPC). Los resultados en la Tabla 8 indican que todos Ios genes relacionados con las células madre neurales (algunos de Ios marcadores de pluripotencia expresados significativamente y Ios marcadores del mesoendodermo también se expresan en las células madre neurales) y el linaje neural se expresaron significativamente en NSLC en lugar de HFF, y el patrón de expresión fue un poco diferente de las hNPC, lo que indican que las NSLC son similares a, pero no idénticas, a las nNPC probadas. Las NSLC 5 de pase 5 tenían una expresión más alta de Ios genes de la entidad madre que las NSLC de pase 1. Las hNPC tenían una expresión más alta de genes neuronalmente comprometidos que las NSLC, Io que indica su estado de neuroprogenitor frente al mayor estado de potencia de NSLC.
Tabla 7: Plásmidos y composición de Ios medios desde el día 1 hasta el día 12.

(continuación)

(continuación)

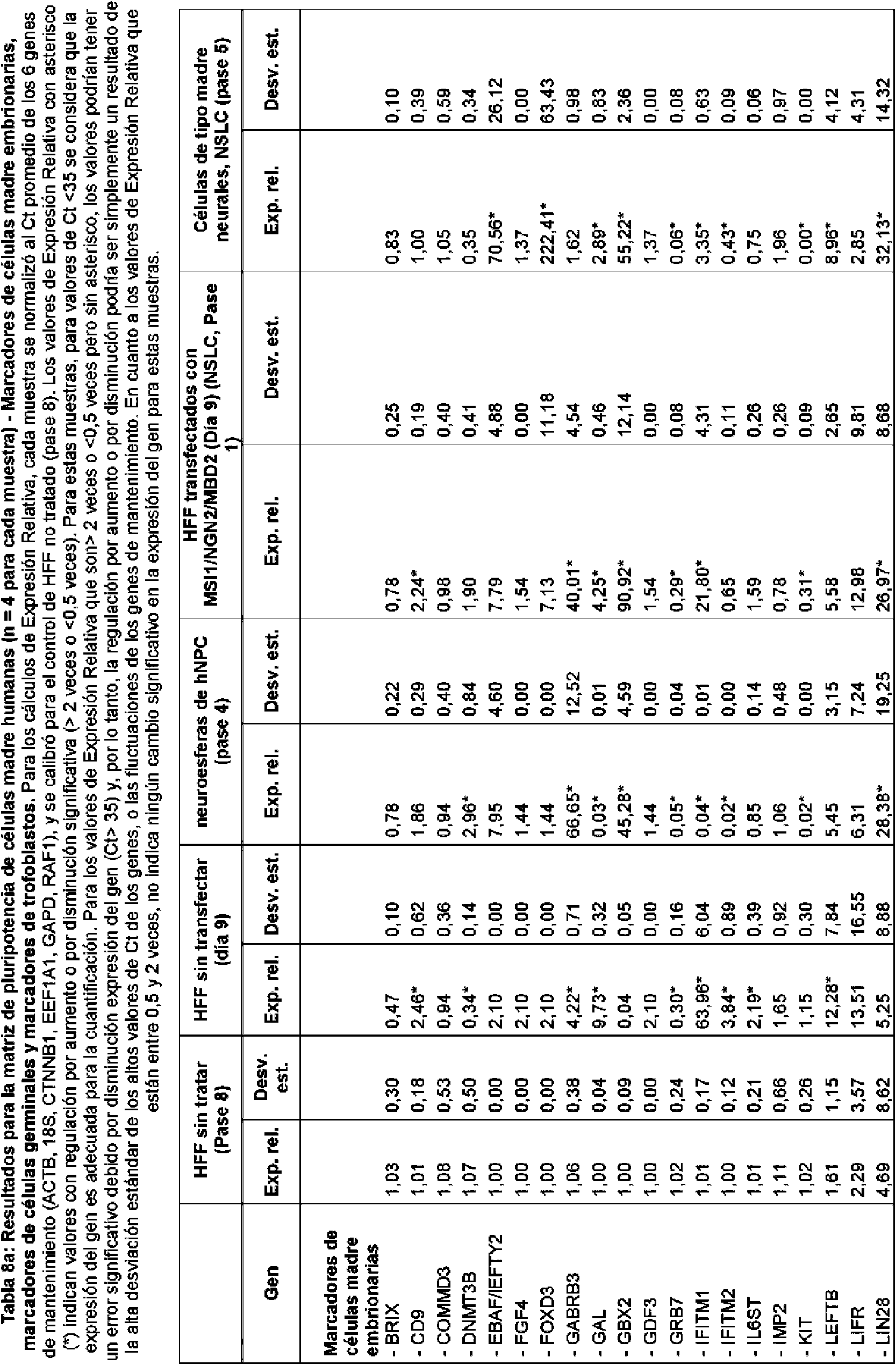


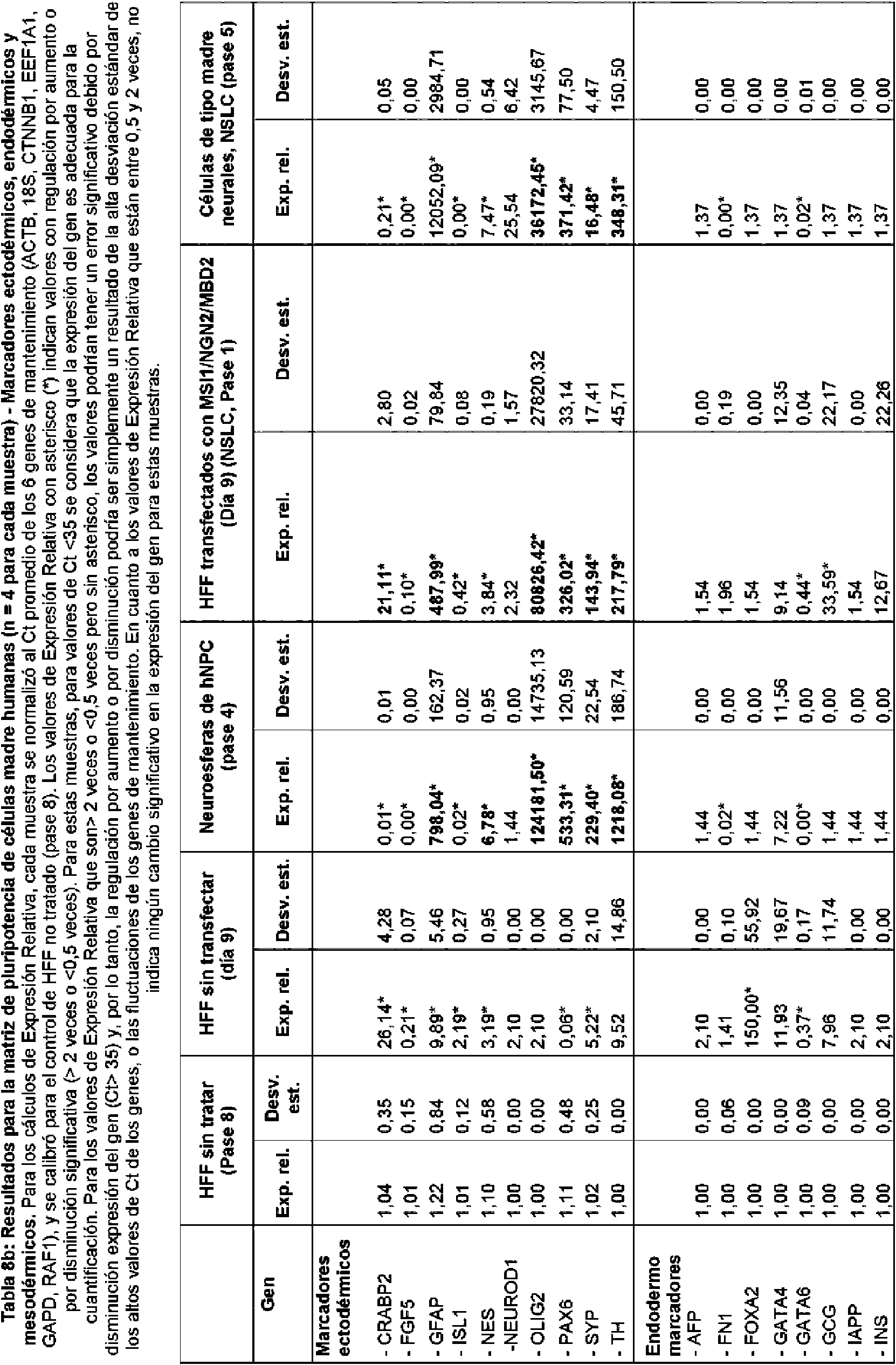




En otra parte del experimento, otro lote de células que se transfectaron con Msi1/Ngn2 pCMV6-XL5-MBD2 se sembraron en placas recubiertas con poli-ornitina (30 min a temperatura ambiente) y laminina (1 ha temperatura ambiente) en medio CDM II en 5 pocilios diferentes. El día 1, el medio en dos de los pocilios se cambió al mismo medio que en la condición la (Tabla 7) hasta el día 12. El medio se cambió diariamente hasta el día 12, en ese momento se cambió al medio de diferenciación NS-A (StemCell Technologies) o al medio NbActive4 (BrainBits™) que se suplementaron con BDNF (20 ng/ml), NT-3 (20 ng/ml), NGF (20 ng/ml), ácido retinoico (5 pM), noggina (20 ng/ml) y forscolina (10 pM). Estas células mostraron una morfología de células de tipo madre neurales para el día 7, y proliferaron hasta el día 12. Durante la exposición a cualquiera de los dos medios de diferenciación, estas NSLC cambiaron a un fenotipo más neuronal y glial como se muestra en las imágenes de campo brillante (Figura 21), pero solo expresaron GFAP el día 17 (Figura 22).
Para los otros tres pocilios, el día 1, el medio se cambió a cualquiera de los medios de diferenciación NS-A (StemCell Technologies), NbActive4 (BrainBits) o medio CDM II; estas dos primeras se suplementaron con las mismas citocinas que se han descrito anteriormente, pero con la adición de Fgf-2 (20 ng/ml). El día 12, el Fgf-2 se eliminó de los dos primeros medios de diferenciación, mientras que las células en el medio CDM II se cambiaron al medio de diferenciación NS-A (StemCell Technologies) suplementado con citocinas sin Fgf-2. Entre el día 12 y el día 17, Ios medios se cambiaron cada dos o tres días. Durante los primeros 12 días de cultivo, las células en los 3 medios se convirtieron en una mezcla de más células con forma de huso en comparación con Ios fibroblastos no transfectados y algunos en células con una morfología NSLC; al eliminar la morfología de las células Fgf-2 se convirtió en una forma neuronal muy pronunciada, así como en células gliales con una red establecida entre las células como se muestra en las imágenes de campo brillante (Figura 21) que expresaban GFAP y plll-tubulina el día 17 (Figura 22).
Se diseñó un estudio adicional para evaluar el efecto de Msil, Ngn2 y MBD2 en sus niveles de proteínas endógenas en células reprogramadas. Las células se transfectaron con el vector MSI1/NGN2 y MBD2 como se ha descrito anteriormente y se cultivaron en condiciones de proliferación a 37 °C, 5 % de CO2 y 5 % de O2. Las muestras se recogieron en varios puntos de tiempo desde el Día 2-10 y se analizaron por RT-PCR para investigar la expresión de genes endógenos y la expresión de células madre neurales y genes neuronales en diferentes puntos de tiempo. La RT-PCR reveló una pérdida gradual de expresión génica de Msil, Ngn2 y MBD2 totales a partir del día 2 hasta el día 10, habiendo perdido casi completamente el aumento en la expresión de MBD2 en relación con el control que el Día 5. Esta disminución se asoció con una activación significativa de Msil y Ngn2 endógenos el Día 5, con otro salto en la expresión génica endógena el día 9 (Tabla 9). Se detectó un aumento significativo en la expresión de Sox2 el día 4 y la expresión de este gen ectodérmico/célula madre neural/neuronal continuó aumentando con cada punto de tiempo posterior (Tabla 10). GFAP (un marcador de célula madre neural y de astrocitos) ya estaba ligeramente elevado desde el Día 2 en adelante, pero aumentó significativamente el día 5 con un gran salto en la expresión de genes en el punto de tiempo del análisis del Día 7 y se mantuvo en este nivel de expresión durante el resto del período de estudio. La expresión del marcador de células madre neurales Nestina también comenzó a aumentar lentamente desde el Día 5 en adelante. La expresión de Ios genes neuronales plll-tubulina (TUBB3) y Map2b ya estaban ligeramente elevados desde el Día 2 en adelante, pero aumentó significativamente el día 5 en adelante. La expresión de un marcador para Ios receptores de acetilcolina (que se encuentra en las neuronas), acetilcolina esterasa (ACHE), también se elevó ligeramente desde el día 2 en adelante, pero no aumentó significativamente hasta el día 7 en adelante. Cabe señalar que entre Ios marcadores de células madre neurales que se analizaron, la expresión relativa de Sox2 fue altamente expresada en forma temprana, lo que luego podría interactuar directa o indirectamente con Ios genes Msi/Ngn2 exógenos y/u otros en la activación de Nestina, GFAP, y Msil y Ngn2 endógenos y otros genes que promueven la reprogramación y el cambio del destino celular, así como la activación de genes neuronales como plll-tubulina (TUBB3), Map2b y ACHE.


Ejemplo IV
Comparación del dispositivo Nucleofector™ ® II y el dispositivo Nucleofector™® de 96 pocilios Shuttle® en la reprogramación de HFF en NSLC en condiciones adherentes y flotantes.
Las células HFF se cultivaron como se describe en el Ejemplo I y se transfectaron utilizando el dispositivo Nucleofector™® II (Lonza) como se ha descrito previamente en el Ejemplo II o el dispositivo Nucleofector™® de 96 pocilios Shuttle® (Lonza). Los HFF se cosecharon con TrypLE™ (Gibco) y 1x106 células/transfección con el dispositivo Nucleofector™® II durante 10 min a 90 g y 6x105 células/transfección con el dispositivo Nucleofector™® de 96 pocilios Shuttle® durante 5 min a 80 x g. Después de la centrifugación, el sedimento celular se resuspendió suavemente en 100 |jl de Solución Nucleofector™ Básica para Nucleofector™ ® II o 20 |jl de Solución SE (kit de línea celular SE, Lonza) para el Nucleofector™® de 96 pocilios Shuttle®. Para el dispositivo Nucleofector™® II, cada 100 j l de suspensión celular se combinó con 2 mezclas diferentes de ADN plasmídico (la muestra 1 se mezcló con 2 jg de pCMv 6-XL5-MsI1 y 2 jg de pCMV6-XL5-MBD2, y la muestra 2 con 2 jg de Msi1/Ngn2 y 2 jg pCMV6-XL5-MBD2). Cada suspensión celular se transfirió a una cubeta certificada por Amaxa y se transfectó con el programa apropiado (U-023). Justo después de la transfección, se añadieron 900 j l de medio CDM1 caliente a cada cubeta y la muestra se transfirió a una placa de cultivo recubierta con Laminina (Stemgent, 10 jg/ml) a una densidad celular de 1x105 a 1,5 x 105células por cm2 o en placas de Petri tratadas con cultivos no celulares para la formación de neuroesferas. Las células se incubaron a 37 0C, 5 % de CO2 durante la noche. Sin embargo, para el dispositivo Nucleofector™® de 96 pocilios Shuttle®, las etapas descritas anteriormente fueron similares con las siguientes excepciones: la suspensión celular se mezcló con 0,6 jg de cada ADN de las mismas 2 mezclas de ADN, la suspensión celular se transfirió a un pocilio de Nucleoplate™ de 96 pocilios (Lonza) y se transfectó con el programa FF-130tm. Después de la transfección, se añadieron 80 j l de medio CDM1 caliente a cada pocilio y las muestras se dejaron durante 10 minutos en la incubadora antes de transferirlas a una placa recubierta con laminina o placas de Petri tratadas con cultivo no celular a la misma densidad celular que se mencionó anteriormente. Para ambos dispositivos, Estas etapas se repitieron para cada muestra que se transfectó. Antes de la transfección, las células se cultivaron en CDM1 como se describe en el Ejemplo I. Después de 24 horas, el medio se cambió a una mezcla de medio CDM al 75 % y medio de proliferación al 25 % que se suplementó con EGF (20 ng/ml), FGF-2 (20 ng/ml), Noggina (20 ng/ml) y citocalasina B (10 jg/ml) y las células se incubaron a 37 oc, 5 % de CO2 y 5 % de O2. El medio se cambió diariamente con una mayor proporción de medio de proliferación neural hasta el 100 % el día 4 y una proporción reducida de citocalasina B que se omitió por completo el día 5. Se añadió forscolina (10 j M) al medio desde el Día 4 en adelante. Las células en condiciones de flotación se sedimentaron por centrifugación y su medio se cambió diariamente como se describe para la condición adherente. Las células se recolectaron los días 3, 7 y 12 para el análisis de la inmunohistoquímica.
Se tomaron imágenes de fluorescencia con un sistema de microscopía Cellomics™ ArrayScan HCS Reader™ para determinar una estimación del porcentaje de células positivas para Sox2, un marcador de células madre neurales. Este análisis reveló que en controles no transfectados y 3 días después de la transfección, no se detectó tinción de Sox2 nuclear. Sin embargo, el día 7 y el día 12, el porcentaje de células Sox2 positivas aumentó progresivamente en todas las condiciones de transfección, excepto la condición pCMV6-XL5-Musashi y pCMV6-XL5-MBD2 Nucleofector™® II. El porcentaje más alto el día l2 se obtuvo con Msi1/Ngn2 y pCMV6-XL5-MBD2 transfectado con el dispositivo Nucleofector™® de 96 pocilios Shuttle® (-80 %). La misma combinación transfectada con el Nucleofector™® II produjo solo -35 % de células positivas. El pcMv6-XL5-Musashi y el pCMV6-XL5-MBD2 con el Shuttle® produjeron -20 % de células positivas, mientras que generalmente no se observó ninguna con el Nucleofector™® II. El porcentaje de células positivas varió fuertemente entre los pocilios. La tinción indicó que la población celular no era homogénea, ya que en todos los casos se pudieron encontrar campos de células Sox2 positivas densamente dispuestas y campos completos con solo células negativas. En general, el Shuttle® fue inicialmente más tóxico para las células que el Nucleofector™® II, sin embargo, al menos en el caso de Msi1/Ngn2 y la lanzadera pCMV6-XL5-MBD2, la población positiva de Sox2 se expandió rápidamente desde el día 7 hasta el día 12 para tener el doble de células positivas Sox2 en comparación con el Nucleofector™® II. Las células en condiciones flotantes no formaron esferas durante el experimento de 12 días en ninguna de las condiciones, lo que sugiere que la formación de neuroesferas requiere la generación de células de tipo madre neurales en condiciones adherentes por primera vez o más tiempo.
La Tabla 11 muestra el porcentaje de células Sox2 positivas con una morfología de células madre neurales típica que utiliza tanto el dispositivo Nucleofector™® II como el dispositivo Nucleofector™® de 96 pocilios Shuttle®. Este último tenía las ventajas de requerir un material de partida más pequeño (menos células y menos ADN requerido) y además dio lugar a un mayor número de células positivas para Sox2. Además, se observó una población muy pequeña de células positivas para Sox2 con el dispositivo Shuttle® solo después de la transfección con un solo factor de transcripción neurogénico (MsI) en presencia del desmetilador de ADN MBD2.
Tabla 11: Porcentaje de células positivas para Sox2 después de la transfecclón de células fibroblastos con diferentes vectores de expresión. Después de la transfecclón, las células se cultivaron en medio de proliferación (StemCell Technologies) suplementado con EGF (20 ng/ml, Peprotech) y FGF (20 ng/ml, Peprotech) durante dos semanas a 37 °C/5 % de CO2/5 % de O2. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± SD (n = 3-5).

Ejemplo V
Ensayo de formación de neuroesferas y análisis de diferenciación celular
Basado en estudios previos que muestran que se logra una mayor reprogramación proporcional mediante la transfección de dos genes neurogénicos, este estudio se diseñó para evaluar el número de células de reprogramación utilizando el vector Msi1/Ngn2, que contenía dos factores de transcripción neurogénicos (MsI 1 y Ngn2) y el papel del desmetilador de ADN o el inhibidor de la metilación del ADN (5-azacitidina) y el inhibidor de la desacetilación de histonas (VPA) en el proceso de reprogramación.
Los HFF se cultivaron y se trataron con citocalasina B como se describe en el Ejemplo III y se trataron simultáneamente con VPA (1 mM) y 5-azacitidina (0,5 |jM). Después de dos días de tratamiento, las células fueron transfectadas por nucleofección como se describe en el Ejemplo II con el vector construido Msi1/Ngn2. Después de preparar las células, se mezclaron con 2 jg de ADN total (Msi1/Ngn2) y las células que no habían sido tratadas con inhibidores químicos (VPA y 5-Aza) se cotransfectaron con MBD2 (2 jg), utilizando el programa apropiado (U023). Las muestras se transfirieron a una placa de cultivo recubierta con laminina (10 jg/ml, Sigma) y se incubaron en una incubadora humidificada a 37 °C/5 % de O2 /5 % de CO2. El medio se cambió a los medios basales de proliferación, medio de proliferación neural (kit de proliferación NeuroCult™, StemCell Technologies), con la presencia de Noggina (20 ng/ml, Peprotech), hFGF recombinante (20 ng/ml, Peprotech) y hEGF recomblnante (2o ng/ml, Peprotech). Después de 6 días de transfecclón, las células se recolectaron usando Accutase™ (Millipore), se centrifugaron (300 x g, 5 mln, RT) y se sembraron en placas de cultivo celular sin recubrimiento en medio de proliferación NeuroCult™ para NSC para investigar la capacidad de cultivar las células en suspensión como neuroesferas o en placas recubiertas con laminina para el cultivo adherente. Para evitar la pérdida de esferas flotantes durante los cambios de medios, las células se sedimentaron mediante centrifugación a 150 x g durante 3 minutos a temperatura ambiente (RT). A continuación, el sedimento se resuspendió en medio fresco y se colocó en placas de cultivo celular de unión baja si recubrir. Los cultivos se incubaron a 37 °C, 5 % de CO2, 5 % de O2 y se alimentaron diariamente durante al menos dos meses.
Para investigar si una sola célula de células precursoras neurales humanas (hNPC) y NSLC humanas era capaz de generar una neuroesfera (una prueba estándar para demostrar que una célula es una célula madre neural), las neuroesferas se disociaron en células individuales y estas células individuales se aislaron y se cultivaron en un medio de proliferación en suspensión y la formación de la neuroesfera se monitorizó tomando imágenes de campo brillante con un microscopio óptico (Nikon, 10X) y por Cellomics™. Estas células comenzaron a proliferar y crecieron como esferas a partir del día 6 al día 10 (Figura 4A). El análisis inmunohistoquímico de estas esferas (Tabla 12 y Figura 4) el día 20 reveló tinción inmunopositiva para los marcadores de células madre neurales Sox2, Musashi, CD133, Nestina y GFAP. Las células también se tiñeron de forma positiva para plll-tubulina (un marcador para las neuronas), 04 (un marcador para oligodendrocitos) y GFAP (un marcador para astrocitos), lo que indica el potencial de diferenciación tripotencial de ambos conjuntos de células (NSLC y hNPC), y negativo para NGFrec y NeuN (marcadores para neuronas diferenciadas) que indica que las células no se diferenciaron terminalmente.
Tabla 12: Porcentaje de células positivas para marcadores de linaje de células madre neurales y neuronales, astrocitos y oligodendrocitos en neuroesferas formadas a partir de nSlC individuales y hNPC cultivadas en medio de proliferación (StemCell Technologies) suplementado con EGF (20 ng/ml, Peprotech) y FGF (20 ng/ml, Peprotech) durante 20 días a 37 °C/5 % de CO2 /5 % de O2. El porcentaje de células positivas se determinó mediante Cellomics™ y se representó como la media ± SD.
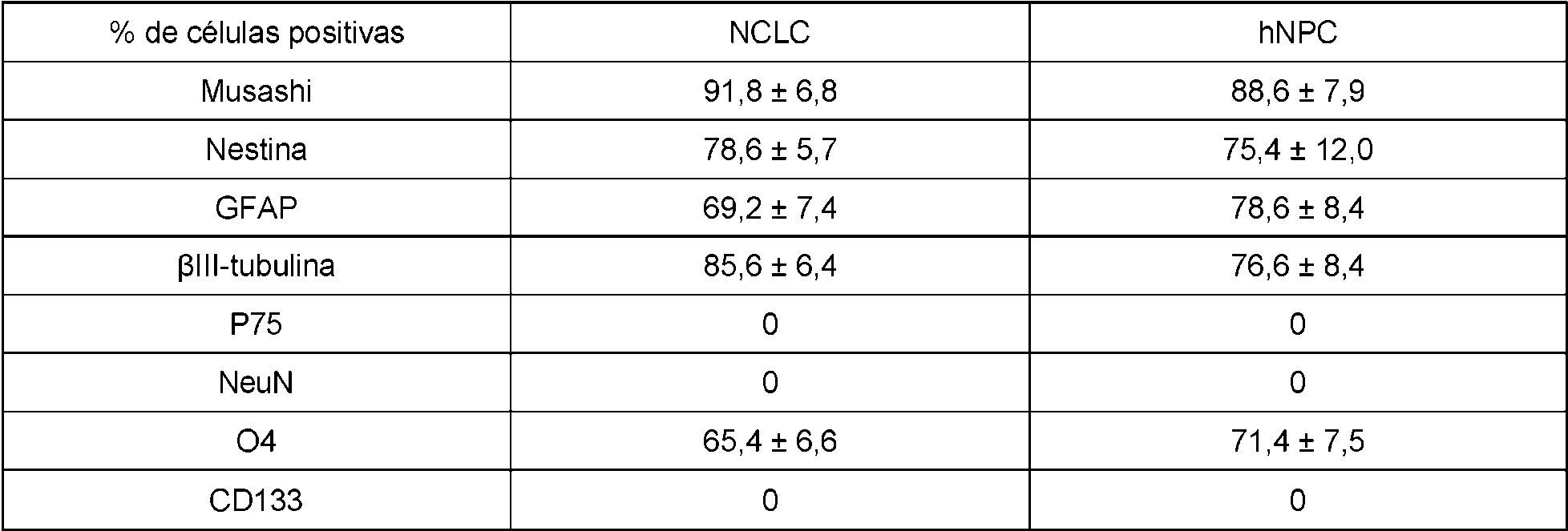
Las células HFF se cultivaron como se describe en el Ejemplo I y se transfectaron utilizando el dispositivo Nucleofector™ II (Lonza) como se describe en el Ejemplo II. Las células se cotransfectaron con pCMV6-XL5-Msi/pCMV6-XL4-Ngn2, pCMV-Msi1-Ngn2 con MBD2 o pretrataron con VPA/5aza. Las células se cultivaron en medio de proliferación como cultivos en suspensión o adherentes. El análisis de la expresión génica en 8 muestras se realizó como se ha descrito anteriormente en el Ejemplo I con el TLDA de Marcadores Neuronales 2 personalizado (Tabla 13) que describió la expresión de 48 genes (incluidos tres genes de mantenimiento: ACTINA, GAPDH y PPIA) en cuatro categorías principales; 1) genes específicos de fibroblastos; 2) genes específicos del linaje neuronal; 3) genes específicos de marcadores de células madre neurales; y 4) Genes para los factores de crecimiento y sus receptores.
Tabla 13: Despliegue de TLDA de los Marcadores neuronales 2 (Applied Biosystems)
Símbolos del gen

Información de la muestra

Tal como se muestra en la Tabla 14, Ios genes específicos de fibroblastos (CoI3A1, Lox, S100A4) se regularon por disminución en células reprogramadas, lo que indica la pérdida de genes específicos de fibroblastos después de la transfección (obsérvese que no todas las células se transfectaron y reprogramaron, por lo que la presencia de la expresión génica específica de Ios fibroblastos en Ios cultivos es principalmente de Ios fibroblastos no programados que quedan en el cultivo). Se observa que la expresión de estos genes aumenta cuando Ios HFF se transfectaron en ausencia de un desmetilador de ADN o el inhibidor de la metilación del ADN, Io que indica que la regulación por disminución de Ios marcadores diferenciados de las células fibroblastos requiere la desmetilación del ADN. La expresión de genes ectodérmicos, tales como MsI1, Sox2 y Nestina aumentó notablemente después de la transfección Junto con la desmetilación del ADN. La expresión de marcadores neuronales, tales como sinaptogamina 1 (una proteína de vesícula sináptica) y NeuroDI se reguló por aumento en células transfectadas con Msi1/Ngn2/MBD2, y aumentó ligeramente en células transfectadas con Msi1/Ngn2/VPA y 5-AZA. Los tres marcadores seleccionados de oligodendrocitos se detectaron en las células transfectadas con un fuerte aumento de Olig2. Dos marcadores para Ios astrocitos, GFAP y ALDH1L1, mejoraron después de la transfección. Los resultados apoyan la idea de que las neuroesferas están compuestas por subtipos de progenitores heterogéneos.
Entre Ios factores neurotróficos, la expresión de CNTF aumentó ligeramente en las células reprogramadas. La expresión de GAP-43 y el neuropéptido Y (NPY) fueron Ios genes más anotados. Se ha reconocido que GAP-43 desempeña un papel fundamental en la plasticidad axonal y se utiliza como marcador de crecimiento de neuritas en regeneración y sinaptogénesis, tanto en el desarrollo embrionario como en la regeneración neuronal en el cerebro y la médula espinal dañados. La expresión de receptores para el crecimiento y factores neurotróficos se incrementó, tal como la expresión del receptor neurotrófico tirosina quinasa.




El análisis y la cuantificación adicionales de la población adherente de NSLC mostraron que las células se tiñeron positivamente para Sox2 (93,43 ± 1,9 %), nestina (60,76 ± 5,7 %) y GABA (37,48 ± 4,9), mientras que estos marcadores no fueron detectables en células no transfectadas (Figura 5, Tabla 15). Además,estas células se tiñeron de forma positiva para p75NTR (31,15 ± 1,6), plll-tubulina (37,55 ± 0,6 %) y GFAP (16,47 ± 0,9). Sin embargo, los HFF no transfectados solo se tiñeron de forma positiva para marcadores de HFF (Figura 5), tales como fibronectina y el marcador de proteína de fibroblastos, mientras que estos marcadores eran indetectables en células reprogramadas, lo que demuestra que las células reprogramadas perdieron los marcadores de las células originales y adoptaron la morfología y los marcadores de células madre neurales y un linaje neuronal.
Tabla 15: El porcentaje de células con tinción positiva para marcadores de células madre neurales y marcadores de fibroblastos en células no transfectadas y células transfectadas con pMsi1/Ngn2/MBD2. Las células transfectadas (NSLC) poseen un alto porcentaje de marcadores de células madre neurales pero un porcentaje muy bajo de marcadores de fibroblastos en comparación con las células no transfectadas. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± Sd (n = 5).

Este estudio también mostró que las NSLC tienen la capacidad de proliferar en el cultivo y exhibir una morfología, expresión de genes y proteínas estable que se mantuvieron durante todo el período de estudio, que fue por más de cinco meses en cultivo (Tabla 16).
Tabla 16. Tiempo de duplicación de NSLC sobre pases en serie. Las NSLC se mantuvieron en condiciones de proliferación durante 35 pases en un incubador a 37 °C, 5 % de CO2 y 5 % de O2. El tiempo requerido para que la población de células se duplicara (g) se calculó para cada pase y se definió como g = (In2)/k, donde k era el número de generaciones que ocurrieron por unidad de tiempo (t) definida como, k = (ln Nf- ln No)/t, donde Nf era el número de células finales y No el número de células sembradas iniciales. El tiempo promedio de generación fue de 25,4 h en más de 35 pases.

continuación

Micromatriz de expresión génica
Se realizó un análisis de expresión en micromatrices para obtener una visión global para comparar el perfil de expresión génica de NSLC de pase 7 con HFF (las células de las que se creó la NSLC) y hNPC. NSLC (n=3), HFF (n = 2) y hNPC (n = 3) se resuspendieron en RNAlater™ (Qiagen) y se enviaron a Genotypics (India) donde se procesaron las muestras y se realizó la micromatriz de expresión génica.
En resumen, los genotípicos extrajeron el ARN de las muestras y realizaron el control de calidad utilizando un Bioanalyzer™ de Agilent. El marcaje se realizó con el kit Quick Amp™ de Agilent (síntesis de ADNc y transcripción in vitro), seguido del control de calidad del marcaje. A continuación se realizó la hibridación utilizando la matriz de 8 x 60 K y el barrido se realizó con un escáner Agilent de alto rendimiento con tecnología SureScan™. El software Agilent Feature Extraction se utilizó para la extracción automática de características, seguido de Raw Data QC e Image QC. A continuación se realizó un análisis de datos avanzado, incluyendo el análisis de ontología de rutas y genes utilizando el GeneSpring GX™ v10.0 de Agilent y el Software Biointerpreter de Genotypic. Las muestras de NSLC se compararon con las muestras de HFF (conjunto 1) y las muestras de hNPC (conjunto 2). Las muestras de NSLC tenían un patrón de expresión génica global que era mucho más cercano a las hNPC que los HFF a partir de los cuales se crearon las nSlC (Figura 23). El análisis de correlación de Pearson reveló que las NSLC están estrechamente relacionadas con las hNPC, incluyendo en términos de marcadores de linaje neuronal, genes regenerativos y genes de migración. Estos datos confirman que las NSLC son similares, pero no idénticas, a las
hNPC.
El análisis de micromatrices reveló una regulación por aumento de Ios genes precursores neurales en las muestras de NSLC en comparación con las muestras de HFF. ACTL6A e PHF10, que pertenecen al complejo de remodelación de la cromatina de Ios progenitores neurales (complejo npbaf) y son necesarios para la proliferación de Ios progenitores neurales, se regularon por aumento 2,9 veces y 2,3 veces, respectivamente. MSI2, que desempeña un papel en la proliferación y el mantenimiento de células madre en el sistema nervioso central, se reguló por aumento por 6 veces (Tabla X I). Los genes Glía estaban regulados por aumento en las muestras de NSLC en comparación con las muestras de HFF. GFAP, es un marcador neuronal específico de células madre y astrocitos que, durante el desarrollo del sistema nervioso central, distingue Ios astrocitos de otras células gliales, está altamente regulado por aumento en la muestra de NSLC en comparación con HFF (690 veces). OLIG1, que favorece la formación y maduración de oligodendrocitos, especialmente dentro del cerebro, también está altamente regulado por aumento en la muestra de NSLC en comparación con HFF (370 veces) (Tabla X2).
La Tabla X3 enumera un subconjunto de genes regenerativos que se regulan por aumento en las muestras de NSLC en comparación con las muestras de HFF. SOX2, un gen crítico para la embriogénesis temprana y para la pluripotencia de las células madre embrionarias, así como para las células madre neurales, está altamente regulado por aumento en las muestras de NSLC en comparación con las muestras de HFF (5000 veces). CCND2, que es esencial para el control del ciclo celular en la transición G1/S (inicio), también está regulado por aumento en muestras de NSLC (70 veces más que en muestras HFF). Como se muestra en la Tabla X4, numerosos genes de fibroblastos estaban regulados por disminución en las muestras de NSLC en comparación con las muestras de HFF. Esto muestra que las NSLC pierden la expresión de numerosos genes de fibroblastos a medida que se reprograman desde HFF a NSLC.
La Tabla X5 muestra que Ios genes precursores neurales también se regularon por aumento en las muestras de NSLC en comparación con las muestras de hNPC. BDNF, que promueve la supervivencia y la diferenciación de poblaciones neuronales seleccionadas de Ios sistemas nervioso central y periférico durante el desarrollo, es incluso más altamente expresado en muestras de NSLC que en muestras de hNpC (34 veces la regulación por aumento). La Tabla X6 muestra que un subconjunto de genes Glía también están regulados por aumento en las muestras de NSLC en comparación con las muestras de hNPC. GFAP, un marcador específico de células madre y de astrocitos que, durante el desarrollo del sistema nervioso central, distingue Ios astrocitos de otras células gliales, está más altamente expresado en muestras de NSLC que en muestras de hNPC (13 veces). PLP1, la proteína principal de la mielina del sistema nervioso central que desempeña un papel importante en la formación o el mantenimiento de la estructura multilamelar de la mielina, también está más altamente expresada en las muestras de NSLC que en las muestras de hNPC (20 veces).
Los genes regenerativos también estaban regulados por aumento en las muestras de NSLC en comparación con las muestras de hNPC (Tabla X7). BMP2, un marcador de la cresta neural, pero que induce el crecimiento, especialmente de la formación de cartílago y hueso y BMP4, que a su vez induce la formación de cartílago y hueso y actúa en la inducción del mesodermo, el desarrollo dental, la formación de las extremidades y la reparación de fracturas, sino también en las células madre neurales, estaban más altamente expresados en muestras de NSLC que en muestras de hNPC (18 veces y 20 veces respectivamente). GAP43, que es un componente principal de Ios conos de crecimiento móviles que forman las puntas de Ios axones en elongación se expresó más altamente en muestras de NSLC que en muestras de hNPC (4 veces). Esto sugiere el potencial regenerativo de NSLC. HOXB4, un factor de transcripción que está involucrado en el desarrollo y también en la expansión de las células madre neurales, así como en las células madre y progenitoras hematopoyéticas in vivo e in vitro, lo que lo convierte en un candidato potencial para la expansión terapéutica de las células madre, también estaba más altamente expresado en NSLC que en hNPC. Estos datos indican que las NSLC son más "de tipo madre" o tienen más "rigidez" que las hNPC.
Tabla X1: Genes precursores neurales regulados por aumento (NSLC frente a HFF)

continuación
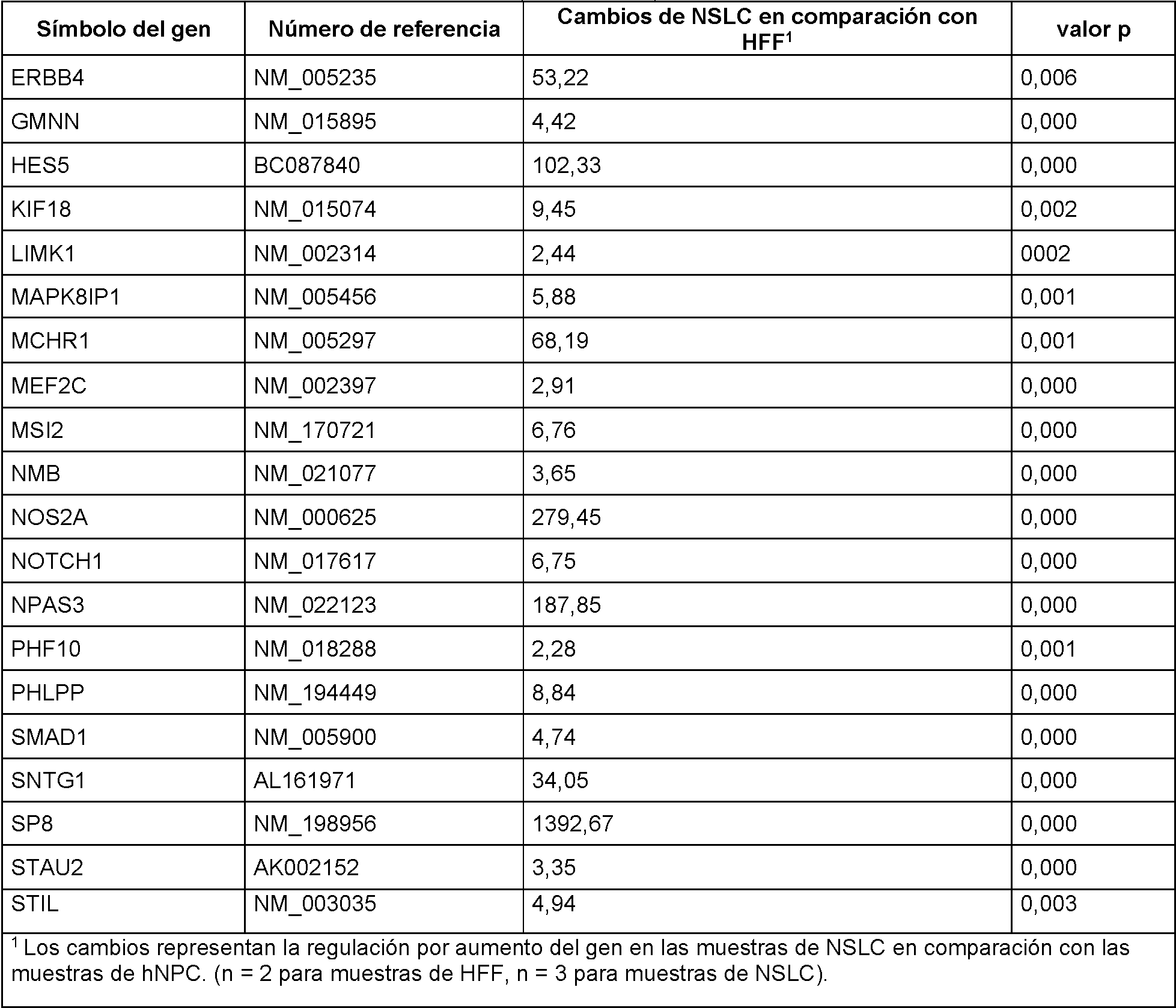
Tabla X2: Genes de glía regulados por aumento (NSLC frente a HFF)

continuación

Tabla X3: Genes regenerativos regulados por aumento (NSLC frente a HFF)

Tabla X4: Genes de fibroblastos regulados por disminución (NSLC frente a HFF)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Tabla X5: Genes precursores neurales regulados por aumento (NSLC frente a hNPC)

continuación

Tabla X6: Genes de Glía regulados por aumento (NSLC frente a hNPC)
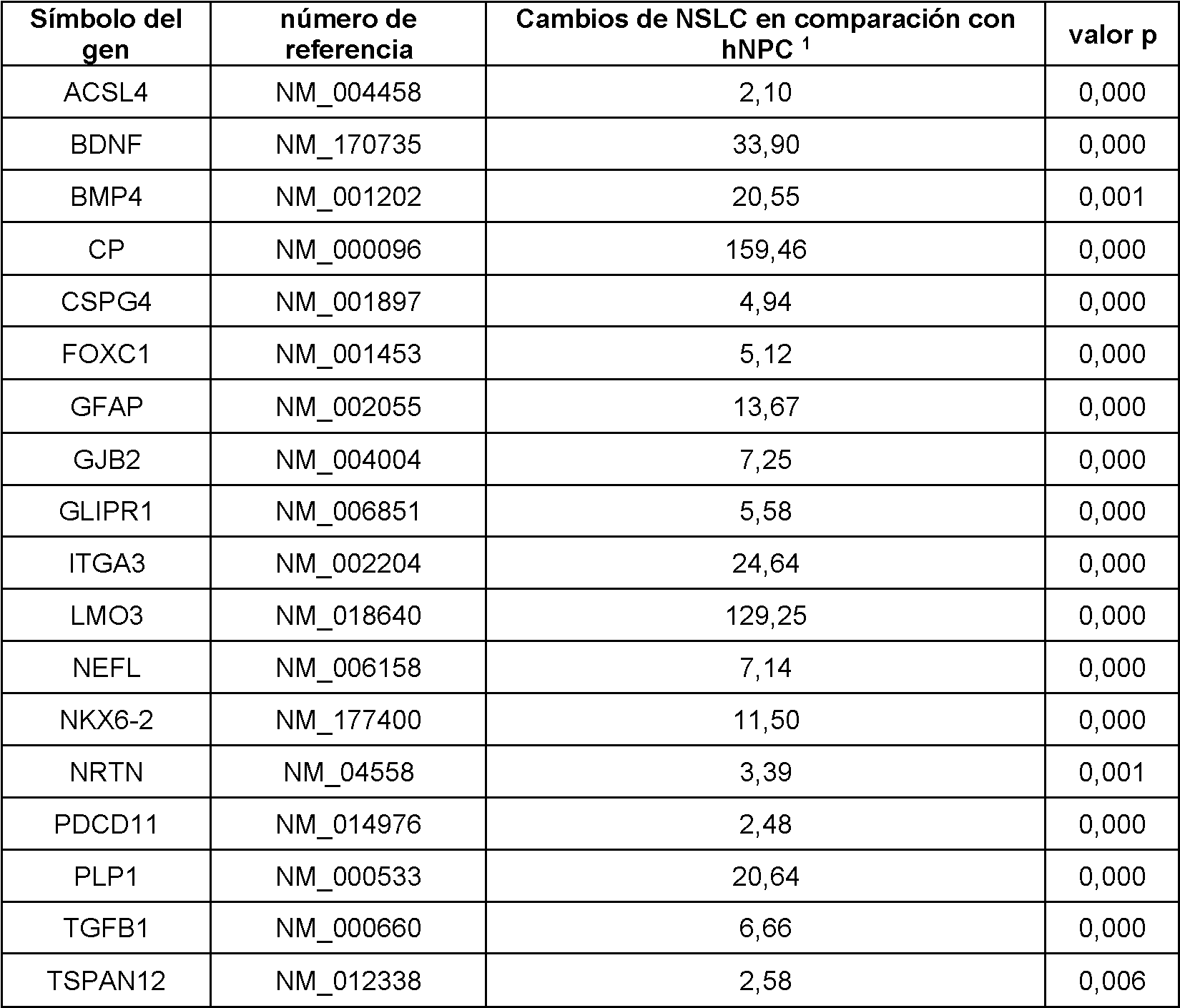
Tabla X7: Genes regenerativos regulados por U (NSLC frente a hNPC)

Para investigar el potencial de diferenciación de las NSLC a linajes neuronales (neuronas, astrocitos y oligodendrocitos), las neuroesferas se disociaron y se sembraron en placas en laminina/poli-D-lisina (10 pg/ml; Sigma) en medio de diferenciación durante dos semanas. La diferenciación hacia el linaje neuronal se realizó utilizando dos medios diferentes: Medio NbActivo (BrainBits™) suplementado con factor de neurotrofina derivado del cerebro (BDNF, 20 ng/ml, Peprotech), ácido todo trans-retinoico (ATRA, 5 pM, Spectrum), y bFGF (40ng/ml, Peprotech) o medio de diferenciación NeuroCult™ (kit de diferenciación NeuroCult™, StemCell Technologies), suplementado con BDNF (20 ng/ml, Peprotech) y bFGF (40 ng/ml, Peprotech). Después de dos semanas de cultivo, las células se tiñeron con el marcador neuronal plll-tubulina, los marcadores de astrocitos GFAP y S1OOp, el marcador de oligodendrocito CNPasa. Las células se fijaron con formaldehído al 4 % y los anticuerpos primarios se añadieron en suero de cabra normal/PBS al 5 % de la siguiente manera: Anticuerpo de ratón pIM-tubulina (1:2OO, Abcam), anticuerpo de conejo SIOOp (1:1OO, Abcam) y anticuerpo de pollo CNPasa (1:50, Abcam). Los anticuerpos secundarios se añaden en un 5 % de suero normal de cabra/PBS de la siguiente manera: Anti-ratón de cabra Alexa546™ (1:200, Invitrogen), Anti-coneJo de cabra Alexa488 ™ (1:200, Invitrogen), y anti-pollo cy5 de cabra (1:1OO, Jackson ImmunoResearch Labs).
El análisis inmunohistoquímico mostró que el medio NbActive promovía la diferenciación por igual al linaje neuronal (48,66 ± 14,07 %, plll-tubulina) y posibles linajes de oligodendrocitos tempranos (5O,O1 ± 4,04 %, CNPasa) y a un menor porcentaje de células de astrocitos (2,68 ± 1,13 %, S1OOp), mientras que el medio de diferenciación NS-A indujo la diferenciación principalmente a las neuronas (64,89 ± 4,11 %, plll-tubulina) y astrocitos (35,94 ± 4,04 %, S1OObeta), y un bajo porcentaje de células de oligodendrocitos tempranos potenciales (8,68 ± 2,71 %, CNPasa). El medio NSC-A se seleccionó sobre NbActive para estudios de diferenciación adicionales. La diferenciación de células en el medio de diferenciación NS-A promueve la diferenciación de hNPC y NSLC de manera similar a como se muestra en la Tabla 17 por la disminución del porcentaje de células positivas para sox2, musashi y nestina. Las NSLC se diferenciaron a linajes neuronales (74,3 ± O,1, GABA), de astrocitos (65,6 ± O,O, S1oObeta) y a un porcentaje menor de células oligodendrocitos (5,2 ± 0,6, CNPasa). El mismo patrón de diferenciación del linaje tripotencial se observó con las hNPC (Tabla 17).
Tabla 17: El porcentaje de células con tinción positiva para células madre neurales y marcadores de linaje neuronal en células transfectadas y no transfectadas. Las NSLC y hNPC se cultivaron en medio de diferenciación NS-A suplementado con BDNF (20 ng/ml) y FGF (40 ng/ml), los cultivos se incubaron a 37 °C, 5 % de CO2, 5 % de O2 durante tres semanas. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± SD (n = 5).

Se utilizaron varios anticuerpos adicionales para antígenos neuronales para caracterizar, con más detalle, la naturaleza de las células diferenciadas. Se usaron anticuerpos contra la proteína asociada a los microtúbulos
(MAP2b), NCAM y sinaptofisina, según lo recomendado por el fabricante del anticuerpo. Después de tres semanas en medio de diferenciación, hubo una reducción inducida por la diferenciación en los marcadores de células precursoras y un aumento en los marcadores neuronales maduros. El porcentaje de marcadores precursores neurales tales como Sox2 disminuyó durante la diferenciación, mientras que p75NTR, plll-tubulina y GABA aumentaron con el tiempo de diferenciación de alargamiento (Figura 6); sin embargo, las células positivas para 04 fueron muy bajas después de 3 semanas de diferenciación de hNPC (6,4 ± 2,9) y NSLC (8,2 ± 0,6). La sinaptofisina, un anticuerpo usado para identificar células neuronales funcionales, se incrementó tras 2 y 3 semanas de diferenciación, lo que indica madurez de las células neuronales. Los marcadores de GABA y acetilcolina aumentaron después de 2 semanas de diferenciación y disminuyeron en la semana 3.
Los cambios morfológicos y la expresión de varios antígenos y genes neuronales muestran que el procedimiento anterior produce células neuronales normales y viables. Adicionalmente, las células neuronales recién formadas tienen los criterios morfológicos de las neuronas. Además de los marcadores anteriores, las células diferenciadas se evaluaron caracterizando marcadores morfológicos de diferenciación de neuritas. Las células de tipo neuronal (células que expresan fuertemente plll-tubulina) mostraron formación de neurita después de la diferenciación, incluyendo un aumento en el número promedio de neuritas por neurona (por ejemplo, 1,38 ± 0,1). Se observó el mismo patrón en células positivas para plll-tubulina. En consecuencia, la longitud promedio de las neuritas (118,3 ± 3,5 |jm) y el número de puntos de ramificación (3,28 ± 0,3) por neurona también aumentaron. Las células de tipo neuronal diferenciadas desarrollaron neuritas largas que tenían más de tres diámetros celulares de longitud con un cono de crecimiento al final, expresaron genes específicos de neuronas y dejaron de proliferar después de la inducción de la diferenciación.
Se realizó una diferenciación adicional utilizando un medio optimizado que promovió la diferenciación hacia el linaje de oligodendrocitos. Las NSLC y hNPC se cultivaron en medio de diferenciación NS-A como se ha descrito previamente suplementado con Fg F-2 (10 ng/ml, Peprotech) y hedgehog sónico (SHH, 100 ng/ml, Peprotech) durante 4 días. Después de 4 días, el medio se cambió a medio de diferenciación NS-A suplementado con T3 (60 ng/ml, Peprotech), IGF1 (10ng/ml, Peprotech), NT-3 (10 ng/ml, Peprotech) y PDGF (10 ng/ml, Peprotech). Las células se cultivaron durante 20 días a 370C, 5 % de CO2.
Tabla 18: El porcentaje de células con tinción positiva para células madre neurales y marcadores de linaje neuronal en células transfectadas y no transfectadas. Las NSLC y hNPC se cultivaron en medio de diferenciación suplementado con SHH (100 ng/ml, Peprotech), T3 (60 ng/ml, Peprotech), IGF1 (10ng/ml, Peprotech), NT-3 (10 ng/ml, Peprotech) y PDGF (10 ng/ml, Peprotech) para inducir la diferenciación hacia oligodendrocitos. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± SD (n = 5).
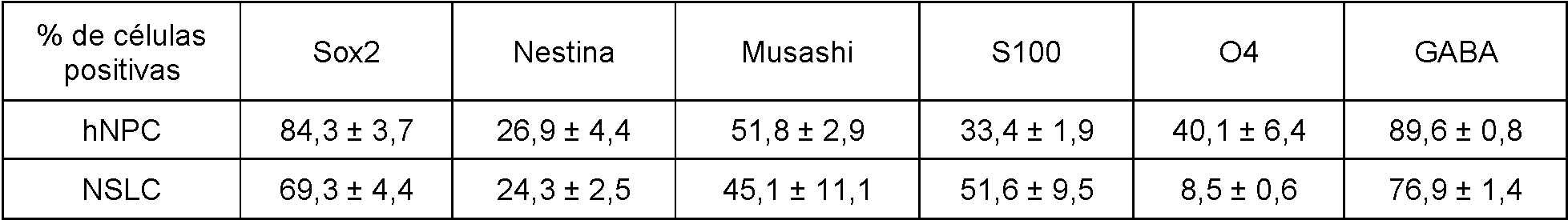
La cuantificación de la diferenciación de hNPC y NSLC reveló una población de células que se tiñeron positivamente para 04. Como se muestra en la Tabla 18, el porcentaje de células positivas para 04 fue más pronunciado en hNPC diferenciadas (40,1 ± 6,4 %) en comparación con las NSLC diferenciadas (8,5 ± 0,6 %) cuando se usó el protocolo de diferenciación anterior.
Este estudio demostró que la transfectación de las células con uno o dos factores de transcripción neurogénicos en presencia de un desmetilador de ADN o moléculas pequeñas para la modificación epigenética logra células estables reprogramadas (NSLC). Como un desmetilador de ADN, la modificación epigenética (inhibición de la acetilación y la metilación) a veces es útil para impulsar el proceso de reprogramación. Estas células poseen y conservan las propiedades de las células madre neurales según lo determinado por: (1) la expresión de genes y proteínas de células madre neurales, (2) la capacidad para generar y crecer como neuroesferas a partir de una sola célula y (3) para diferenciarse en linajes neuronales en condiciones de diferenciación. Cuando se diferencian en neuronas, las células muestran una o más características morfológicas, fisiológicas y/o inmunológicas específicas neurales asociadas con un tipo de célula neuronal. Los criterios útiles incluyen características morfológicas (procesos largos o neuritas), características fisiológicas y/o inmunológicas, tales como la expresión de un conjunto de marcadores o antígenos neuronales específicos. Adicionalmente, las NSLC se convierten fácilmente en una célula precursora de tipo tripotencial con potencial de diferenciación a un alto porcentaje de poblaciones neuronales, de astrocitos y menor porcentaje de poblaciones de oligodendrocitos.
Ejemplo VI
Implicación de la vía de señalización e la BMP en la reprogramación de HFF
Este estudio fue diseñado para evaluar el papel de Noggina en el proceso de desdiferenciación de HFF hacia NSLC. Los HFF se cultivaron y se trataron con citocalasina B como se describe en el Ejemplo lll. Después de dos días de
tratamiento, las células se transfectaron mediante nucleofección como se describe en el Ejemplo II con el vector construido Msi1/Ngn2. Brevemente, después de preparar las células, se mezclaron con 2 |jg de ADN total (Msi1/Ngn2) y se cotransfectaron con MBD2 (2 jg), por el Nucleofector ™ de Amaxa de acuerdo con el protocolo del fabricante. A continuación, las muestras se transfirieron a una placa de cultivo recubierta con laminina (10 jg/ml, Sigma) y se cultivaron en presencia de medio de proliferación neural (Kit de proliferación NeuroCult™, StemCell Technologies) con hFGF recombinante (20 ng/ml, Peprotech), hEGF recombinante (20 ng/ml, Peprotech), y con o sin la presencia de Noggina (20 ng/ml, Peprotech). Las muestras se recolectaron en diferentes puntos de tiempo (1, 3, 4, 6 y 8 días) para analizar la expresión génica neuronal mediante RT-PCR y los niveles de expresión de proteínas mediante inmunohistoquímica.
Se realizó tinción imunohistoquímica fluorescente en muestras después de 4 días de transfección como se describe previamente en el Ejemplo I. Las células transfectadas se tiñeron y se analizaron para determinar la expresión de Sox2, el porcentaje de Sox2 fue de 33,3 ± 1,00 % en presencia de Noggina en comparación con 27,5 ± 0,5o % sin la presencia de noggina el día 4. El análisis por RT-PCR de la expresión relativa de marcadores de células neuronales precursoras como la nestina y Sox2 después de la transfección de HFF con pCMV-Msi1-2A-Ngn2 y pCMV6-XL5-MBD2 con o sin la presencia de Noggina (20 ng/ml) se asoció con un aumento de nestina y Sox2 a partir del día 3 y se mantuvo hasta el día 8 (Tabla 19). No se notó ninguna diferencia en la expresión en ausencia de Noggina. La inhibición de la vía de señalización de la BMP por parte de la Noggina mejoró así la reprogramación, pero no tuvo ningún efecto de reprogramación en sí mismo.
Tabla 19: Análisis por RT-PCR de la expresión relativa de marcadores de células neuronales precursoras, tales como nestina y Sox2, después de la transfección de HFF con pCMV-Msi1-2A-Ngn2 y pCMV6-XL5-MBD2 con o sin Noggina (20 ng/ml). La expresión relativa de Sox2 y nestina aumentó después de la transfección con y sin Noggina.

continuación

Ejemplo Vil
Las NSLC creadas a partir de células HFF no son precursoras derivadas de la piel (SKP)
Se sabe que las células denominadas precursoras derivadas de la piel (SKP) pueden residir en la piel humana adulta (Fernandes y col., 2004). Estas células son capaces de proliferar en respuesta a EGF y bFGF y expresan nestina, versican y fibronectina, y pueden diferenciarse en progenie neuronal y mesodérmica. Con el fin de verificar que las NSLC son distintas de las SKP, se realizó diferenciación hacia células adipocitarias. Las células madre derivadas de tejido adiposo (ADSC) se mantuvieron en medio libre de suero StemPro™ MSC (Invitrogen) en matraces recubiertos con CellStart™ (Invitrogen). CellStart™ se diluyó a 1:100 en dPBS/Ca2+/Mg2+ y el matraz se incubó durante 2 horas a 37 °0C. Las células se pasan cada 3 a 4 días utilizando Accutase™ y el medio se cambia cada 2 días. De tres a cuatro días antes de iniciar la diferenciación, se sembraron ADSC y Ns LC en placas de 6 pocilios en CellStart™ (1: 100 en dPBS/Ca2+/Mg2+/ 2 horas a 37 °0C) placas de cultivo tisular recubiertas. Cuando las células alcanzaron la confluencia (después de 3 a 4 días), los medios de proliferación se reemplazaron por medio de diferenciación consistente en DMEM/F12 (50:50), ITS (1:100), HEPES (1:100), GlutaMAX™ (1:100), T3 (0,2 nM), Rosiglitasona (0,5 pg/ml), IBMX (100 pM) y Dexametasona (1 pM). Tres días después, se retiraron el IBMX y la dexametasona del medio de diferenciación. El día 10, las células se fijaron con una solución de formaldehído al 4 % durante 10 minutos y se tiñeron con una solución de tinción con Oil Red O (Invitrogen) durante 15 minutos. Se eliminó la tinción y las células se lavaron dos veces con PBS. Las células adiposas aparecieron rojas con gotitas de lípidos teñidas específicamente con Oil Red O, sin embargo, las NSLC e tiñeron negativas, sin presencia de gotitas de lípidos en las células, y las células adoptaron la morfología de las células neuronales.
El análisis inmunohistoquímico confirmó que las NSLC eran distintas de los SKP (Figura 24): Las NSLC se tiñeron positivamente para p75NTR y negativamente para fibronectina y versican, mientras que la SKP expresan fibronectina y versican y no expresan p75NTR (Fernandes y col., 2004). Este estudio indica que las NSLC representan una célula precursora de tipo tripotencial y no son una subpoblación de SKP.
Ejemplo Vlll
Liberación de BDNF a partir de células de tipo neuronal (NLC)
Las células de tipo madre neurales (NSLC) diferenciadas en células neuronales y gliales se mantuvieron en cultivo durante 55 días y el BDNF liberado en el medio condicionado se midió mediante ELISA de captura de antígeno en diferentes puntos de tiempo y se comparó con la liberación en neuronas maduras (ScienCell), células precursoras normales humanas neurales no diferenciadas (NHNP, Lonza), así como a NSLC no diferenciadas y células no transfectadas (HFF). Se recogió medio acondicionado de cada grupo, se centrifugó, y, a continuación, se almacenó a -80 °C hasta su ensayo. Las concentraciones de BDNF se midieron con kits ELISa (Sistema de inmunoensayo
Emax BDNF, Promega Corporation, Estados Unidos), de acuerdo con las instrucciones del fabricante. En resumen, las inmunoplacas de ELISA de 96 pocilios se recubrieron con Anti-BDNF (N.0 cat. G700B) diluido 1/1000 en tampón de carbonato (pH 9,7) y se incubaron a 4 °C durante la noche. Al día siguiente, todos los pocilios se lavaron con TBS-Tween™ al 0,5 % antes de la incubación con tampón Block/Sample IX a temperatura ambiente durante una hora sin agitación. Después de bloquear, se añadieron a las placas los patrones y las muestras, y se incubaron y se agitaron (450 ± 100 rpm) durante 2 horas a temperatura ambiente. Posteriormente, después del lavado con tampón de lavado TBS-Tween ™, las placas se incubaron durante 2 h con pAb anti-BDNF humano (dilución 1:500 en tampón de bloqueo y muestras IX) a 4 °C. Después de la incubación, las placas se lavaron cinco veces con tampón de lavado TBS-Tween™ al 0,5 % y se añadieron 100 mi de conjugado de anti-IgYHRP diluido a cada pocilio (dilución 1:200 en IX tampón de bloqueo y muestra) y se incubaron durante 1 hora a temperatura ambiente con agitación (450 ± 100 rpm). Después, las placas se lavaron cinco veces con tampón de lavado TBS-Tween™ al 0,5 % y se añadieron lOo mi de TMB One Solution a cada pocilio. Después de 10 minutos de incubación a temperatura ambiente con agitación (45O ± 1OO rpm) para la placa BDNF, se formó un color azul en los pocilios. Después de detener la reacción añadiendo 1OO mi de ácido clorhídrico 1N, la absorbancia se leyó a 45O nm en un lector de microplacas (Synergy 4™) en los 30 minutos posteriores a la detención de las reacciones. La concentración de BDNF liberado en los sobrenadantes se determinó de acuerdo con las curvas estándar.
Los resultados de ELISA revelaron que se liberó BDNF a la misma concentración de células de tipo neuronas diferenciadas (NLC diferenciadas de NSLC) y células neuronas humanas normales a partir del día 11 y se mantuvo hasta el día 55 (Tabla 20), mientras que no se liberó BDNF (excepto pequeñas cantidades en el grupo de HFF no transfectados) en los otros grupos.
Tabla 20: Cuantificación de la liberación de BDNF por células de tipo neural (NLC) que se habían diferenciado durante 55 días a partir de células de tipo madre neurales (NSLC) que se habían creado a partir de HFF transfectados. La liberación de BDNF desde los NLC al medio, en diferentes puntos de tiempo, se midió mediante ELISA de captura de antígeno y se comparó con la liberación de BDNF de neuronas humanas maduras normales
(ScienCeli).

Además de adoptar criterios de morfología neuronal, las NLC eran funcionales y poseían la capacidad de liberar factor neurotrófico (BDNF). La generación de líneas celulares de tipo neuronal reprogramadas que pueden liberar localmente estos factores neurotróficos podría usarse como procedimiento para tratar varias afecciones neurológicas y podría ofrecer beneficios cruciales en la regeneración y la recuperación funcional del cerebro y otras lesiones. Ejemplo IX
Reprogramación de diferentes tipos celulares hacia NSLC: Este estudio se realizó para investigar la capacidad de los queratinocitos (Invitrogen), las células madre derivadas de adipocitos humanos (ADSC, Invitrogen) y las células madre hematopoyéticas humanas (CD34+, Invitrogen) en células de tipo madre neurales.
Preparación de células CD34+ humanas, ADSC humanos y queratinocitos humanos: Las células CD34+ de sangre periférica movilizadas humanas se adquirieron en StemCeil Technologies y se expandieron como cultivo flotante en placas Petri en medio completo StemPro™®-34 sin suero (Invitrogen) suplementado con factor de células madre (SCF, 15O g/ml, Peprotech), factor estimulante de colonias de granulocitos (GM-CSF, 37,5 ng/ml, Peprotech) e IL-3 (75 ng/ml, Peprotech). El medio suplementado con citocinas se cambió todos los días 2-3 días después de la centrifugación de la suspensión celular a 3OO xg durante 10 min. Cada dos días, las citocinas se agregaban directamente al cultivo sin cambiar los medios. Las células se incubaron a 37 oc, 5 % de CO2. Para su pase, las células se centrifugaron, se resuspendieron en el medio anterior más citocinas y se colocaron en el número adecuado de placas Petri.
Las células madre derivadas de tejido adiposo humano (ADSC) se adquirieron en Invitrogen y se expandieron en matraces recubiertos con medio StemPro™ MSC sin suero (Invitrogen) en CelIStart™™ (Invitrogen) (diluido a 1:1OO en PBS con Ca2+/ Mg2+) a una densidad celular de 1x104 células/cm2. El medio se reemplazó cada dos días con StemPro ™ MSC SFM completo precalentado recién preparado. Las células se incubaron a 37 oc, 5 % de CO2. Las
células se sub-pasaron cuando el 80 % fue confluente por Incubación durante 3-5 minutos en TrypLE™™ (Invitrogen) precalentado y, a continuación, se recolectaron en medio StemPro™ MSC. Después de la centrifugación a 1500 rpm durante 5 min, las células se sembraron en matraces recubiertos con CellStart™™ como se ha descrito anteriormente.
Los queratinocitos humanos primarios se adquirieron en Invitrogen y se expandieron en medio sin suero de queratinocitos definidos en matraces recubiertos (Invitrogen) de matriz de recubrimiento (Invitrogen) a una densidad celular de 5x103 células/cm2 Las células se incubaron a 370C, 5 % de CO2. El medio se reemplazó con medio de crecimiento completo recién preparado cada dos o tres días hasta el subcultivo. Una vez que las células habían alcanzado un 70-80 % de confluencia, el medio se eliminó y las células se incubaron en Versene™ (Invitrogen) durante 3-5 minutos a temperatura ambiente. Se retiró Versene™ y se añadió a los matraces con tripsina al 0,05 %-EDTA (Invitrogen) precalentado. Después de 5-10 minutos de incubación, se añadió medio de crecimiento que contenía inhibidor de tripsina de soja (Invitrogen) a los matraces y las células se trituraron suavemente. Después de la centrifugación a 100 x g durante 10 min, las células se resuspendieron en el volumen deseado de medio de crecimiento completo precalentado en matraces recubiertos como se ha descrito anteriormente.
Antes de la transfección, las células se digirieron con tripsina y se cotransfectaron transitoriamente con pCMV-Msil-Ngn2 y pCMV6-XL5-MBD2 como se ha descrito anteriormente en el Ejemplo IV utilizando la lanzadera y se sembraron en placas de cultivo recubiertas con laminina (Sigma, 10 |jg/ml). Comenzando un día después de la transfección, las células se trataron con VPA (1 mM) durante 4 días y el medio se cambió gradualmente a medio de proliferación suplementado con FGF (20 ng/ml) y EGF (20 ng/ml) y se cultivaron durante 18 días a 37 oc, 5 % de CO2 y 5 % de O2, A continuación, las células se analizaron para detectar marcadores de células madre neurales mediante RT-PCR e inmunohistoquímica.
El análisis y la cuantificación adicionales de las células reprogramadas revelaron una población de NSLC engendradas a partir de queratinocitos y células CD34+. El análisis por RT-PCR reveló un aumento de la expresión relativa de marcadores de células madre neurales, tales como Sox2, nestina, GFAP y plll-tubulina después de la transfección de queratinocitos y CD34+ por MsI1 y Ngn2. La expresión relativa de nestina y GFAP se mejoró en las NSLC creadas a partir de queratinocitos y células CD34+ en comparación con las NSLC de HFF; sin embargo, lo contrario fue cierto para la expresión de Sox 2 y ACHE. La expresión de plll-tubulina (TUBB3) y Map2b fue más alta en las NSLC creadas a partir de células CD34+, seguidas de las NSLC creadas a partir de HFF (Tabla 21). Estos datos muestran que se pueden crear diferentes tipos de NSLC con diferentes perfiles de expresión génica (y características) a partir de diferentes tipos de células de partida/origen (y se ha observado lo mismo para crear algunos otros tipos de células de tipo madre analizadas en esta solicitud). Los datos también son intrigantes, ya que no se esperaba que los queratinocitos (que derivan del ectodermo al igual que las células madre neurales endógenas) tuvieran una expresión más baja que los HFF para todos los genes analizados, excepto para la nestina (se esperaba que los queratinocitos fueran más fáciles de reprogramar en NSLC, ya que derivan del ectodermo).
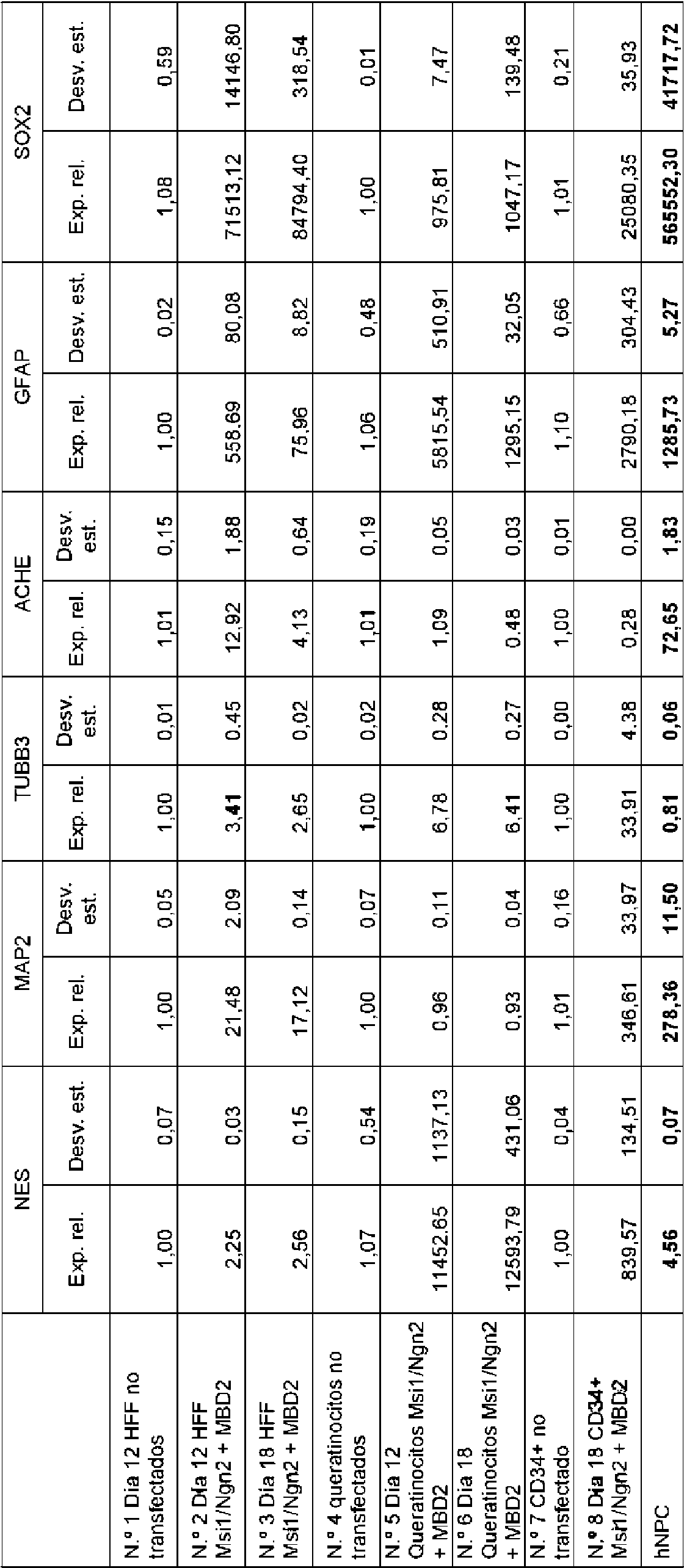
La inmunohistoquímica reveló tinción positiva para GFAP, Sox2 y nestina como se muestra en la Figura 7. Las NSLC desarrolladas a partir de HFF producen un mayor porcentaje de tinción positiva para Sox2 y GFAP (55,8 ± 3,8 y 78,1 ± 2,4) en comparación con las células CD34+ (42,8 ± 2,7 y 24,2 ± 4,4), y queratinocitos (47,1 ± 2,1 y 43,4 ± 8,9). El porcentaje de células positivas para nestina fue alto en queratinocitos (77,6 ± 10,7) y HFF (68,45 ± 12,9) y menor en células CD34+ (15,5 ± 2,7) (Tabla 22). La tinción positiva para Sox2 y Nestina no fue detectable en las a d s c .
Tabla 22: El porcentaje de células positivas para Sox2 y nestina para marcadores de células madre neurales después de la transfección de fibroblastos, queratinocitos y células CD34+ con pCMV-Msi1-Ngn2 en presencia de MBD2 y VPA. Las células se cultivaron en placas de cultivo recubiertas en medio de proliferación (StemCell Technologies) suplementado con EGF (20 ng/ml) y FGF (20 ng/ml) durante 18 días. Las células no transfectadas se consideraron como control negativo. El porcentaje de células inmunopositivas se determinó mediante Cellomics™ y se representó como la media ± SD (n = 5).

Las NSLC generadas a partir de queratinocitos y células CD34+ se analizaron para determinar su capacidad tripotencial. Se realizaron estudios de diferenciación adicionales para inducir la diferenciación de estas NSLC hacia el linaje neuronal, utilizando el medio de diferenciación NeuroCult™ (kit de diferenciación NeuroCult™, StemCell Technologies) suplementado con BDNF (20ng/ml, Peprotech) y bFGF (40ng/ml, Peprotech) como se describe en el Ejemplo V. Las NSLC generadas a partir de HFF y hNPC se utilizaron como controles, los cultivos se incubaron a 37 °C, 5% de CO2, 5% de O2 durante tres semanas. Las muestras se recolectaron o se fijaron los días 14 y 28 después de la diferenciación para un análisis adicional. El análisis por RT-PCR reveló una disminución de los genes no diferenciados (Nestina y Sox2) y un aumento de los genes diferenciados (Map2, pIM-tubulina, CNPasa y GFAP) como se muestra en las Tablas 23A, 23b , 23C y 23D.




La tinción inmunohistoquímica fluorescente se realizó en muestras después de 14 días y 28 días de diferenciación. La expresión de Sox2 y Nestina disminuyó el tiempo de forma dependiente en células diferenciadas (HFF, queratinocito y CD34+). Esta disminución se asoció con un aumento de marcadores diferenciados el día 28 como GFAP (68,51 ± 11,87 para HFF-NC, 59,55 ± 9,12 para queratinocitos NC y 61,70 ± 1,48 para CD34+ -NC). Se generó un alto porcentaje de células positivas para plll tubulina a partir de NSLC diferenciadas generadas a partir de HFF (57,83 ± 4,49) en comparación con células positivas a plll tubulina generadas a partir de queratinocitos (23,27 ± 2,91) y células CD34+ (39,15 ± 7,99) (Tabla 24)
Tabla 24: El porcentaje de células con tinción positiva para marcadores de células madre neurales y marcadores de linaje neuronal en hNPC (Lonza) y queratinocitos transfectados, células HFF y CD34+ con pMsi1/Ngn2/MBD2. Las células transfectadas (NSLC) se cultivaron en medio de proliferación o medio de diferenciación durante 28 días a 37 0C, 5 % de CO2, 5 % de O2. El porcentaje de células inmunopositivas (Sox2, nestina, GFAP, S100beta y pllltubulina) se determinaron mediante Cellomics™ y se representaron como media ± SD (n = 5).

El % de células positivas para Sox2 disminuyó más rápido, el % de células positivas para nestina disminuyó generalmente más despacio y el % de células que expresan uno de los marcadores de diferenciación (S100p, pllltubulina, GFAP) aumentó generalmente más despacio en hNPC que en las NSLC durante la diferenciación. De los tres tipos de líneas de NSLC creadas, el % de células que expresan uno de los marcadores de diferenciación (S100P, plll-tubulina, GFAP) generalmente aumentó más despacio en las NSLC creados a partir de queratinocitos y más rápido en las NSLC creadas a partir de HFF.
Este estudio indica que se pueden crear NSLC a partir de queratinocitos y células sanguíneas CD34+ y estas células comparten morfología y marcadores de manera similar a las NSLC generadas a partir de HFF. Al igual que las hNPC, las NSLC creadas a partir de queratinocitos, las células CD34+ y los HFF tenían una tendencia a diferenciarse más hacia un linaje de astrocitos que a un linaje neuronal (excepto las NSLC creadas a partir de los HFF tenían un número casi similar de células positivas para plll-tubulina y positivas para GFAP) como lo muestra el
alto porcentaje de células positivas para GFAP durante la diferenciación, lo que se confirmó mediante tinción SlOObeta. Sin embargo, la proporción de astrocitos y células neuronales generadas a partir de hNPC fue menor en las mismas condiciones de cultivo, lo que indica que las NSLC generadas a partir de HFF, queratinocitos y células CD34+ pueden dar lugar a un mayor número de células neuronales y astrocíticas en comparación con las hNPC. Las NSLC, ya sea creadas a partir de HFF, queratinocitos o células CD34+ (o potencialmente incluso alguna otra célula), son células tripotenciales y poseen la capacidad de diferenciarse en neuronas, astrocitos y oligodendrocitos de manera similar a las hNPC. Sin embargo, el análisis de RT-PCR e inmunohistoquímica de a Ds C transfectadas no reveló ninguna expresión significativa de los genes de células madre neurales, lo que indica la necesidad de optimizar las condiciones para convertir las ADSC en NSLC o investigar el efecto de otros factores neurogénicos que podrían convertirlos en nSlC.
Ejemplo X
Fabricación de matriz extracelular 3D (CDM)
Las células fibroblastos se cultivaron en medio DMEM en presencia de FCS al 10 % como se describe en el Ejemplo I, seguido de siembra en placas de 12 pocilios prerrecubiertas con laminina (10 pg/ml) a una concentración de 2x1o6 células/ml en medio CDM definido que consiste en una relación 3:1 de medio Eagle modificado por Dulbecco (DMEM, alto contenido de glucosa (4,5 g/l) con L-glutamina y piruvato de sodio) y medio F-12 de Ham suplementado con los siguientes componentes: eGf (4,2 x lo -10 M), bFGF (2,8 x 10-10M), ITS (8,6x10'5 M), dexametasona (1,0x10' 7M), fosfato de ácido L-ascórbico, sal de magnesio n-hidrato (3,2 x 10'4M), L-3,3',5-triyodotironina (2,0 x lo -10 M), etanolamina (1O'4M),GlutaMAX™ (4x10'3M), glutatión (3,3x10'6M) y 1 % de penicilina/estreptomicina/anfotericina B. Al cultivar las células fibroblastos a una densidad hiperconfluente en este medio completamente químicamente definido, esto hace que entren en una fase sintética alta con una desaceleración en la proliferación, lo que lleva a la producción de un equivalente de tejido vivo (LTE) que consiste en múltiples capas de fibroblastos dentro de la matriz extracelular 3D de novo (CDM) que se sintetiza completamente por los mismos fibroblastos.
Transdiferenciación y reprogramación de células dentro de CDM
Las muestras de CDM del día 14 se trataron con citocalasina B (10 pg/ml, Calbiochem), con la concentración de citocalasina B reducida de 10 pg/ml a O pg/ml (nada) durante 5 días, mientras que al mismo tiempo se cambia el medio de medio CDM a medio NbActive. Las muestras se cultivaron durante otros 12 días a 370C, 5 % de CO2 y el medio se cambió todos los días. Las muestras se fijaron para realizar inmunohistoquímica como se ha descrito anteriormente para detectar marcadores neuronales. Se usaron los anticuerpos siguientes: anti-nestina 647 de ratón (1:1OO, BD) y anti-pIN-tubullna (1:2OO, Neuromics). No se observaron cambios morfológicos claros de las células en el CDM y el análisis inmunohistoquímico no pudo detectar células positivas para plll-tubullna. Así, la inducción de la transdiferenciación de las células utilizando solo citocalasina B y medio neural definido químicamente no fue suficiente para reprogramar las células.
A continuación, muestras de CDM del día 6 cultivadas en placas prerrecubiertas con LAS a 37 °C y 5 % de CO2, se expusieron simultáneamente a citocalasina B (1O pg/ml) durante 5 días, inhibidor de la desacetilación de histonas (VPA, 4 mM, Calbiochem) e inhibidor de la metilación del ADN (5-azacitidina, 5 pM, Sigma). Cuatro días después el medio se cambió a medio de diferenciación que consiste en una relación 3:1 de medio CDM sin la presencia de EGF y medio activo Nb (BrainBits™) suplementado con NT-3 (20 ng/ml, Peprotech) y BDNF (20 ng/ml, Peprotech). La relación del medio de diferenciación se incrementó gradualmente día tras día hasta alcanzar el 1OO % del medio de diferenciación completo. Después de dos semanas de tratamiento, las células se fijaron para un análisis inmunohistoquímico para investigar la identidad de las células. La Figura 18 muestra células inmunoteñidas con pllltubulina el día 7, lo que indica la desdiferenciación de las células fibroblastos en neuronas. Sin embargo, una semana después, estas células transdiferenciadas volvieron a las células fibroblastos y se perdió la expresión de pIM-tubulina (Figura 8). La pérdida de la morfología y la expresión de pIN-tubulina después de la retirada de los agentes de cebado indican que no se produjo una conversión completa en células reprogramadas funcionales y estables.
A continuación, el CDM se trató con VPA (4 mM), 5-Aza (5 pM) y citocalasina B (1O pg/ml) como anteriormente. Después de 2 días de tratamiento químico, las células fibroblastos dentro del CDM se transfectaron con ADN usando el reactivo Lipofectamine (Invitrogen) según el protocolo del fabricante. Se usaron 15 pg de los vectores de expresión de ADN eucarióticos pCMV6-XL5-Pax6, Se utilizaron pCMV6-XL5-MsI1 y pCMV6-XL4-Ngn2 (Origene) para transfectar las células. 24 horas después, el medio se cambió a medio basal progenitor neural (Lonza) suplementado con Noggina (50 ng/ml), EGF (20 ng/ml) y bFGF (20 ng/ml) y las células se cultivaron a 37 oc, 5 % de CO2 y 5 % de O2 y el medio se cambió todos los días. El día 6, la diferenciación se inició mediante la adición gradual de medio NBActive (BrainBits™) suplementado con NT-3 (20 ng/ml, Peprotech), ácido todo trans-retinoico (ATRA, 5 pM, Spectrum), BDNF (20ng/ml, Peprotech) y bFGF (40ng/ml, Peprotech). Para caracterizar las células reprogramadas, el análisis Inmunohistoquímico y la RT-PCR se realizaron en varios puntos de tiempo de acuerdo con los procedimientos descritos en el Ejemplo II utilizando cebadores para nestina., pIN-tubulina, g Fa P, MAP2b y ACHE. De acuerdo con estudios previos, las células no transfectadas y las células transfectadas con Pax6 no expresaron genes específicos para linajes neuronales (Tabla 25). Por otro lado, tras la transfección con MsI1, Ios niveles de nestina y ACHE se incrementaron a 4 veces y 8 veces, respectivamente, y esta expresión se mantuvo
durante el período de 12 días. Asimismo, Ios niveles de ARNm de GFAP aumentaron el tiempo de forma dependiente en aproximadamente 14 veces. Asimismo, se observó el mismo patrón en células transfectadas con Ngn2. Mientras que la expresión de plll-tubulina y MAP2b aumentaron moderadamente después de la transfección con un factor de transcripción neurogénico, la regulación de la expresión génica después de transfectar las células con dos factores neurogénicos, Msil o Ngn2 con Pax6, no aumentó adicionalmente la expresión de Ios genes neuronales. La Figura 19 muestra que la expresión de estos genes se mejoró cuando las células se transfectaron con Msil y Ngn2, con pMI-tubulina aumentada a casi 6 veces el día 12.
„ ° 0 i
D) i u - ” -n ro ® 112 B O í „ O c = ° y a. E ¿ i :e 3 -- -S a) O
co O O QJ f/¡ £ ^ TJ
q> uS p .33
0Jd ^ « *“ E ^ . - ” 03 > CD 8 O ■
:‘ o Q. ~ § 3 "O ^ en 5 Q. ü 1> ( wD 2 co " -Oo urj 5 tu v ^-t . uE ¿ E



Se observó el mismo patrón de expresión génica al transfectar las células con tres factores de transcripción (MsI1, Ngn2 y Pax6), pero la expresión fue menos pronunciada que en las células que se transfectan solo con Msil y Ngn2. En términos de análisis inmunohistoquímico después de los 12 días de la transfección, las células mostraron marcadores neuronales después de la transfección con Msil o Ngn2, como lo indica la expresión de nestina y MAP2b (Figura 9). Las células transfectadas con pCMV-XL-PAx6 no se tiñeron para nestina y MAP2b.
Este estudio muestra que la transfección de células dentro de CDM con un solo factor neurogénico (Msil o Ngn2) induce cambios morfológicos y la expresión de uno o más marcadores de células madre neurales y células neuronales. Dado que las células reprogramadas expresaron un factor neurogénico clave, un marcador neuronal precursor y un marcador neuronal maduro a bajo porcentaje (10 %), esto sugiere que las células dentro del CDM se transformaron en NSLC y, a continuación, comenzaron a diferenciarse a través de varias fases del programa de determinación y diferenciación neuronal inducido en células madre neurales.
Ejemplo XI
Análisis de la expresión génica de células reprogramadas en CDM
Este estudio fue diseñado para probar el efecto de transfectar células con Msil y Ngn2 en presencia de MBD2 en el proceso de reprogramación. Las células se transfectaron después de dos días de tratamiento previo con citocalasina B con los vectores de expresión de ADN utilizando el reactivo Lipofectamine como se describe en el Ejemplo X. Se usaron 15 |jg de vectores de expresión de ADN eucarióticos pCMV6-XL5-Musashi o pCMV6-XL4-Ngn2 y pCMV6-XL5-MBD2 (Origene) para cotransfectar las células. Después de 24 horas, el medio se cambió a medio de mantenimiento CDM: progenitores neuronales (1:1) suplementado con Noggina (50 ng/ml), EGF (20 ng/ml) y bFGF (20 ng/ml). El medio se cambió todos los días aumentando el porcentaje de NPBM y disminuyendo el medio CDM. Las células se cultivaron durante 6 días a 37 0C, 5 % de CO2 y 5 % de O2. Después de una semana, la diferenciación se inició suplementando gradualmente el medio NPBM con NT-3 (20 ng/ml, Peprotech), ácido a // -trans-retinoico (ATRA, 5 j M, Spectrum), BDNF (20ng/ml, Peprotech) y bFGF (40ng/ml, Peprotech). Las muestras se recolectaron al final del estudio (día 14) y los datos se analizaron mediante una matriz de genes para identificar los genes que se encontraron de forma reproducible que son específicos para los linajes neuronales.
Análisis de la expresión génica
El análisis de la expresión génica en 8 muestras se realizó como se ha descrito anteriormente en el Ejemplo I con los marcadores neuronales personalizados 2 TLDA para identificar la expresión de genes relacionados con las células madre neurales, células neuronales y células gliales, y factores de crecimiento expresados por las células después de la transfección. La expresión de los genes oligodendrocitos, tales como NKx2.2, olig2 y MAG se incrementó en Msil y Ngn2; sin embargo, el aumento fue más pronunciado con Msil en comparación con Ngn2 (Tabla 26). Dos marcadores para astrocitos (GFAP y AQP4) se expresaron altamente después de la transfección con Msil y Ngn2 en presencia del desmetilador de AdN MBD2. De forma interesante, se mejoraron varios marcadores de células neuronales tempranas; 12 días después de la transfección, los datos de TDLA revelaron aumentos en marcadores específicos para interneuronas, tal como somatostatina y calbindinal. La doblecortina (DCX), que se expresa en células inmaduras en migración durante el desarrollo, y acetilcolina (ACHE), un marcador temprano de células neuronales, se expresaron considerablemente en células reprogramadas (Tabla 26). La transfección con Msil o Ngn2 aumentó la expresión de dihidropirimidinasa 3 (DPYSL3), un marcador temprano de neuronas recién formadas a cinco veces con Msil y siete veces con Ngn2. La expresión de la proteína 2 asociada a los microtúbulos (MAP2), un marcador esencial para el desarrollo y mantenimiento de la morfología neuronal temprana y la molécula de adhesión de células neuronales (NCAM) se expresaron altamente con Msil y Ngn2. La expresión de enolasa-2, un marcador de neuronas maduras, aumentó 20 veces con Msil y Ngn2. El miembro de la familia NeuroD NeuroDI se expresó altamente después de la transfección con Msil a 84,22 veces y a 34,27 por Ngn2. La expresión génica de factores de crecimiento tales como IGF-1, IGF-2, NPY y CSF-3 se mejoraron después de la transfección con Msil o Ngn2. La expresión de los genes VEGF y GDNF se incrementó a casi cinco veces y siete veces por Msil y Ngn2, respectivamente. Sin embargo, en las células transfectadas, la expresión de BDNF, EGF y bFGF no se activó e incluso se reguló por disminución en comparación con las células no transfectadas. La expresión de la proteína asociada al crecimiento (GAP-43), un marcador asociado al crecimiento y la regeneración de la extensión de las neuritas, y la expresión de netrina, implicada en el desarrollo y la orientación neuronal, e expresaron considerablemente en células transfectadas (Tabla 26). La expresión de receptores para el crecimiento y factores neurotróficos se incrementó, tales como el receptor de tipo III tirosina quinasa, el receptor de tirosina quinasa neurotrófico y la tirosina quinasa neurotrófica. Los marcadores específicos de fibroblastos vimentina y fibronectina estaban regulados por disminución en las células reprogramadas.
La transfección de HFF solo con Msil y Ngn2 en presencia de MBD2 aumentó la expresión de células gliales y marcadores de células neuronales.
Tabla 26: Conjunto de genes de CDM transfectados con pMsil y pNgn2 después del tratamiento previo con citocalasina B (10 pg/ml), VPA (4 mM) y 5-azacitidina (5 pM). Las células transfectadas se cultivaron en medio de diferenciación (NbActive, BrainBits™) suplementado con ATRA (5 pM), bFGF (40ng/ml) y BDNF (20ng/ml).

continuación

Ejemplo XII
Reprogramación de células en CDM mediante Lipofectamine y nucleofección
Este estudio se diseñó ara mejorar la transfección de CDM combinando lipofectamine y nucleofección y usando dos vectores pCMV6-XL5-Msí1 y pCMV6-XL4-Ngn2 individualmente o en combinación con pCMV-XL5- MBD2. Las células el día 4 en CDM se lipotransfectaron durante 6 horas con Msí1/MBD2, Ngn2/MBD2 o Msi/Ngn2/MBD2 después de 2 días de tratamiento previo con o sin citocalasina B. En paralelo, la transfección se realizó en HFF
recién preparados después de las 6 horas usando Nucleofección como se describe en el Ejemplo II y se transfirió a la parte superior del CDM cuando el medio de lipofectamina se cambió al medio CDM recién preparado. Después de 24 horas, el medio se cambió a medio basal progenitor neural (NPBM, Lonza) con la presencia de Noggina (50 ng/ml, Peprotech), hFGF recombinante (20 ng/ml, Peprotech) y hEGF recombinante (20 ng/ml, Peprotech). El día 7 se indujo diferenciación, añadiendo medio de diferenciación NSA-A (StemCell Technologies) durante 21 días.
Análisis de la expresión génica
Las muestras se recogieron a los 8, 15 y 21 días para evaluar la naturaleza de las células recién formadas mediante el análisis de la expresión de varios genes marcadores neuronales utilizando RT-PCR de acuerdo con los procedimientos descritos previamente en el Ejemplo I. Como se muestra en la Tabla 27, las células transfectadas con un factor de transcripción neurogénico (Msil o Ngn2) expresan niveles altos de nestina y pMI-tubulina el día 8. El mismo patrón de expresión se observó los días 15 y 21, mientras que la expresión disminuyó ligeramente en ausencia de citocalasina B en células transfectadas con Ngn2. La expresión de todos los genes, excepto el marcador neuronal maduro MAP2b, aumentaron notablemente en las células transfectadas con ambos factores de transcripción neurogénicos. La regulación por aumento de estos genes se redujo ligeramente en ausencia de citocalasina B, lo que indica su papel en la mejora de la reprogramación.




Análisis imunohistoquímico
Las muestras se recogieron a Ios 4, 8, 14 y 21 días para evaluar la naturaleza de las células reprogramadas mediante el análisis de la expresión de varios marcadores neuronales utilizando análisis inmunohistoquímico de acuerdo con Ios procedimientos descritos previamente en el Ejemplo I. El análisis inmunohistoquímico en varios puntos de tiempo reveló que dentro de Ios primeros 8 días se indujo la expresión de nestina en una gran proporción de células y disminuyó de forma dependiente del tiempo después de inducir la diferenciación (Figura 10).
Este estudio indica que al transfectar las células con uno o dos genes neurogénicos en presencia de citocalasina B y MBD2, las células reprogramadas fueron estables en cultivo, respondieron a Ios cambios ambientales (proliferación frente a diferenciación) y expresaron marcadores neuronales durante al menos 24 días en cultivo.
Ejemplo Xlll
Actividad telomerasa de las NSLC
La telomerasa está activa en las células precursoras neurales y sugiere que su regulación es un parámetro importante para que ocurra la proliferación celular en el cerebro de Ios mamíferos (Caporaso GL et, 20o3). Este estudio se realizó para evaluar la actividad telomerasa en extractos celulares de NSLC adherentes (NSLC cultivadas en placas recubiertas con laminina), así como NSLC en neuroesferas flotantes (NSLC cultivadas en placas con una superficie de unión baja) en el pase temprano (P7) y tardío (P27). La actividad telomerasa de las 4 muestras se midió mediante el protocolo de amplificación de repetición de telómeros (TRAP) basado en PCR utilizando el kit de detección de telomerasa TRAPeze® (Chemicon). En resumen, las células se cultivaron en placas de 24 pocilios, se lavaron en PBS y se homogeneizaron durante 30 minutos en hielo en un tampón que contenía Tris-HCl 10 mM, pH 7,5, MgCÍ21 mM, EGTA 1 mM, benzamidina 0,1 mM, p-mercaptoetanol 5 mM, CHAPS al 0,5 % y glicerol al 10 % (IX tampón de lisis CHAPS, incluido en el kit) e inhibidor de la ARNasa. Las muestras se centrifugaron y la concentración de proteína del sobrenadante se determinó mediante el ensayo BCA. Se añadieron 900 ng de proteína de cada extracto celular directamente a la mezcla de reacción TRAP que contenía el tampón de reacción TRAP, dNTP, cebador del sustrato molde (TS), mezcla de cebadores TRAP y Taq polimerasa. Las mezclas de reacción se incubaron a 30 °C durante 30 minutos para la síntesis del molde, seguido de un procedimiento de PCR (95 °C/15 min para la desnaturalización inicial, 94 oC/30 s, 59 °C/30 s, 72 °C/1 min durante 32 ciclos) para la amplificación de Ios productos de telomerasa extendida. Para detectar la actividad telomerasa, la electroforesis en gel de poliacrilamida (PAGE) se realizó para Ios productos de reacción en un gel TBE no desnaturalizante al 10 %. Después de la electroforesis, el gel se tiñó con SYBR® Green I Nucleic Acid Gel Stain durante 30 minutos, seguido de la captura de imágenes mediante un sistema de documentación en gel (Alpha Innotech).
Las 4 muestras fueron positivas para la telomerasa (como lo indica la escala de productos TRAP) como se muestra en la Figura 11. Como era de esperar, el control tratado con calor (H) no mostró actividad de telomerasa (control negativo). Se usa una banda de control interno de 36 pb (S-IC) para monitorizar la amplificación de la PCR (para distinguir Ios resultados falsos negativos). Esta banda S-IC se observó para todas las muestras, excepto para las muestras de prueba. Esto puede deberse a la actividad telomerasa excesivamente alta en las muestras de prueba; la amplificación de Ios productos TRAP y la banda de control S-IC son semicompetitivas. Todos Ios controles dieron Ios resultados esperados (no hay productos TRAP para el control de CHAPS, y la escala TRAP de productos para las células de control positivo y el control TSR8).
Ejemplo XIV
Ensayo de formación de tumores
Las células transformadas de manera maligna muestran requerimientos reducidos de factores que promueven el crecimiento extracelular, no están restringidas por el contacto célula-célula y son, a menudo, inmortales. El crecimiento y la proliferación independientes del anclaje es una de las características de la transformación maligna, que se considera el ensayo in vitro más preciso y riguroso para detectar la transformación maligna de las células. Las NSLC adherentes y de neuroesferas en el pase temprano y tardío (P7 y P25), así como células neuroprogenitoras humanas normales (hNPC), se investigaron para determinar el crecimiento independiente del anclaje. Los HFF se utilizaron como control negativo y las células HeLa de carcinoma cervical se usaron como control positivo. Las células se sedimentaron mediante centrifugación a 150 x g durante 3 minutos a temperatura ambiente (RT). El ensayo se realizó utilizando el ensayo de transformación de células de 96 pocilios CytoSelect™ (CellBiolabs). La capa de agar base (1,2 %) se disolvió en 2X DMEM/solución de PBS AL 20 % y se añadieron 50 |jI de la solución de agar a la placa y se incubaron durante 30 minutos a 4 °C para solidificar. Antes de añadir la capa de agar celular, la placa se dejó calentar durante 15 minutos a 37 oc. Las células se resuspendieron a diferente densidad (20.000 y 5000 células/pocillo), excepto que las hNPC se resuspendieron solo a 5000 células/pocillo debido a la falta de suficientes células. Las células se mezclaron con la solución de agar al 1,2 %, 2X DMEM/PbS al 20 % suspensión celular (1:1:1) y 75 j l de la mezcla se transfirieron a Ios pocilios que ya contenían la capa de agar base solidificada y, a continuación, se colocaron a 4 °C durante 15 minutos para permitir que la capa de agar celular solidificara. Se añadieron 100 j l de medio de proliferación (StemCell Technologies) y la placa se incubó durante 8 días a 37 °C y 5 % de CO2 antes de solubilizarse, se lisó y detectó con el colorante CyQuant ™ GR en un lector de
placas de fluorescencia. La medición de fluorescencia se realizó utilizando el Flexstation™ (Molecular Devices) con un filtro de 485/538 nm.
Tabla 28: La medición de la fluorescencia (Unidad de Fluorescencia Relativa, RFU) indica que, en las mismas condiciones, solo las células HeLa de carcinoma crecen como una colonia independiente del anclaje, mientras que tanto las hNPC como las NSLC (neuroesferas adherentes y flotantes) fueron negativas para el crecimiento tumoral en el ensayo estándar de formación de tumores en placa de agar (kit de transformación de células CytoSelect™, Cell Biolabs Inc.).
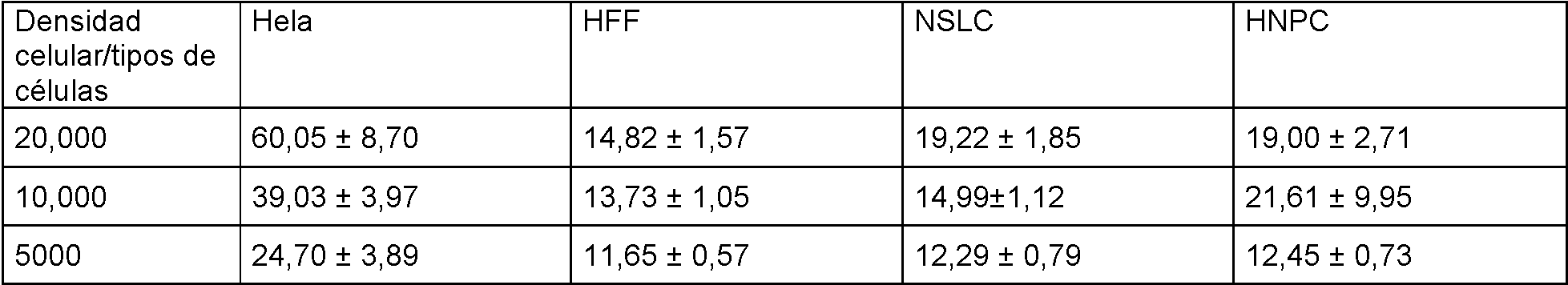
Tal como se muestra en la Tabla 28, la medición de fluorescencia indicó que en las mismas condiciones, solo las células HeLa de carcinoma crecieron y proliferaron significativamente como colonias independientes de anclaje, mientras que tanto las hNPC como las NSLC (neuroesferas adherentes y flotantes) fueron negativas para el crecimiento tumoral (el mismo valor que los HFF (control negativo) para 5.000 y 10.000 células) en el ensayo de formación de tumores en placa de agar estándar mediante observación visual de células mediante observación con microscopio óptico usando campo brillante a 10X confirman la medición de fluorescencia. Por lo tanto, el procedimiento de transfección transitoria y los genes utilizados permiten la reprogramación de células sin la transformación neoplásica que generalmente ocurre con la transfección estable o ciertos genes a través de una serie de alteraciones genéticas y epigenéticas que producen una población de células que es capaz de proliferar independientemente de las señales tanto externas e como internas que normalmente frenan el crecimiento.
Ejemplo XVI
Ausencia de integración genómica de ADN plasmídico en NSLC de transfección transitoria
El plásmido de ADN Msi1/Ngn2 (diseñado y construido de forma interna) se usó en la transfección transitoria para la generación de NSLC junto con MBD2 (para la muestra 1) o 5-Aza y VPA (para la muestra 2). Dos semanas después de la transfección, se realizó una transferencia Southern para probar la posible integración genómica del ADN plasmídico. 3 |jg de ADN genómico extraído de las muestras NSLC, así como de HFF (una línea celular de fibroblastos humanos) utilizada como control negativo, se digirieron con varias enzimas de restricción, incluyendo Bglll, Pstl y Stul, se sometieron a electroforesis en un gel de agarosa al 1 % y se transfirieron a una membrana de nailon con carga positiva (Roche). La membrana se hibridó en el tampón DIG Easy Hyb™ (Roche) a 42 °C durante la noche con una sonda de PCR marcada por Dig de 1,2 kb amplificada del ADN plasmídico usando un conjunto de cebadores. La membrana se lavó dos veces a temperatura ambiente con 2 x SSC, SDS al 0,1 % durante 5 minutos por lavado, dos veces con 0,5 x SSC, SDS al 0,1% a 65 °C durante 15 minutos por lavado. Las señales de hibridación de la membrana se detectaron utilizando el sustrato CDP-Star™ (Roche). La membrana se expuso a una película de rayos X para su análisis. Las señales se eliminaron de la membrana utilizando un tampón de extracción (NaOH 0,2 M, SDS al 0,1 %). La membrana se volvió a hibridar con una sonda de PCR marcada con Dig de 0,9 kb amplificada a partir del ADN plasmídico utilizando un conjunto de cebadores.
El análisis de transferencia Southern (Figura 12) con la sonda de PCR marcada con Dig de 1,2 kb reveló distintas señales en las muestras de control positivo en las que el ADN plasmídico Msi1/Ngn2 se introdujo en el ADN genómico de HFF para la equivalencia de 1, 10 o 100 integraciones por genoma. Hubo algunas bandas débiles e idénticas que aparecieron en el ADN genómico digerido con enzimas de restricción procedente de HFF, NSLC muestras N.01 y N.02, lo que sugiere que no había integración de ADN plasmídico en el ADN genómico de las NSLC. Estas bandas pueden representar el gen Ngn2 endógeno ya que la sonda de PCR marcada con Dig de 1,2 kb contiene una pequeña parte del gen Ngn2. Este dato muestra que ninguna, o solo un pequeño número de ellas, NSLC presentaba integración de plásmidos en el genoma del huésped después de la transfección transitoria y que los genes transfectados solo están presentes en la célula durante un corto período de tiempo (menos de dos semanas).
Ejemplo XVII
Efecto neuroprotector de las hNSLC transplantadas en:
1) Modelo animal de esclerosis múltiple.
La esclerosis múltiple (EM) es una enfermedad inflamatoria desmielinizante inflamatoria del sistema nervioso central (SNC) (Frohman EM et al 2006). Las terapias para la EM se basan en la manipulación del sistema inmunológico, pero con una efectividad a menudo modesta en la reducción de episodios clínicos o discapacidad neurológica
permanente, requiriendo inyecciones frecuentes y con efectos secundarios a veces significativos (Langer-Gould A et al 2004). La encefalomielitis alérgica experimental (EAE) es un modelo animal de EM usado habitualmente para estudiar mecanismos de enfermedades y probar terapias potenciales. La EAE puede inducirse en diversas especies y cepas de animales [ratones, rata, mono tití, macacos rhesus] utilizando diversos antígenos del SNC [glicoproteína oligodendrocítica de mielina (MOG), proteína proteolipídica (PLP) y proteína básica de mielina (MBP)].
Después de obtener todas las aprobaciones de animales apropiadas para los experimentos, se compraron ratones hembra C57BL/6 de 7 a 8 semanas de edad de Charles Rivers y se alojaron en las instalaciones de animales MISPRO durante una semana antes de la experimentación para la adaptación al nuevo entorno. Se inyectaron en los ratones C57BL/6 s.c. 100 |jg de MOG 35-55 en CFA (Sheldon Biotechnology, McGill University) que contiene 5 mg/ml de Mycobacterium tuberculosis H37Ra (Difeo, inc), en 2 sitios en la parte posterior. Todos los ratones recibieron 20o ng de toxina pertussis (List Biologieal Laboratories, Inc) i.p. los días 0 y 2, mientras que las puntuaciones clínicas se calcularon a ciegas diariamente durante un período de 43 días, según la escala 0-5 de la siguiente manera: 1, cola floja o marcha de pato con tonicidad de la cola; 2, marea de pato con cola floja (ataxia); 2,5, ataxia con parálisis parcial de las extremidades; 3, parálisis completa de 1 extremidad; 3,5, parálisis completa de 1 extremidad con parálisis parcial de la segunda extremidad; 4, parálisis completa de 2 extremidades; 4,5, moribundo; y 5, muerte.
Tratamiento del modelo animal de EAE con y sin células:
Se administraron hNSLC y hNPC (1,5 x 106 células en 200 j l de PBS/eada ratón) mediante inyección única í.v. a través de la vena de la cola cuando los animales comenzaron a mostrar síntomas de EAE (día 13 í.v). Ambos grupos de animales recibieron ciclosporina (10 rng/kg/día) un día antes de la inyección de células y diariamente desde el día del trasplante para evitar cualquier rechazo de las células humanas. Se usaron ratones del mismo sexo, la misma edad, de cepa equivalente y tratados de forma simulada, a los que se inyectó i.p. PBS solo, como controles. Todos los grupos de animales se sometieron a observación durante 43 días. Se sacrificó a los animales a los 43 del tratamiento, se extrajeron los cerebros y la médula espinal en sacarosa al 30 % en PBS. El análisis estadístico de las puntuaciones clínicas reveló que los signos clínicos de la EAE se atenuaron significativamente en los animales a los que se inyectó NSLC en comparación con los animales de control y a los que se inyectó hNPC. Las puntuaciones acumulativas se redujeron significativamente en los animales trasplantados con NSLc (Figura 13) y el tratamiento no tiene efecto sobre el peso corporal.
2) Modelo animal hemipléjico (ablación unilateral de la corteza sensoríomotora izquierda en ratas adultas).
Después de obtener todas las aprobaciones de animales apropiadas para los experimentos, Se anestesió a 8 ratas por grupo (Sprague-Dawley, 250-300 g, Charles River) con ketamina (Bimeda-MTC)/xílazína (50/10 mg/kg, Novopharm) y se colocaron en un mareo estereotáxieo. Se realizó una incisión craneal en la línea media con una hoja de bisturí quirúrgica estéril, se expuso la bóveda craneal expuesta y se identificó el bregma. Se abrió el cráneo sobre la corteza sensoríomotora y el área de la corteza sensoríomotora [0,5 - 4,0 mm caudal a bregma y 1,8 - 3,8 mm lateral a la línea media (Paxinos y Watson 1986)] se aspiró cuidadosamente. Después de la ablación, los tratamientos (alginato, alginato hNPC, alginato NSLc , RMx NSLc , RMx solo, gel de fibrina, o solución salina) se aplicaron directamente en el cerebro después de la ablación. A continuación, la abertura en el cráneo se rellenó con cera de hueso. En caso de sangrado, se insertaron pequeños trozos de tejido homeostátieo estéril en la lesión para detener el sangrado. Las suturas se realizaron utilizando la forma de aguja de 1/2 círculo de sutura de monofilamento Ethicon™. Las cirugías se realizaron en salas limpias estériles y se aplicaron antibióticos tópicos (Cieatrin®, GlaxoSmithKIÍne) al cráneo y cuero cabelludo expuestos para limitar la infección local. Las ratas se inmunosuprimieron mediante inyección diaria i.p. de ciclosporina A (10 mg/kg/día) a partir del día anterior a la cirugía hasta el final del período de estudio. El fin de la inyección de ciclosporina A era reducir la reacción inmune de la rata al tratamiento. La ínmunosupresión se mantuvo hasta el final del estudio para garantizar que cualquier fallo potencial de regeneración (sí se produce) no se debió a la reacción inmunitaria contra el tratamiento. Las puntuaciones funcionales se realizaron semanalmente en todos los grupos, El deterioro sensorímotor se evaluó según las pruebas de comportamiento que se describen a continuación.
Prueba de Rotarod: La velocidad del rotarod se calibró manualmente para la velocidad de 10 y 20 RPM antes de todos los procedimientos. Los animales debían posarse en la barra estacionaria durante 30 segundos para aclimatarse al medio ambiente. Durante este tiempo, si caía algún animal, se volvía a colocar en la barra hasta que alcanzara las capacidades estacionarias durante un período de 30 segundos. Se permitió a los animales 3 pruebas. Los animales que se sentían cómodos permaneciendo en la barra estacionaria durante 30 segundos se dejaron correr con una velocidad constante de 10 y 20 RPM durante 60 segundos y el número de caídas se registró electrónicamente.
Viga para caminar: La viga para caminar mide la coordinación de las extremidades posteriores por medio de la distancia recorrida a través de una viga de 100 em (2,3 em de diámetro, 48 em del suelo). Las ratas fueron entrenadas sistemáticamente para caminar a lo largo de la viga elevada de principio a fin con el objetivo de completar la tarea. Se coloca un lugar seguro, es decir, una caja plana, se coloca al final de la viga para motivar a la rata a completar la tarea.
Escala utilizada para evaluar el rendimiento en la viga para caminar
ESCala Característica de rendimiento
1 Los animales no atraviesan la viga y no colocan el miembro posterior sobre la superficie horizontal de la viga
2 Los animales no atraviesan la viga, pero colocan la extremidad posterior en la superficie horizontal de la viga y mantienen el equilibrio
3 Los animales atraviesan la viga mientras arrastran el miembro posterior
Los animales atraviesan la viga y colocan el miembro posterior al menos una vez durante la 4 travesía
Los animales atraviesan la viga utilizando el miembro posterior para ayudar a menos del 50 % de 5 sus pasos en la viga
6 Los animales atraviesan la viga utilizando el miembro posterior izquierdo para ayudar a más del 50 % de sus pasos en la viga
7 Los animales atraviesan la viga con no más de dos resbalones
8 Animales normales
Antes de la cirugía, todos los animales se cayeron al menos una vez del rotarod, no porque tuvieran un problema al caminar o de coordinación, sino porque la velocidad era alta. Después de la cirugía (2 días), todos los animales mostraron signos de problemas significativos de marcha y coordinación que llevaron a un aumento en el número de caídas del rotarod. Tres semanas después de la cirugía, el número de caídas se redujo claramente para los animales que recibieron NSLC como tratamiento en comparación con los controles (Figura l4 ).
Los animales pasaron la prueba de marcha en la viga antes de la cirugía sin ninguna dificultad. Las ratas cruzaron la viga de 100 cm y llegaron al lugar seguro sin caerse de la viga. Dos días después de la cirugía, todos los grupos fracasaron completamente en pasar la prueba y los animales no pudieron mantenerse en equilibrio en la viga. Una semana después de la cirugía, todos los animales mostraron alguna mejora en su capacidad de caminar, pero no se observó diferencia significativa entre los diferentes grupos tratados. Desde la semana 4 hasta la semana 26, los animales tratados con NSLC y RMx mostraron mejoras significativas en su capacidad de caminar en comparación con los controles (Figura 15).
Ejemplo de referencia XVIII
Transfección de HFF por varias combinaciones de genes con el dispositivo Shuttle® y tratamiento con diferentes moléculas pequeñas para reprogramar células de tipo mesoendodérmico
Las células HFF se cultivaron como se describe en el medio CDM II como se describe en el Ejemplo I con solo modificar EGF (5 ng/ml) y FGF (10 ng/ml), y se transfectaron utilizando el dispositivo Shuttle® de 96 pocilios Nucleofector™® (Lonza) después del procedimiento descrito en el Ejemplo IV. Las células se transfectaron con diversas combinaciones de clones de ADNc como se describe en la Tabla 29. Después de la transfección, las células se sembraron en placas recubiertas con gelatina al 0,1 % y se incubaron a 370C, 5 % de CO2, 5 % de O2. El medio se cambió cada dos días de acuerdo con la Tabla 30. Las células se analizaron el día 4 mediante PCR cuantitativa en tiempo real.
Tabla 29: Varias combinaciones de plásmidos con potencial para transfectar las células hacia el linaje
mesoendodérmico.

continuación
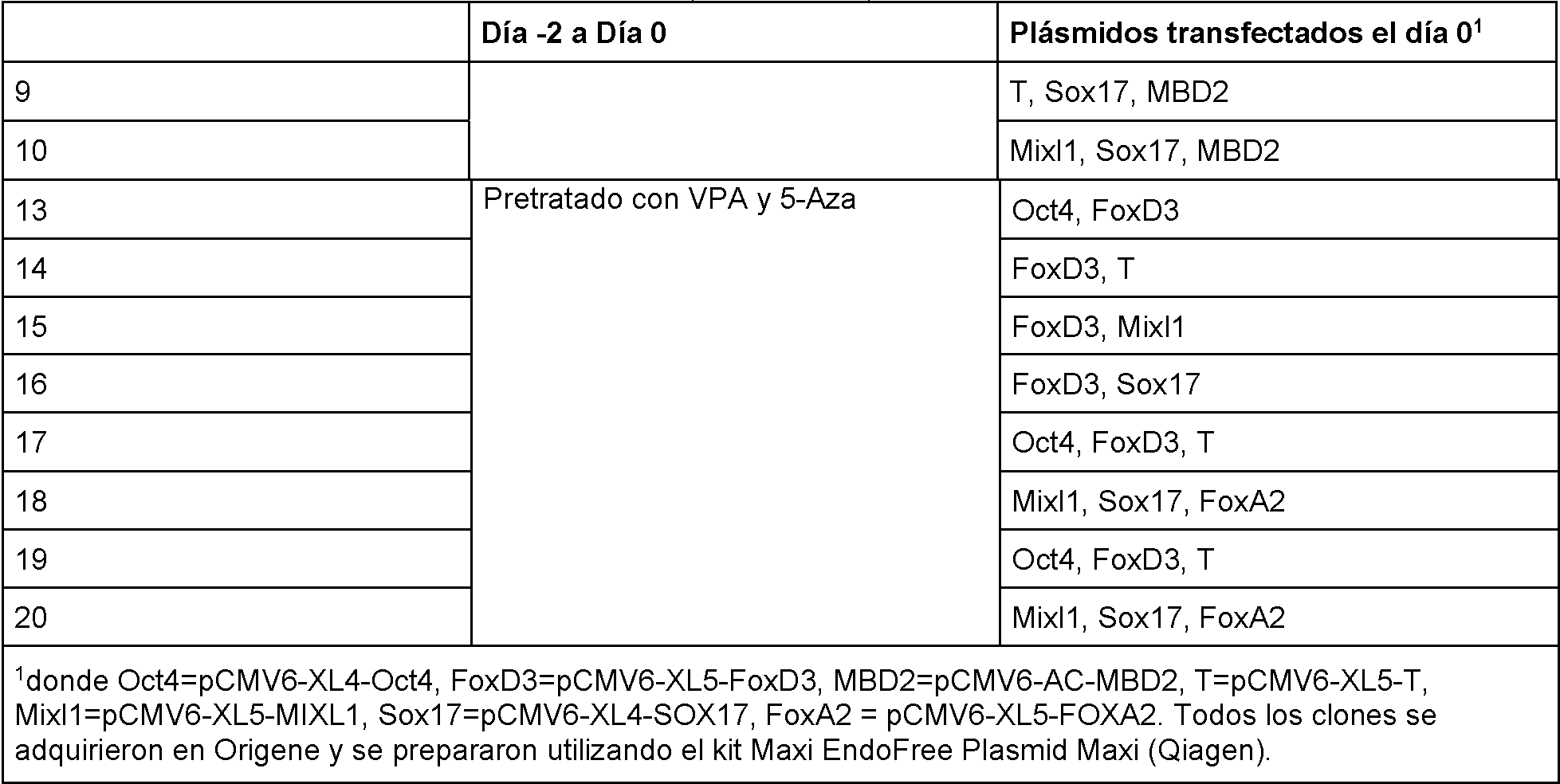
Tabla 30: Composición media desde el día -2 hasta el día 10

Las células se recolectaron el día 4 al separarlas con TrypLE™, seguido de centrifugación a 80 x g durante 5 minutos. El sobrenadante se aspiró y el sedimento celular se congeló a -86 °C hasta que estuvo listo para el aislamiento del ARN. El aislamiento y la cuantificación del ARN se realizaron como se ha descrito previamente en el Ejemplo I. Se preparó un ADNc y se realizó una PCR cuantitativa en tiempo real como se ha descrito previamente en el Ejemplo II, excepto que se usaron los siguientes ensayos de expresión de genes Taqman™® (Applied Biosystems):

continuación

Tabla 31: Expresión relativa de FoxA2, Sox17 y Cxcr4 después de transfectar HFF una vez con varias combinaciones de genes con potencial para reprogramar células en células tipo mesoendodérmico. Los valores exactos no son significativamente exactos debido al bajo rendimiento de a Rn . sin embargo, se detectó una tendencia de aumento de la expresión génica para FqxA2. Sox17 y CXCR4.

continuación

Tabla 32: Expresión de GATA4, CDH1 (E-cadherina), p63 y SOX2 en relación con el control de HFF no tratado 4 días después de la transfección de células HFF con varias combinaciones de genes con potencial para reproaramar células en células tipo mesoendodérmico.


La identificación de combinaciones de genes que pueden inducir la formación de células de tipo mesoendodérmico se investigó mediante transfección con combinaciones de Oct4, Sox17, FoxD3, T, MíxII, FoxA2 y MBD2. Como se muestra en la Tabla 25 y 26, la expresión relativa de CXCR4 y GATA4, ambos marcadores mesoendodérnico/endodérmico/mesodérmico, parecen estar regulados por aumento en varias combinaciones, más notoriamente en FoxD3/MíxI1/MBD2 y FoxD3/Sox17/MBD2. De forma similar, FOXA2, un marcador para endodermo y mesodermo, se reguló por aumento FoxD3/Sox17-muestra transfectada, Aunque la expresión sigue siendo muy baja. Cuatro días después de la transfección, SOX17 aún se expresa altamente en las muestras transfectadas con s Ox 17 (50.000 a 400.000 veces en comparación con la muestra de HFF no tratada). La expresión del gen SOX17 representa el ADN plasmídico sobrante (SOX17 exógeno) que aún permanece 4 días después de la transfección y cualquier expresión de SOX17 endógeno que pueda haberse inducida. Los marcadores ectodérmicos CDH1, p63 y Sox2 también se regularon por aumento en algunas muestras (por ejemplo, OcT4/FoxD3/MBD2, Oct4/Sox 17/m BD2).
Reprogramación de HFF en células de tipo progenitoras pancreáticas: Las células HFF se cultivaron como se describe en el Ejemplo I y se transfectaron utilizando el dispositivo Shuttle® (Lonza) de 96 pocilios Nucleofector™® siguiendo el procedimiento descrito en el Ejemplo IV. Las células se transfectaron con diversas combinaciones de clones de ADNc como se describe en la Tabla 27. Después de la transfección, las células se sembraron en placas de gel de colágeno recubiertas con fibronectina y se incubaron a 370C, 5 % de CO2, 5 % de O2. Las placas de gel de colágeno recubiertas con fibronectina se prepararon antes de la transfección. El colágeno I de rata (Gibco) se diluyó a 1,125 mg/ml utilizando 10X PBS y agua destilada, donde se añadieron 125 |jI a cada pocilio de una placa de 24 pocilios y se incubaron a 37 °C durante 40 minutos. Después de aclarar con IX PBS, se añadió fibronectina (BD Biosciences) en la parte superior del gel a una concentración de 1,9 jg/pocillo. Los medios se cambiaron cada dos días de acuerdo con la Tabla 33. Las células se analizaron el día 7 mediante PCR cuantitativa en tiempo real.
Tabla 33: Plásmidos y composición de medios del día 0 al día 14

continuación

Las células se recolectaron el día 7 y el aislamiento y la cuantificación del ARN se realizaron como se ha descrito anteriormente en el Ejemplo I. Se preparó un ADNc y se realizó una PCR cuantitativa en tiempo real como se ha descrito anteriormente en el Ejemplo II, excepto que se usaron los siguientes ensayos de expresión de genes Taqman™® (Applied Biosystems):

La identificación de combinaciones de genes que pueden inducir la formación de células de tipo progenitoras pancreáticas se investigó mediante transfección con combinaciones de FoxD3, Sox17, Pdxl, Ngn3, Mixll e MBD2. FoxA2, un marcador para endodermo y mesodermo, estaba regulado por aumento ligeramente para la muestra transfectada con FoxD3/Sox17/Ngn3/MBD2 en comparación con la muestra de control transfectada de manera simulada con GFP. De forma similar, CXCR4, también un marcador tanto para el endodermo como para el mesodermo, estaba regulado por aumento ligeramente (3 veces en comparación con GFP-ctrI) para la muestra transfectada con FoxD3/MixI1/Ngn3/MBD2. 7 días después de la transfección, SOX17 aún puede detectarse para las
muestras transfectadas con SOX17 en niveles variables (4 a 570 veces regulación por aumento en comparación con la GFP-ctrl). La expresión de la regulación por aumento de SOX17 más alta se detecta para la muestra transfectada con Sox17/Mixl1/Pdx1/Ngn3 (570 veces en comparación con GFP-ctrl), lo que puede sugerir que esta combinación de genes puede aumentar la cantidad de ARN de SOX17 en las células.
Ejemplo de referencia XIX
Reprogramación de células madre derivadas de adipocitos humanos (ADSC) en células madre de tipo pluripotencial (PLSC): Se cultivaron ADSC (Invitrogen Corporation) en matraces de cultivo celular con medio StemPro™ -43 completo (Invitrogen) a 370C, 5 % de CO2 y el medio se cambió 3 veces por semana. Después de 3 días en cultivo, las células (pase 5) se digirieron con tripsina y se contaron para ser transfectadas. Las células se transfectaron transitoriamente con un plásmido: pCMV6-Oct4-2A-Klf4-2A-Nanog, pCMV-Sall4-2A-Oct4-2A-Klf4-2A-Nanog, pCMV-Dax1-2A-Oct4-2A-klf4, pCMV-FoxD3-2A-Oct4-2A-klf4, pCMV-Oct4-2A-Klf4-2A-Sall4, pCMV-MBD2-2A-Oct4-2A-Klf4-2A, pCMV-AGR2-2A-Oct4-2A-Klf4-2A o Rex1-EF-Oct4-2A-Klf4 (2 jg); o por dos plásmidos: pEF-Oct4nuc-IRES2-MBD2 con pCMV-Sox2nuc-IREC-Lin28 o pCMV-Klf4nuc-IRES2- Tptlnuc o pEF-Stella-IRES2-NPM2, utilizando Nucleofector™ como se describe en el Ejemplo II. Después de la transfección, las células se cultivaron en placas de 6 pocilios en suspensión con una relación 50:50 de medio completo de adipocitos (StemPro™ -43) y medio de células madre embrionarias (mTeSRI). Después de dos días de cultivo, las células se volvieron a transfectar con los mismos plásmidos enumerados anteriormente y las células se sembraron en placas de 96 pocilios recubiertas con Matrigel™ (BD Biosciences) en presencia de medio completo mTesR suplementado con tiazovivina (0,5 |j M), un inhibidor de ALK-5 (SB 341542, Stemgent, 2 j M), e inhibidor de MEK (Pd 0325901, Stemgent, 0,5 j M). El medio se cambió todos los días y las células se cultivaron durante 22 días a 37 oc, 5 % de CO2, 5 % de O2. Se realizó un kit de detección de fosfatasa alcalina (AP, Millipore) e inmunohistoquímica para analizar la expresión de marcadores de pluripotencia. La tinción de ALP se realizó utilizando el kit de detección de AP (Millipore) de acuerdo con las instrucciones del fabricante.
Cellomics™ realizó la observación visual de las células reprogramadas utilizando una tinción en vivo para SSEA-4647 (BD Biosciences) y TRA-I-8 I 555 (BD Biosciences) a partir del día 6 después de la transfección y cada 5 días a partir de entonces. Las colonias reprogramadas de PLSC, teñidas positivamente con SSEA-4 y TRAl-81, se observaron solo con el plásmido pCMV-Sall4-2A-Oct4-2A-Klf4-2A-Nanog, pEF-Rex1-EF-Oct4-2A-Klf4-2A-RFP, pEF-Oct4nuc-IRES1-MBD2 con pC-MV-Sox2nuc-IRES1-Lin28, y pEF-Oct4nuc-IRES1-MBD2 con pCMV-Klf4nuc-IRES2-Tpt1nuc. Estas colonias emergieron alrededor del Día 6 y se mantuvieron en cultivo hasta el final del período de estudio (Día 22) con una morfología estable. Entre los plásmidos citados anteriormente, pCMV-Sall4-2A-Oct4-2A-Klf4-Nanog y pEF-Rex1-EF-Oct4-2A-Klf4-2A-RFP dieron el mayor número de colonias. La tinción en vivo mostró que estas colonias expresaban marcadores típicos de pluripotencia, incluyendo SSEA-4 y TRAl-81, y el análisis adicional de estas colonias mostró que las colonias también expresaban otros marcadores ESC, tales como la fosfatasa alcalina y Oct4 (Figura 16). Cuando los cultivos se trataron con PD0325901 y SB431542 durante hasta 22 días, se obtuvo una mejora de 4 veces en la eficiencia sobre el procedimiento convencional después de la transfección de ADSC con pCMV-Sall4-2A-Oct4-2A-Klf4-Nanog y pEF-Rex1-EF-Oct4-2A-Klf4-2A-RFP.
Basado en el estudio anterior, la mayor eficiencia de reprogramación se observó utilizando pEF-Rex1-EF-Oct4-2A-Klf4-2A-RFP y pCMV-Sall4-2A-Oct4-2A-Klf4-2A-Nanog. Se diseñó otro estudio para determinar el efecto de pEF-Rex1-EF-Oct4-2A-Klf4-2A-RFP en la eficiencia de reprogramación e investigar el efecto de genes pluripotenciales individuales Rexl, Oct4, y Klf4 en diferentes combinaciones. Las ADSC se transfectaron como antes con pEF-Rexl-EF-Oct4-2A-Klf4-2A-RFP, pCMV6 = XL5-Rex1, pCMV6-XL4-Oct4/pCMV6-XL5-Klf4, pCMV6-XL5-Rex1/pCMV6-XL4-Oct4 o pCMV6 - XL5-Rex1/pCMV6-XL5-Klf4. Después de la segunda transfección, se cultivaron ADSC en placas de 96 pocilios recubiertas con Matrigel™ durante 24 días en presencia de medio mTeSRI suplementado con SB341542 y PD 0.325901 a 37 oc, 5 % de CO2, 5 % de O2. Con el fin de caracterizar subpoblaciones de células después de la transfección, se usaron tinción en vivo, inmunohistoquímica y tinción AP para seguir el cambio en los marcadores pluripotenciales. 1-5 % del total de células transfectadas con Rex1/Oct4 o Rex1/Klf4 mostraron un fenotipo SSEA4+ y t RA-1-81+ y este patrón fue estable hasta el final del período de estudio (día 2 2 ). La observación a lo largo del tiempo mostró que el fenotipo de estas colonias pasó de un fenotipo SSEA-4+ anterior a un fenotipo Oct4 /Sox2/Nanog tardío el día 22, que está más cerca del estado reprogramado final de una célula madre pluripotencial (Figura 17).
Se probaron varios genes por su efecto en la reprogramación de la eficiencia hacia células de tipo pluripotencial. Se cultivaron células ADSC como se describe en el Ejemplo IX con 2 días de VPA y tratamiento previo con 5-AZA (1 mM y 0,5 j M respectivamente) en medio StemPro™ MSC SFM. Las células se transfectaron utilizando el dispositivo Nucleofector™® de 96 pocilios Shuttle® siguiendo el procedimiento descrito en el Ejemplo IV y utilizando el programa de transfección EW-104 con las mezclas de ADN descritas en la Tabla 34. Después de la transfección, las células se sembraron en placas en medio StemPro™ MSC SFM descrito en el Ejemplo A en placas de 24 pocilios recubiertas con Matrigel™ (BD Biosciences) y se incubaron a 37 °C, 5 % de CO2, 5 % de O2. El día 1, el medio se cambió a una mezcla de 75 % de StemPro™ MSC y 25 % de medio de células hES; el porcentaje de StemPro™ MSC se redujo todos los días durante cuatro días para tener un medio de células con 100 % hES el día 4. A partir de entonces se cambió el medio cada dos días. El medio de células hES consistió en medio Eagle modificado por Dulbecco (DMEM, Invitrogen) suplementado con un 20 % de Knockout™ Serum Replacement (KSR, Invitrogen), GlutaMAX™ 1 mM, aminoácidos no esenciales 100 j M, 100 j M p-mercaptoetanol y 10 ng/ml Fgf-2. A lo largo del
experimento se añadieron diferentes inhibidores y factores de crecimiento; estos se enumeran en la Tabla 34. Las células se analizaron el día 7 y el día 14 mediante análisis de inmunohistoquímica y el día 7 mediante RT-PCR.
Tabla 34: Plásmidos y composición de los medios desde el día 1 hasta el día 14.

continuación

Con el fin de caracterizar subpoblaciones de células después de la transfección, tinción en vivo, inmunohistoquímica y tinción AP se realizaron para seguir el cambio en los marcadores pluripotenciales. Las células transfectadas con Oct4/UTF1/MBD2, Oct4/Dppa4/MBD2, FoxD3/Dppa4/MBD2, Oct4/FoxD3/Dppa4 o Sox2/FoxD3/UTF1 mostraron colonias positivas para t RA1-60, TRA1-81 e s SeA4. Esta observación indicó que MBD2 generalmente no tuvo
ningún efecto por sí mismo en la reprogramación hacia células de tipo pluripoteneial, excepto en el caso de la transfección de OcT4/FoxD3/MBD2. Las colonias comenzaron a formarse el día 7 y continuaron formándose hasta el día 14 (Figura 18) (el final del período de estudio). Estas colonias también fueron positivas para AP.
Estos resultados se confirmaron mediante un análisis de RT-PCR que mostró una regulación por aumento de la expresión de Oct4 como se muestra en la Tabla 35. La expresión relativa para SOX2 también aumentó ligeramente la regulación por aumento el día 7 después de transfeetar las células con Oct4/Foxd3/MBD2. También hay una tendencia de regulación por aumento de Sox2 después de la transfección con Oct4/Sox2/Foxd3 y Oet4/Foxd3/Utf1.
Tabla 35: Expresión relativa de genes pluripoteneiales después de transfeetar ADSC con diversas combinaciones de vectores como se describe en la Tabla 34

Reprogramación de la eficiencia de los factores de pluripotencia definidos en HFF después de la triple transfección (una transfección cada 3 días)
Las células HFF se cultivaron como se describe en el Ejemplo I, con la excepción de las concentraciones de VPA y 5-AZA que fueron respectivamente 2 mM y 2,5 |jM. Las células se transfeetaron utilizando el dispositivo Nucleofector™® II (Lonza) siguiendo el procedimiento descrito en el Ejemplo II, con la excepción de la cantidad de ADN: se usó 1 jg de cada uno de los 3 plásmidos de ADN. Las células que habían sido pretratadas con VPA y 5-Aza y las células no tratadas se transfeetaron ambas con una mezcla de pCMV-Oct4nuc-IRES2-Sox2nuc, pCMV-Klf4nuc-IRES2-Cmycnuc o pCMV-Nanognue-IRES2-Lin28. Después de la transfección, las células se sembraron en placas en el medio de fibroblastos descrito en el Ejemplo I, suplementado con o sin VPA y 5-AZA en placas de 6 pocilios recubiertas con Matrigel™ (BDBioseienees) recubierto y se incubaron a 370C, 5 % de CO2. Los días 1 y 2, el medio se cambió a medio 100 % mTeSRI (StemCell Technologies) suplementado con o sin VPA y 5-AZA. El día 3 y el día 6, las células de cada condición se separaron mediante incubación en TrypLE™ durante 5 minutos, se contaron y se centrifugaron. Las células se volvieron a transfeetar como antes y se sembraron en placas recubiertas con Matrigel™ en medio mTeSRI suplementado con o sin VPA y 5-AZA. Los medios se cambiaron diariamente como se describe para los días 1 y 2. El medio se suplementó en Y27632 (Stemgent, 10 j M) desde el día 7 hasta el día 14 para promover la viabilidad y la expansión elonal de posibles células reprogramadas. Las células se analizaron el día 20 utilizando el kit de detección de fosfatasa alcalina (Millipore) y mediante análisis de inmunohistoquímiea.
Este análisis reveló que después de tres transfeeeiones, se observaron tres clones positivos para la actividad de la fosfatasa alcalina y mostraron una morfología redondeada de células/colonias. La tinción con anticuerpos contra los
marcadores de células madre embrionarias (ES) SSEA-4 y TRA-1-81 confirmó que estos clones eran de tipo pluripotenclal (Figura 19). Las células HFF circundantes fueron negativas para estos marcadores. Estos clones se obtuvieron solo en la condición que no contenía inhibidores (es decir: VPA y 5-AZA). Inesperadamente, no se observaron clones para la condición tratada con estos inhibidores.
Reprogramación de NSLC en pluripotencia
Las NSLC y células madre neuronales derivadas de BG-01, una línea de células humanas ES que expresa marcadores que son característicos de las células ES, incluyendo SSEA-3, SSEA-4, TRA-1-60, TRA-1-81 y OCT-3/4, se reprogramaron para que fueran pluripotenciales. Las células BG-01 se habían cultivado previamente en condiciones para inducir la diferenciación hacia células madre neurales, tal como se describe en Chambers SM y col., 2009. Las NSLC y BG-01-NSC se cultivaron en medio de proliferación suplementado con FGF (20 ng/ml) y EGF (20 ng/ml). Las NSLC y BG-01-NSC se transfectaron como se ha descrito previamente en el Ejemplo II por dos vectores episomales, pEF-Oct4nuc-IRES2-MBD2 (NC1) o pCMV-FoxD3-2A-Oct4-2A-Klf4 (F72). Después de la transfección, se recogieron las células y se sembraron en placas Petri sin recubrimiento en presencia de medio de proliferación y medio mTeSRI (50:50) en condiciones de proliferación a 370C, 5 % de C02. Después de 48 horas, las células se volvieron a transfectar con el mismo plásrnido y se sembraron en placas de 96 pocilios recubiertas con Matrigel™ y se cultivaron en presencia de medio mTeSRI complementado por las pequeñas moléculas BIX01294 (Stemgent, 2 |j M) y BayK8644 (Stemgent, 2 |j M) a 37 oc, 5 % de O2 durante 22 días. Se realizaron tinciones en vivo e inmunohistoquímica para caracterizar subpoblaciones de células para marcadores de pluripotencia.
Las NSLC y BG-01-NSC se tiñeron positivamente con SSEA-4 comenzando el día 7 y se mantuvieron hasta 22 días en cultivo (el final del estudio) (Figura 20). En diez días, se observaron células que eran morfológicamente similares a las ESC y expresaron un amplio panel de marcadores de pluripotencia, incluyendo SSEA-4, TRA1-81, Nanog y Oct4 (Figura 20). Este estudio identificó otra manera de obtener células de tipo pluripotencial a partir de células somáticas a través de las células de tipo madre neurales (NSLC). La utilidad de las nSlC podría ofrecer múltiples ventajas para la reprogramación hacia células tipo pluripotenciales. Por ejemplo, la omisión del requisito de genes tumorigénicos como c-Myc reduce el riesgo de inducir células cancerosas. Para aplicaciones neurodegenerativas y neurodegenerativas, estas células podrían representar una fuente valiosa de células para investigar además la inducción de células pluripotenciales humanas, y también representan una fuente potencial de células para derivar células madre multipotenciales y pluripotenciales específicas del paciente para modelar enfermedades humanas. Ejemplo XX
Ensayo de formación de teratomas en ratones SCID
El trasplante de células madre pluripotenciales (SC) humanas en ratones "inmunodeficientes con compromiso grave" (SCID) conduce a la formación de tumores diferenciados que comprenden las tres capas germinales para células madre pluripotenciales, que se asemejan a teratomas humanos espontáneos, y tejido especializado para células madre multipotenciales. Estos ensayos se consideran los estándares en la literatura para demostrar el potencial de diferenciación de las células madre pluripotenciales y son prometedores como estándar para evaluar la seguridad entre las poblaciones de células derivadas de SC destinadas a aplicaciones terapéuticas
Después de obtener todas las aprobaciones de animales apropiadas para el experimento, se adquirieron 24 ratones de Charles Rivers y se alojaron en las instalaciones para animales de MISPRO durante una semana sin ningún tipo de experimentación para adaptarse al nuevo entorno. Se inyectó un millón de NSLC humanas, células neuroprogenitoras humanas normales (hNPC) o células madre embrionarias humanas (ES) en 100 j l de solución salina tamponada con fosfato, libre de calcio y magnesio (CMF-PBS) por vía intramuscular con una aguja de 21-G en la extremidad posterior derecha de ratones SCID-beige machos de 4 semanas de edad con anestesia con ketamina/xilazina (8 ratones por grupo ). Después de la inyección, la jeringa se aspiró hacia arriba y hacia abajo un par de veces en una placa de cultivo que contenía medio para verificar que se habían inyectado las células y que no se habían quedado dentro de la jeringa.
Los ratones se mantuvieron durante 12 semanas y se controlaron los signos clínicos y el crecimiento de cualquier tumor regularmente. Cualquier tejido especializado o crecimiento de teratoma se monitorizó mediante exploración externa y un aumento en el tamaño del músculo en relación con el mismo músculo en la extremidad trasera izquierda. Cuando se identificó un tejido especializado o teratoma, se midió la ubicación y el tamaño del crecimiento (utilizando calibradores de medición) y se registraron. El tejido especializado o teratoma generalmente se identifica primero como un pequeño crecimiento del tamaño muscular en comparación con el músculo de control izquierdo. Los animales se controlaron semanalmente hasta la aparición de cualquier crecimiento tumoral y diariamente después de que aparecieran los tumores. Después de l2 semanas, se sacrificó a los ratones mediante eutanasia con CO2. Se observó a cada animal completo para detectar cualquier crecimiento tumoral en cualquier lugar del animal, y el músculo inyectado y el control muscular izquierdo comparable se midieron (con calibradores de medición) (véase la tabla de resultados a continuación) y, después, se extrajeron y almacenaron en una solución de paraformaldehído al 4 % para análisis histológico. Los tamaños de los músculos fueron los siguientes:

La medición del tamaño de Ios músculos reveló que todos Ios músculos humanos inyectados con células madre embrionarias eran más grandes que Ios controles musculares izquierdos comparables, lo que indica crecimiento de teratoma en Ios músculos a Ios que se inyectaron células ES. Aproximadamente la mitad de todos Ios músculos humanos inyectados con células neuroprogenitoras eran más grandes que Ios controles musculares izquierdos comparables, mientras que Ios ratones a Ios que se inyectó NSLC no mostraron ninguna diferencia entre Ios músculos (tratados con las células o no). Los ratones a Ios que se inyectó NSLC no mostraron ninguna evidencia de crecimiento tumoral o teratoma.
Referencias
Zeitlow R, Lañe EL, Dunnet SB, Rosser AE.Human stem cells for CNSrepair Cell Tissue Res.2008;331(1):301-22. Mimeault, M., Hauke, R. y Batra, S. K. 2007. Células madre: a revolution in therapeutics-recent advances in stem cell biology and their therapeutic applications in regenerative medicine and cáncer therapies. Clin Pharmacol Ther,82, 252-64.
Levesque, MF y Neuman T. Transdiffentiation of transfected epidermal basal cells into neural progenitor cells, neuronal cells and/or glial cells. Patente, fecha de presentación 2000.
Shea TB. Neuritogenesis in mouse NB2a/d1 neuroblastoma cells: triggering by calcium influx and involvement of actin and tubulin dynamics. Cell Biol Int Rep. 1990;14(11):967-79.
Yeomans ND, Trier JS, Moxey PC y Markezin ET. Maturation and differentiation of cultured fetal stomach. Effects of corticosteroids, pentagastrin, and cytochalasin B. Gasteroenterology 1976;71(5):770-7.
Paterson FC, Rudland PS. Microtubule-disrupting drugs increase the frequency of conversión of a rat mammary epithelial stem cell line to elongated, myoepithelial-like cells in culture. J Cell Phsiol. 1985;125(1):135-50. Bouwens L. Transdifferentiation versus stem cell hypothesis for the regeneration of islet beta-cells in the páncreas. Micro Res Tech. 1998;43(4):332-6.
Bouwens L. Cytokeratins and cell differentiation in the páncreas. J Pathol. 1998b; 184(3):234-9.
Theise ND, Nimmakayalu M, Gardner R, lllei PB, Morgan G, Teperman L, Henegariu O, Krause DS. Liver from bone marrow in humans. Hepatology 2000; 32 (1): 11-6.
Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61(4):364-70.
Brunet, JF; Ghysen, A. Deconstructing cell determination: proneural genes and neuronal identity. Bioessays.
1999;21:313-318.
Bertrand N, Castro DS y Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3(7):517-30.
McCormick MB, Tamimi RM, Snider L, Asakura A, Bergstrom D, Tapscott SJ. NeuroD2 and neuroD3: distinct expression patterns and transcriptional activation potentials within the neuroD gene family. Mol Cell Biol. 1996; 16( 10):5792-800.
Guillemot F, Lo LC, Johnson JE, Auerbach A, Anderson DJ, Joyner Al. Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 1993;75(3):463-76.
Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. The bHLH protein NEUROGENIN 2 is a determination factor for epibranchial placode-derived sensory neurons. Neuron 1998;20(3):483-94.
Fernandes KJL, McKenzie IA, Mill P, Smith KM, Akhavan M, Barnabé-Heider F, Biernaskie J, Junek A, y coI. A dermal niche for multipotent adult skin-derived precursor cells. Nature Cell Biology 2004;6:1082-1093.
Jacobsen F, Hirsch T, Mittler D, Schulte M, Lehnhardt M, Druecke D, Homann HH, Steinau HU, Steinstraesser L.Polybrene improves transfection efficacy of recombinant replication-deficient adenovirus in cutaneous cells and burned skin. J Gene Med. 2006;8(2):138-46.
Kearns CM, Gash DM. GDNF protects nigral dopamine neurons against 6-hydroxydopamine in vivo. Brain Res.
1995;672(1-2): 104-11.
Gash DM, Zhang Z, Ovadia A, Cass WA, Yi A, Simmerman L, Russel D, Martin D, Lapchak PA, Collins F, Hoffer BJ, Gerhardt GA. Functional recovery in parkinsonian monkeys treated with GDNF.Nature 1996;380(6571):252-5. Lindner MD, Winn SR, Baetge EE, Hammang JP, Gentil FT, Doherty E, McDermott PE, Frydel B, Ullman MD, Schallert T y coI. Implantation of encapsulated catecholamine and GDNF-producing cells in rats with unilateral dopamine depletions and parkinsonian symptoms. Exp Neurol. 1995; 132(1)-. 62-76.
Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, y col. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science 2000;290(5492):767-73.
Martinez-Serrano A, BJorklund A. Immortalized neural progenitor cells for CNS gene transfer and repair. Trends Neurosci. 1997;20(11):530-8.
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversión of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275-80.
En el presente documento, los encabezados se han incluido por referencia y para ayudar a localizar determinadas secciones. No se pretende que estos encabezados limitar el ámbito de los conceptos descritos bajo los mismos, y estos conceptos pueden tener aplicabilidad en otras secciones a lo largó de la totalidad de la memoria descriptiva.
Claims (8)
1. Un procedimiento de obtención de una célula madre de tipo neural (NSLC), que comprende:
1) poner en contacto el ADN y/o la cromatina de una célula proporcionada de un primer tipo que no es una NSLC con un acetilador de histonas, un inhibidor de la desacetilación de histonas, un desmetilador de ADN, y/o un inhibidor de la metilación del ADN;
2) aumentar los niveles intracelulares en la célula de un primer tipo de un polipéptido Musashil (Msí1) y/o un polipéptido Neurogenina 2 (Ngn2); y
3) colocar la célula de la etapa (2 ) en condiciones de cultivo que apoyan la transformación de la célula de un primer tipo en una NSLC durante un período de tiempo suficiente para permitir una expresión estable de una pluralidad de genes cuya expresión es característica de las propiedades fenotípicas y/o funcionales de una NSLC;
por lo que al final de dicho período de tiempo se obtiene una NSLC y en el que dicha NSLC obtenida se caracteriza además por una represión estable de una pluralidad de genes expresados en la célula de un primer tipo.
2. El procedimiento de la reivindicación 1, en el que los niveles intracelulares crecientes del uno o más polipéptidos comprenden transfectar transitoriamente la célula de un primer tipo con un vector de expresión que permite la expresión de un polipéptido Musashil (Msí1) y/o Neurogenina 2 (Ngn2); un polipéptido Musashi 1 (Msí1) y proteína 2 de dominio de unión a metilo-CpG (MBD2); neurogenina 2 (Ngn2) y proteína 2 del dominio de unión a metil-CpG (MBD2); y un polipéptido Musashil (Msi 1), neurogenina 2 (Ngn2) y polipéptido de la proteína 2 del dominio de unión a metil-CpG (MBD2).
3. El procedimiento de la reivindicación 1 o 2, en el que el NSLC expresa uno o más de Sox2, nestina, GFAP, Msil y Ngn2.
4. El procedimiento de cualquiera de las reivindicaciones 1-3, en el que la célula de un primer tipo se selecciona del grupo que consiste en: células madre embrionarias, células madre de adulto, células progenitoras, células derivadas de mesodermo, endodermo o ectodermo, células madre derivadas de tejido adiposo, célula madre mesenquimatosa, célula madre hematopoyética, célula precursora derivada de la piel, célula del folículo piloso, fibroblasto, queratinocito, célula epidérmica, célula endotelial, célula epitelial, célula epitelial granulosa, melanocito, adipocito, condrocito, hepatocito, linfocitos B, linfocitos T, granulocito, macrófago, monocito, célula mononuclear, células de los islotes pancreáticos, célula de Sertoli, neurona, célula glial, célula muscular cardíaca y otra célula muscular.
5. El procedimiento de cualquier reivindicación precedente, en el que el inhibidor de la metilación del ADN se selecciona del grupo que consiste en 5-azacitidina, 5-aza-2'-desoxicitidina, 1-p-D-arabinofuranosil-5-azacitosina, dihidro-5-azacitidina, zebularina y RG108; el inhibidor de la desacetilación de histonas se selecciona del grupo que consiste en ácido valproico, fenilbutirato, tricostatina A, butirato de sodio, 5 benzamidas y tetrapéptidos cíclicos; y el desmetilador de ADN es la proteína 2 del dominio de unión a metil-CpG (MBD2).
6. El procedimiento de cualquier reivindicación anterior, en el que la NSLC así obtenida posee todas las siguientes características:
i) capacidad de autorrenovación o proliferación;
ii) no es una célula cancerosa;
iii) es estable y no se mantiene artificialmente por la expresión génica forzada o por medios similares y se puede mantener en medios de células madre neurales estándar;
iv) puede diferenciarse en al menos una de una célula progenitora, una célula precursora, una célula somática y otro tipo de célula más diferenciada del mismo linaje, excepto si la célula es una célula somática diferenciada terminalmente;
v) no exhibe crecimiento descontrolado, formación de teratoma y formación de tumores in v iv o .
7. El procedimiento de las reivindicaciones 1,2 o 4 a 6, en el que la NSLC así obtenida posee una o más de las siguientes características:
- expresión de uno o más marcadores de células madre seleccionados del grupo que consiste en Sox2, nestina, GFAP, Msil y Ngn2;
- disminución de la expresión de uno o más genes específicos de la célula del primer tipo;
- forma neuroesferas en el ensayo de formación de colonias de neuroesferas;
- que se puede cultivar en suspensión o como un cultivo adherente;
- capaz de proliferar sin la presencia de un agente de reprogramación exógeno durante más de 1 mes;
- capaz de dividirse cada 36 horas a un pase bajo;
- positiva para la actividad telomerasa;
- capaz de diferenciarse en una célula de tipo neuronal, célula de tipo astrocito, célula de tipo oligodendrocito y combinaciones de las mismas;
- disminución de la expresión de la telomerasa y uno o más marcadores de células madre neurales;
- tener uno o más procesos morfológicos similares a neuritas (axones y/o dendritas) de más de un diámetro de
célula de longitud;
- expresión de al menos un antígeno neural específico seleccionado del grupo que consiste en tubulina neural específica, proteína 2 asociada a los microtúbulos, NCAM y marcador para un neurotransmisor;
- expresión de uno o más marcadores neurales funcionales;
- capaz de liberar uno o más factores neurotróficos;
- negativa en un ensayo de formación de colonias de tumores;
- negativa para el crecimiento de tumores en ratones SCID;
- negativa para el crecimiento de teratoma en ratones SCID;
- capaz de mejorar significativamente una o más medidas funcionales después de la colocación de un número adecuado de NSLC en el hueco en un modelo de ablación cerebral;
- capaz de mejorar significativamente o mantener una o más medidas funcionales después de inyectar un número adecuado de NSLC en un modelo de EAE; o
- capaz de mejorar una o más medidas funcionales más significativamente que las hNPC en lesiones del SNC o modelos neurodegenerativos.
8. El procedimiento de una cualquiera de las reivindicaciones anteriores, en el que se obtiene una pluralidad de NSLC y en el que dicha pluralidad de NSCL se organiza dentro de una estructura tridimensional.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US25696709P | 2009-10-31 | 2009-10-31 | |
| PCT/CA2010/001727 WO2011050476A1 (en) | 2009-10-31 | 2010-11-01 | Methods for reprogramming cells and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2739672T3 true ES2739672T3 (es) | 2020-02-03 |
Family
ID=43921216
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES10825907T Active ES2739672T3 (es) | 2009-10-31 | 2010-11-01 | Procedimientos de reprogramación de células y usos de los mismos |
Country Status (17)
| Country | Link |
|---|---|
| US (7) | US9528087B2 (es) |
| EP (2) | EP3567103B1 (es) |
| JP (5) | JP5982286B2 (es) |
| KR (3) | KR101874463B1 (es) |
| AU (3) | AU2010312240B2 (es) |
| BR (1) | BR112012009921B1 (es) |
| CA (1) | CA2779310C (es) |
| DK (1) | DK2494039T3 (es) |
| ES (1) | ES2739672T3 (es) |
| HR (1) | HRP20191213T1 (es) |
| IL (3) | IL219453A (es) |
| LT (1) | LT2494039T (es) |
| MX (1) | MX353245B (es) |
| SG (1) | SG10201702120VA (es) |
| SI (1) | SI2494039T1 (es) |
| WO (1) | WO2011050476A1 (es) |
| ZA (1) | ZA201203902B (es) |
Families Citing this family (68)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7642091B2 (en) | 2005-02-24 | 2010-01-05 | Jau-Nan Lee | Human trophoblast stem cells and use thereof |
| US9528087B2 (en) * | 2009-10-31 | 2016-12-27 | Genesis Technologies Limited | Methods for reprogramming cells and uses thereof |
| US9453205B2 (en) | 2009-10-31 | 2016-09-27 | Genesis Technologies Limited | Methods for reprogramming cells and uses thereof |
| WO2011096825A1 (en) * | 2010-02-05 | 2011-08-11 | Auckland Uniservices Limited | Cell programming |
| AU2011329002C1 (en) * | 2010-11-15 | 2017-05-25 | Accelerated Biosciences Corp. | Generation of neural stem cells from human trophoblast stem cells |
| CN114164167A (zh) | 2010-12-22 | 2022-03-11 | 菲特治疗公司 | 用于单细胞分选与增强ipsc重新编程的细胞培养平台 |
| KR101376635B1 (ko) * | 2011-01-14 | 2014-03-24 | 한국생명공학연구원 | Rex1을 포함하는 세포 리프로그래밍 조성물 및 이를 이용한 유도만능줄기세포 제조방법 |
| EP2734622B1 (en) * | 2011-07-19 | 2018-09-05 | Vivoscript, Inc. | Compositions and methods for re-programming cells without genetic modification for repairing cartilage damage |
| US20130224782A1 (en) * | 2012-02-29 | 2013-08-29 | Chenzhong Kuang | Neurogenesis screening method and system using adipose tissue derived stem cells |
| US9428730B2 (en) | 2012-02-29 | 2016-08-30 | Mead Johnson Nutrition Company | Coatings and culture media for promoting neurogenesis in adipose tissue derived stem cells |
| GB201118964D0 (en) * | 2011-11-03 | 2011-12-14 | Ucl Business Plc | Method |
| WO2013086570A1 (en) * | 2011-12-13 | 2013-06-20 | Flinders University Of South Australia | Method of producing multipotent stem cells |
| CN102604894B (zh) * | 2012-02-29 | 2014-07-30 | 中国科学院广州生物医药与健康研究院 | 用于制备神经干细胞的培养基及其用途 |
| US20150297674A1 (en) * | 2012-03-12 | 2015-10-22 | Loma Linda University Medical Center | Substances and methods for the treatment of cerebral amyloid angiopathy related conditions or disease |
| CN113559282A (zh) | 2012-07-19 | 2021-10-29 | 宾夕法尼亚州研究基金会 | 再生用于在神经系统中治疗疾病和损伤的功能性神经元 |
| US20140134139A1 (en) * | 2012-11-07 | 2014-05-15 | Northwestern University | Cd34+ cells and methods of use |
| EP3401391A1 (en) * | 2012-11-29 | 2018-11-14 | Takara Bio Europe AB | Maturation of hepatocyte-like cells derived from human pluripotent stem cells |
| CA3137404A1 (en) | 2012-11-30 | 2014-06-05 | Accelerated Biosciences Corp. | Pancreatic progenitor cells expressing betatrophin and insulin |
| JP2016502844A (ja) * | 2012-12-14 | 2016-02-01 | アントフロゲネシス コーポレーション | アノイキス抵抗性胎盤幹細胞及びその使用 |
| US9982232B2 (en) * | 2012-12-17 | 2018-05-29 | John PIMANDA | Methods of generating cells with multilineage potential |
| AU2014240603B2 (en) | 2013-03-14 | 2017-11-09 | Massachusetts Institute Of Technology | Compositions and methods for epithelial stem cell expansion and culture |
| WO2014153115A2 (en) * | 2013-03-14 | 2014-09-25 | Children's Medical Center Corporation | Compositions and methods for reprogramming hematopoietic stem cell lineages |
| WO2014170488A1 (en) * | 2013-04-19 | 2014-10-23 | Universita' Degli Studi Di Milano | Methods for the conversion of somatic cells into pancreatic-hormone secreting cells |
| KR101655383B1 (ko) | 2013-07-27 | 2016-09-08 | 고려대학교 산학협력단 | 소분자 화합물을 포함하는 만능성 줄기세포의 염색체 안정성 유지용 조성물 |
| CA2926372C (en) * | 2013-10-10 | 2023-09-26 | Mcmaster University | Diagnosis and monitoring of endometriosis through bdnf and full-length ntrk2 levels |
| WO2015084908A1 (en) * | 2013-12-03 | 2015-06-11 | The Johns Hopkins University | Method for highly efficient conversion of human stem cells to lineage-specific neurons |
| US9512406B2 (en) | 2013-12-20 | 2016-12-06 | The J. David Gladstone Institute, a testamentary trust established under the Will of J. David Gladstone | Generating hepatocytes |
| FR3016372B1 (fr) * | 2014-01-13 | 2018-01-19 | Centre Leon Berard | Procede de preparation de cellules pluripotentes induites |
| US10752881B2 (en) * | 2014-01-29 | 2020-08-25 | Helmholtz Zentrum München—Deutsches Forschungszentrum für Gesundheit und Umwelt (GmbH) | Trans-differentiation of differentiation cells |
| SG11201606934SA (en) | 2014-03-04 | 2016-09-29 | Fate Therapeutics Inc | Improved reprogramming methods and cell culture platforms |
| US20150353623A1 (en) * | 2014-04-03 | 2015-12-10 | Loma Linda University | Substances and methods for the treatment of cerebral amyloid angiopathy related conditions or diseases |
| US9259357B2 (en) | 2014-04-16 | 2016-02-16 | Loma Linda University | Composition, preparation, and use of chitosan shards for biomedical applications |
| KR101731624B1 (ko) * | 2014-07-01 | 2017-05-04 | 광주과학기술원 | 세포 리프로그래밍 유도용 조성물 |
| KR102493376B1 (ko) | 2014-09-03 | 2023-01-27 | 더 브리검 앤드 우먼즈 하스피털, 인크. | 청력 손실의 치료를 위해 내이 털세포를 생성하기 위한 조성물, 시스템, 및 방법 |
| CN109251894B (zh) | 2014-11-25 | 2022-10-04 | 宾州研究基金会 | 人神经胶质细胞转化成用于大脑和脊髓修复的神经细胞的化学重编程 |
| WO2016086132A1 (en) | 2014-11-26 | 2016-06-02 | Accelerated Biosciences Corp. | Induced hepatocytes and uses thereof |
| MA41296A (fr) * | 2014-12-30 | 2017-11-07 | Orgenesis Ltd | Procédés de transdifférenciation et procédés d'utilisation de ceux-ci |
| JP6841609B2 (ja) | 2015-07-10 | 2021-03-10 | 3スキャン インコーポレイテッド | 組織学的染色の空間的多重化 |
| CN105062953A (zh) * | 2015-08-06 | 2015-11-18 | 深圳爱生再生医学科技有限公司 | 三维诱导间充质干细胞转化为胰岛细胞的方法 |
| CN117737124A (zh) | 2015-10-16 | 2024-03-22 | 菲特治疗公司 | 用于诱导和维护基态多能性的平台 |
| AU2017205194A1 (en) | 2016-01-08 | 2018-07-19 | Massachusetts Institute Of Technology | Production of differentiated enteroendocrine cells and insulin producing cells |
| US10973930B2 (en) | 2016-02-18 | 2021-04-13 | The Penn State Research Foundation | Generating GABAergic neurons in brains |
| US10213511B2 (en) | 2016-03-02 | 2019-02-26 | Frequency Therapeutics, Inc. | Thermoreversible compositions for administration of therapeutic agents |
| US11260130B2 (en) | 2016-03-02 | 2022-03-01 | Frequency Therapeutics, Inc. | Solubilized compositions for controlled proliferation of stem cells / generating inner ear hair cells using a GSK3 inhibitor: IV |
| US10201540B2 (en) | 2016-03-02 | 2019-02-12 | Frequency Therapeutics, Inc. | Solubilized compositions for controlled proliferation of stem cells / generating inner ear hair cells using GSK3 inhibitors: I |
| JP7005528B2 (ja) * | 2016-06-03 | 2022-01-21 | 中国人民解放軍軍事医学科学院野戦輸血研究所 | 消化管由来上皮細胞を内胚葉幹細胞/前駆細胞に初期化するための低分子化合物の組み合わせ、初期化方法および適用 |
| CN105861447B (zh) * | 2016-06-13 | 2017-12-19 | 广州市搏克生物技术有限公司 | 一种非病毒iPSCs诱导组合物及其试剂盒 |
| AU2017282153B2 (en) | 2016-06-20 | 2022-08-04 | Genesis Technologies Limited | Automated cell processing systems and methods |
| SG10201910821XA (en) | 2016-12-30 | 2020-01-30 | Frequency Therapeutics Inc | 1h-pyrrole-2,5-dione compounds and methods of using them to induce self-renewal of stem/progenitor supporting cells |
| US20190352604A1 (en) * | 2017-02-24 | 2019-11-21 | Koji Tanabe | Nerve cell production method |
| CN111500538A (zh) * | 2017-03-01 | 2020-08-07 | 中国科学院动物研究所 | 一种将非神经元细胞转化为神经元细胞的方法 |
| WO2019144968A1 (zh) * | 2018-01-29 | 2019-08-01 | 中国科学院动物研究所 | 一种细胞诱导的方法 |
| CN110093305B (zh) * | 2018-01-29 | 2021-07-02 | 中国科学院动物研究所 | 一种诱导肝细胞体外扩增的方法 |
| CN110093309B (zh) * | 2018-01-29 | 2021-07-02 | 中国科学院动物研究所 | 一种诱导成纤维细胞转分化为脂肪细胞的方法 |
| CN110093310B (zh) * | 2018-01-29 | 2021-07-02 | 中国科学院动物研究所 | 一种将成纤维细胞转化为永生化细胞的方法及其应用 |
| US20210032300A1 (en) * | 2018-02-02 | 2021-02-04 | The Penn State Research Foundation | Methods and materials for treating brain injuries |
| CN110305846A (zh) * | 2018-03-20 | 2019-10-08 | 中山大学中山眼科中心 | 视网膜节细胞的制备方法 |
| WO2019241846A1 (en) * | 2018-06-22 | 2019-12-26 | The University Of Sydney | Expansion and differentiation of neuronal precursor cells |
| WO2020037326A1 (en) | 2018-08-17 | 2020-02-20 | Frequency Therapeutics, Inc. | Compositions and methods for generating hair cells by downregulating foxo |
| CN113195707A (zh) | 2018-08-17 | 2021-07-30 | 频率治疗公司 | 用于通过上调jag-1来生成毛细胞的组合物和方法 |
| WO2020061366A1 (en) * | 2018-09-19 | 2020-03-26 | Regeneration Biomedical, Inc. | Wnt-activated adipose-derived stem cell apparatuses, methods and systems |
| US11518971B2 (en) | 2018-11-27 | 2022-12-06 | Research Triangle Institute | Method and apparatus for spatial control of cellular growth |
| KR102450790B1 (ko) * | 2019-01-02 | 2022-10-06 | 주식회사 셀라퓨틱스바이오 | 체세포로부터 분화된 신규한 유사신경교세포, 이의 제조 방법, 이의 제조용 칵테일 조성물, 이를 포함하는 신경 질환 예방 또는 치료용 세포 치료제 및 이를 투여하여 신경 질환을 예방 및 치료하는 방법 |
| JP2022517784A (ja) * | 2019-01-11 | 2022-03-10 | フィジーン、エルエルシー | 線維芽細胞再生細胞 |
| KR102183208B1 (ko) * | 2019-07-25 | 2020-11-25 | 한국과학기술연구원 | Ngn2(neurogenin-2)를 이용하여 척수 손상 모델에서 반응성 성상세포를 신경세포로 리프로그래밍하는 방법 |
| JP2023504573A (ja) * | 2019-12-09 | 2023-02-03 | ザ ブリガム アンド ウィメンズ ホスピタル インコーポレイテッド | 造血幹細胞を生成するための方法 |
| CN111471651A (zh) * | 2020-04-24 | 2020-07-31 | 中国人民解放军第四军医大学 | 一种人脂肪干细胞无血清培养基及其制备方法 |
| CN113862218A (zh) * | 2021-10-12 | 2021-12-31 | 广州医科大学附属第一医院(广州呼吸中心) | 一种肿瘤相关性血管周细胞亚群及制备方法与应用 |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6087168A (en) | 1999-01-20 | 2000-07-11 | Cedars Sinai Medical Center | Conversion of non-neuronal cells into neurons: transdifferentiation of epidermal cells |
| US7621606B2 (en) | 2001-08-27 | 2009-11-24 | Advanced Cell Technology, Inc. | Trans-differentiation and re-differentiation of somatic cells and production of cells for cell therapies |
| US20020136709A1 (en) * | 2000-12-12 | 2002-09-26 | Nucleus Remodeling, Inc. | In vitro-derived adult pluripotent stem cells and uses therefor |
| US20030217378A1 (en) | 2002-04-05 | 2003-11-20 | The University Of Georgia Research Foundation, Inc | Cloning using rapidly matured oocytes |
| AU2003252559A1 (en) * | 2002-08-17 | 2004-03-03 | Hae-Young Suh | A method for transdifferentiating mesenchymal stem cells into neuronal cells |
| WO2006086746A2 (en) | 2005-02-09 | 2006-08-17 | Burnham Institute For Medical Research | Homogeneous neural precursor cells |
| WO2007016037A2 (en) | 2005-07-27 | 2007-02-08 | Board Of Regents, The University Of Texas System | Methods for trans-differentiating cells |
| CN101389761A (zh) | 2006-02-27 | 2009-03-18 | 银怎株式会社 | 使用bmi-1使星形胶质细胞去分化成为神经干细胞 |
| EP2099290A4 (en) * | 2006-12-07 | 2010-01-13 | Teva Pharma | PROCESS FOR GENERATING AND EXPANDING PROGENITOR CELLS OF TISSUES AND TISSUE CELLS MILLED FROM INTACT BONE MARROW OR UMBILICAL INTACT CORD TISSUE |
| WO2009018832A1 (en) | 2007-08-09 | 2009-02-12 | Rigshospitalet | Method for increasing the plasticity level of a cell |
| MX2010002242A (es) * | 2007-08-31 | 2010-06-01 | Whitehead Biomedical Inst | Estimulación de ruta wnt en reprogramacion de celulas somaticas. |
| WO2009057831A1 (ja) * | 2007-10-31 | 2009-05-07 | Kyoto University | 核初期化方法 |
| US20090136461A1 (en) * | 2007-11-28 | 2009-05-28 | Moon Suk Kim | Neuronal differentiation method of adult stem cells using small molecules |
| US20090162329A1 (en) * | 2007-11-30 | 2009-06-25 | Piero Anversa | Compositions comprising hdac inhibitors and methods of their use in restoring stem cell function and preventing heart failure |
| AU2008338989A1 (en) | 2007-12-17 | 2009-06-25 | Gliamed, Inc. | Stem-like cells and method for reprogramming adult mammalian somatic cells |
| CN102027105B (zh) * | 2008-03-17 | 2015-03-11 | 斯克里普斯研究所 | 用于产生诱导的多能干细胞的组合化学遗传方法 |
| WO2009126250A2 (en) * | 2008-04-07 | 2009-10-15 | Nupotential, Inc. | Reprogramming a cell by inducing a pluripotent gene through rna interference |
| WO2010052904A1 (en) | 2008-11-05 | 2010-05-14 | Keio University | Method for producing neural stem cells |
| US20120121546A1 (en) | 2009-02-05 | 2012-05-17 | Vishal Bhasin | Method of Producing Progenitor Cells from Differentiated Cells |
| WO2010124143A1 (en) * | 2009-04-23 | 2010-10-28 | Nevada Cancer Institute | Reprogramming of somatic cells with purified proteins |
| AU2010249821B2 (en) * | 2009-05-20 | 2015-07-02 | Cardio3 Biosciences Sa | Method for determining the cardio-generative potential of mammalian cells |
| US9453205B2 (en) * | 2009-10-31 | 2016-09-27 | Genesis Technologies Limited | Methods for reprogramming cells and uses thereof |
| US9528087B2 (en) * | 2009-10-31 | 2016-12-27 | Genesis Technologies Limited | Methods for reprogramming cells and uses thereof |
| CN102947444A (zh) * | 2010-06-23 | 2013-02-27 | 帷幄生物技术公司 | 用于治疗心血管疾病的非遗传修饰性重编程细胞的组合物和方法 |
| EP2750509B1 (en) * | 2011-08-30 | 2016-12-28 | The J. David Gladstone Institutes | Methods for generating cardiomyocytes |
| US9540612B2 (en) | 2012-01-30 | 2017-01-10 | Icahn School Of Medicine At Mount Sinai | Methods for programming differentiated cells into hematopoietic stem cells |
-
2010
- 2010-11-01 US US13/504,988 patent/US9528087B2/en active Active
- 2010-11-01 WO PCT/CA2010/001727 patent/WO2011050476A1/en active Application Filing
- 2010-11-01 BR BR112012009921-3A patent/BR112012009921B1/pt not_active IP Right Cessation
- 2010-11-01 KR KR1020127014081A patent/KR101874463B1/ko active IP Right Grant
- 2010-11-01 DK DK10825907.8T patent/DK2494039T3/da active
- 2010-11-01 CA CA2779310A patent/CA2779310C/en active Active
- 2010-11-01 AU AU2010312240A patent/AU2010312240B2/en active Active
- 2010-11-01 KR KR1020197024507A patent/KR102134240B1/ko active IP Right Grant
- 2010-11-01 EP EP19170686.0A patent/EP3567103B1/en active Active
- 2010-11-01 SI SI201031910T patent/SI2494039T1/sl unknown
- 2010-11-01 EP EP10825907.8A patent/EP2494039B1/en active Active
- 2010-11-01 ES ES10825907T patent/ES2739672T3/es active Active
- 2010-11-01 KR KR1020187008318A patent/KR102014977B1/ko active IP Right Grant
- 2010-11-01 MX MX2012004881A patent/MX353245B/es active IP Right Grant
- 2010-11-01 SG SG10201702120VA patent/SG10201702120VA/en unknown
- 2010-11-01 LT LTEP10825907.8T patent/LT2494039T/lt unknown
- 2010-11-01 JP JP2012535565A patent/JP5982286B2/ja active Active
-
2012
- 2012-04-29 IL IL219453A patent/IL219453A/en active IP Right Grant
- 2012-05-29 ZA ZA2012/03902A patent/ZA201203902B/en unknown
-
2016
- 2016-03-24 JP JP2016059620A patent/JP2016146841A/ja active Pending
- 2016-10-19 US US15/297,972 patent/US10017737B2/en active Active
- 2016-10-19 US US15/298,034 patent/US10260046B2/en active Active
- 2016-10-19 US US15/298,006 patent/US10131879B2/en active Active
-
2017
- 2017-03-15 AU AU2017201774A patent/AU2017201774B2/en active Active
- 2017-08-24 IL IL254144A patent/IL254144A0/en unknown
-
2018
- 2018-07-04 JP JP2018127585A patent/JP2018183152A/ja active Pending
- 2018-11-19 US US16/195,005 patent/US10557123B2/en active Active
-
2019
- 2019-04-09 AU AU2019202451A patent/AU2019202451B2/en active Active
- 2019-07-03 HR HRP20191213TT patent/HRP20191213T1/hr unknown
-
2020
- 2020-01-24 US US16/752,462 patent/US20200165570A1/en not_active Abandoned
- 2020-02-23 IL IL272848A patent/IL272848B/en active IP Right Grant
- 2020-05-07 JP JP2020082207A patent/JP2020124219A/ja active Pending
-
2022
- 2022-06-16 JP JP2022097154A patent/JP7407865B2/ja active Active
-
2023
- 2023-03-22 US US18/188,421 patent/US20230227785A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2739672T3 (es) | Procedimientos de reprogramación de células y usos de los mismos | |
| US11795439B2 (en) | Methods for reprogramming cells and uses thereof | |
| US20190024056A1 (en) | Methods for reprogramming cells and uses thereof | |
| AU2014250190B2 (en) | Method for producing reprogrammed derivative neuronal stem cell from non-neuronal cell by using HMGA2 | |
| Tseng et al. | Substrate-mediated reprogramming of human fibroblasts into neural crest stem-like cells and their applications in neural repair |