Noninflammatory myopathies are heterogeneous mostly hereditary rare disorders. Prevalence is increasing because of genetic testing advances. Individuals with noninflammatory myopathy may present with proximal weakness, myalgia, and, sometimes, systemic involvement (eg, cardiac, endocrine, audiologic, and ocular symptoms). Onset can occur at any age and presentation varies with age.
This article summarizes epidemiology, clinical features, and diagnostic approach to the most common noninflammatory myopathies seen in general neurology practice. Dystrophinopathies (Becker’s and Duchenne’s muscular dystrophy), nutritional myopathies, and endocrine myopathies are outside the scope of this review.
Myotonic Dystrophy
Type 1 Myotonic Dystrophy
Individuals with type 1 myotonic dystrophy (DM1) typically present as youth with distal extremity weakness that may progress proximally. Specific neck flexor involvement may be apparent early, and typical facial features include temporalis atrophy and a tent-shape mouth caused by facial muscle atrophy. Delayed relaxation (action myotonia) of hand-grip or eyelid closure is seen. Percussing bellies of affected muscles (eg, abductor pollicis brevis) results in prolonged muscle contraction with delayed relaxation (percussion myotonia). A systemic disorder, DM1 affects the heart, eyes, muscles of respiration, and gastrointestinal tract. Cardiac conduction defects may lead to sudden cardiac death, and all persons with DM1 should be followed by a cardiologist; some benefit from a pacemaker. Cataracts, gastrointestinal motility issues, sleep apnea, testicular atrophy, and high rates of fetal loss to mothers with DM1 are common. Individuals with adult-onset DM1 may have mild intellectual disability and those with congenital DM1 born to mothers with DM1 may have severe intellectual disability.
Diagnostic test results usually show normal to mildly elevated creatine kinase (CK) levels, normal sensory and motor nerve conduction studies, and myotonic discharges on needle EMG of several muscles. Diagnosis is with genetic testing results that show a CTG trinucleotide repeat expansion (> 50 repeats) of DMPK on chromosome 19q13.2.1,2,3-11 The size of the unstable CTG repeat expansion is directly related to the severity of clinical weakness. Genetic anticipation is seen, with children of affected parents having a higher number of repeats and more severe clinical phenotype. Typical facial features and examination findings should prompt a focused genetic workup for these individuals.
Type 2 Myotonic Dystrophy
Type 2 (DM2) also affects many organ systems causing weakness, cardiac conduction defects, cataracts, hypogammaglobulinemia, and testicular atrophy. Onset is typically in early to middle adulthood. Individuals with DM2 typically present with intermittent stiffness and proximal leg muscle pain that may be described as debilitating or burning. Proximal and distal weakness progresses slowly and clinical myotonia may not be as prevalent as in DM1. Symptoms and severity of clinical weakness are heterogeneous, even within a family. Intellectual disability is less common in adults with DM2 compared with DM1 (Table 1). Diagnostic test results show mildly elevated CK, normal motor and sensory nerve conduction studies, and needle EMG myotonic discharges even in patients without clinical myotonia. Muscle biopsy shows nonspecific myopathic features (eg, increased internal nuclei, fiber size variability with type 2 fiber atrophy, small angulated fibers, and atrophic fibers with pyknotic nuclear clumps).12-14 Genetic test results show CCTG repeat expansion NF9 intron 1 on chromosome 3.12,15
Nondystrophic Myotonias and Periodic Paralysis
Myotonia Congenita
Inherited in autosomal dominant (Thomsen’s disease) or autosomal recessive patterns (Becker’s disease), myotonia congenita is a chloride channelopathy that causes slowly progressing limb stiffness, usually first in the lower extremities and progressing to the upper extremities. Muscle stiffness typically improves with activity, termed the warm-up phenomena. Individuals with myotonia congenita usually do not complain of muscle pain. Cold temperatures or pregnancy may worsen clinical myotonia. Anesthesia may cause malignant hyperthermia.
People with myotonia congenita often have a muscular build, even without regular exercise, action myotonia and percussion myotonia, and mild proximal weakness on manual strength testing. Thomsen’s disease usually presents in infancy; whereas, Becker’s disease typically presents after age 5 years and has worse muscle weakness.
Laboratory test results include normal CK levels, normal sensory and motor nerve conduction studies, and predominant myotonic discharges on needle EMG. On short exercise tests, individuals with Becker’s disease have decreased compound muscle action potential (CMAP) amplitude immediately after exercise that returns to normal after 30 to 40 seconds. In contrast, people with Thomsen’s disease have decreased CMAP amplitude after the limb muscle cools down. However, short exercise tests have been rendered obsolete by genetic testing for causative CLCN1 mutations on chromosome 7q35in both forms.16-18 Mutations in the chloride ion channel result in decreased chloride conductance and decreased rate of muscle membrane repolarization.
Paramyotonia Congenita
Paramyotonia congenita is an autosomal dominant disorder that typically presents before age 10 years, with eyelid opening weakness after crying or eyelid closure. Cold temperatures or potassium intake may trigger myotonia and weakness. In paramyotonia congenita there is worse muscle stiffness with exercise or prolonged activity. Muscle pain is less prominent than in DM2, and muscle weakness may progress.
Results of laboratory testing show mildly elevated CK, normal to elevated potassium levels during exacerbations, and normal sensory and motor nerve conduction studies in the absence of exacerbations. After brief exercise, repetitive afterdischarges occur with a single supramaximal stimulus, termed postexercise myotonic potentials (PEMPs). There may be a decrement with repetitive stimulation at 5 Hz. Short exercise test results are normal or show small increments in a warm muscle; in a cooled muscle there will be a marked drop in CMAP amplitude and very slow recovery over 1 hour. On needle EMG, there are prominent myotonic discharges. Genetic testing shows SCN4A mutations.19
Periodic Paralyses
Marked by episodic attacks of flaccid muscle weakness, periodic paralyses are typically associated with fluctuating serum potassium levels—high or low. Between exacerbations, serum potassium levels and sensory and motor nerve conductions are normal. After long exercise there is an initial increase in CMAP followed by gradual drop, then recovery to baseline. Exercise testing shows a drop in CMAP amplitude after sustained contraction of the abductor digiti minimi, maximally approximately 25 minutes after exercise. Treatment for periodic paralyses includes acetazolamide or other carbonic anhydrase inhibitors and avoidance of triggers.
Hyperkalemic Periodic Paralysis. Typically presenting before age 10 years, attacks of flaccid paralysis in hyperkalemic periodic paralysis last minutes to hours. Triggers include fasting, rest after exercise, and intake of potassium-rich foods. Ictal areflexia with preserved sensation is typical. Some people with hyperkalemic periodic paralysis progress to fixed proximal weakness. During attacks, serum potassium may be less than 5 mmol per L or increased by more than 1.5 mmol per L above baseline. Genetic test results show mutations in SCN4A on chromosome 17q23. Preventative measures, including a low-potassium/high carbohydrate diet and avoiding fasting, intense exercise, and cold temperatures may be useful. Acetazolamide may be used preventatively.
Hypokalemic Periodic Paralysis. Presenting in young adulthood, hypokalemic periodic paralysis has the highest frequency of attacks before age 35. The severity of weakness may vary from mild weakness to frank flaccid paralysis. Exacerbations last hours to days, significantly longer than with hyperkalemic periodic paralysis. Triggers include alcohol, carbohydrate-rich foods, stress, and rest after exercise. Serum potassium levels may drop to less than 3.0 mmol per L during an attack. Genetic test results show CACNA1S mutations on chromosome 1q32 or SCN4A mutations on chromosome 17q23.20-22 Treatment with acetazolamide or potassium-sparing diuretics can be tried and trigger avoidance is useful.
Andersen-Tawil Syndrome. Marked by episodes of flaccid muscle weakness, Andersen-Tawil syndrome occurs in the setting of any potassium, ventricular arrhythmia, and prolonged QT interval. Individuals with Anderson-Tawil syndrome have dysmorphic features, including short stature, fifth digit clinodactyly, and syndactyly of the second and third toes. Genetic test results show KCNJ2 mutation, Kir 2.1 on chromosome 17q23 in the majority of patients.23
Congenital Myopathies
Although some present in later childhood or early adulthood, most congenital myopathies present in infancy with hypotonia and generalized weakness, and motor development is often delayed. Even within the same family with the same known genotype, clinical phenotype varies. Classification is based on clinical presentation and electron microscopic muscle biopsy structural findings (Table 2).
Metabolic Myopathies
The metabolic myopathies are genetic disorders that impair intermediary metabolism in skeletal muscle; most fall into 1 of 3 categories, including the glycogen storage diseases, fatty acid oxidation defects, and mitochondrial myopathies. Metabolic myopathy typically presents with exercise intolerance and weakness.24
Glycogen Storage Diseases. Presenting during brief periods of high-intensity exercise, glycogen storage diseases cause muscle cramps within seconds to minutes of exercise. Many people with glycogen storage disease also have pigmenturia (ie, dark or red urine from myoglobin caused by rhabdomyolysis during exercise). Most glycogen storage diseases are autosomal recessive with a negative family history. Neurologic examination findings are typically normal between episodes.
McArdle’s disease, a disorder of glycolysis, is the most common metabolic myopathy, occurring in 1 in 100,000 people. Many individuals may have a second-wind phenomenon, unique to McArdle’s disease, in which pain may resolve after onset of exertional myalgias or cramps, allowing the individual to resume exercising.
In glycogen storage diseases, CK can be—but is not always—elevated, and EMG and nerve conduction studies are often normal and not of diagnostic value with the exception of Pompe’s disease. An autosomal recessive disease, Pompe’s disease has a classic infantile form presenting with hypertrophic cardiomyopathy and a late-onset juvenile/adult form without cardiomyopathy. The late-onset form should be suspected in people with progressive proximal weakness in a limb-girdle distribution with or without respiratory involvement. In Pompe’s disease, EMG and nerve conduction study results show characteristic myotonic and complex repetitive discharges. Gene sequencing is the preferred test for diagnosis.25
The classic diagnostic test for glycogen storage myopathies is the forearm exercise test in which a blood pressure cuff is inflated beyond arterial pressure while isometric rhythmic exercises are performed for 1 minute, followed by release of the cuff. Lactate and ammonia values collected before inflating and immediately after deflation are collected and compared. In glycolytic and glycogenolytic diseases, ammonia is elevated threefold, but lactate shows no significant rise.26 Genetic testing can be done with next generation sequencing or targeted analysis of a known genetic mutation. Muscle biopsy is not necessary after these steps but can show high glycogen, absent phosphorylase, or absent phosphofructokinase.
Fatty Acid Oxidation Defects. Fatty acid oxidation defects present during long duration or short intensity activities, fasting, or stressful events (eg, surgery, fever, or flu). In fatty acid oxidation defects, CK levels may be elevated during acute rhabdomyolysis, and EMG and nerve conduction study results are often normal. The most sensitive and specific diagnostic test is a serum acylcarnitine profile, performed under fasting conditions.26 Next generation sequencing panels or target analysis of the known genetic mutation based on the serum acylcarnitine profile can be done afterwards. Muscle biopsy is not necessary after genetic testing; when done it may show nonspecific increase in neutral lipid.
Mitochondrial Myopathies. A heterogeneous group of disorders with a range of phenotypes and genotypes, mitochondrial myopathies also present during long duration or short intensity exercise, fasting, or stressful events. Rhabdomyolysis and pigmenturia are uncommon and findings on the neurologic examination are variable owing to the multisystem involvement of the disease (eg, ptosis, ophthalmoplegia, or deafness). The serum lactate level is elevated in 65% of patients with mitochondrial myopathies, and EMG and nerve conduction study results are often normal and not of diagnostic value.25 Muscle biopsy is more helpful than in the glycogen storage diseases and fatty acid oxidation defects with characteristic histologic features of ragged red fibers on the modified-Gomori trichrome stain. As with the other myopathies, next generation sequencing or target analysis of the known genetic mutation is available for diagnosis.
Muscular Dystrophy
The primary symptom of muscular dystrophy, an inherited disorder, is muscle weakness. Dystrophinopathies (Becker and Duchenne muscular dystrophy) are outside the scope of this review. Here we focus on facioscapulohumeral and limb girdle muscular dystrophies (FSHD and LGMD).
Facioscapulohumeral Muscular Dystrophy
There are 2 types of genetically distinct FSHD with similar clinical features; 95% of cases are type 1 and 5% are type 2. Both are autosomal dominant, thought to be secondary to abnormal expression of toxic DUX4 protein. Among the most common muscular dystrophies, FSHD has prevalence of 1 in 15,000. The classic distribution of muscle weakness involves the facial, scapular, upper arm, lower leg, and abdominal muscles, with asymmetric involvement (Figure).
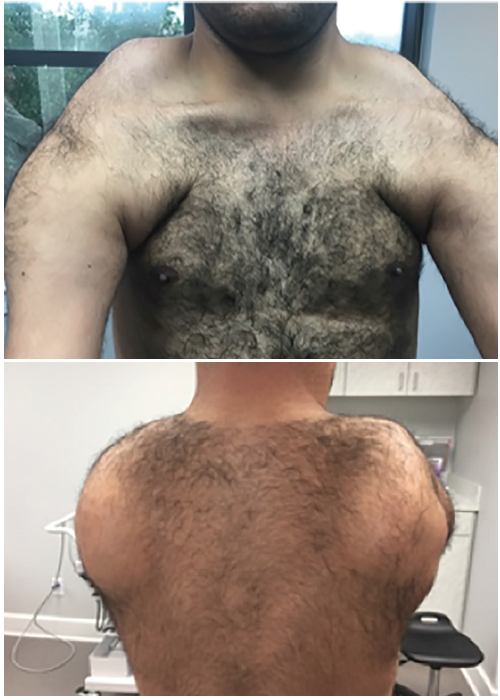
The most frequent initial symptoms are inability to lift arms over shoulder height or weakness of the facial muscles. Characteristic facial and upper torso appearance is seen on physical examination with decreased brow furrow, inability to close eyes, flattened smile, scapular winging, trapezius hump, and significant weakness and atrophy of the biceps brachii and triceps. The forearm muscles are relatively spared, producing “Popeye arms.” A positive Beevor’s sign may be elicited (umbilicus will move up or down a few centimeters when the patient is supine and attempts to flex the head).27 Extramuscular involvement including retinal vasculopathy, hearing loss, cardiac arrhythmia, cognitive impairment, and epilepsy may occur.
Diagnosis is usually based on clinical history, exam, and family history. Molecular genetic testing confirms the diagnosis. Individuals with type 1 FSHD have reduced D4Z4 repeats in chromosome 4q, and those with type 2 have SMCHD1 mutations on chromosome 18. Both types cause reduced methylation in the D4Z4 region, resulting in the expression of toxic DUX4 protein.
Limb Girdle Muscular Dystrophy
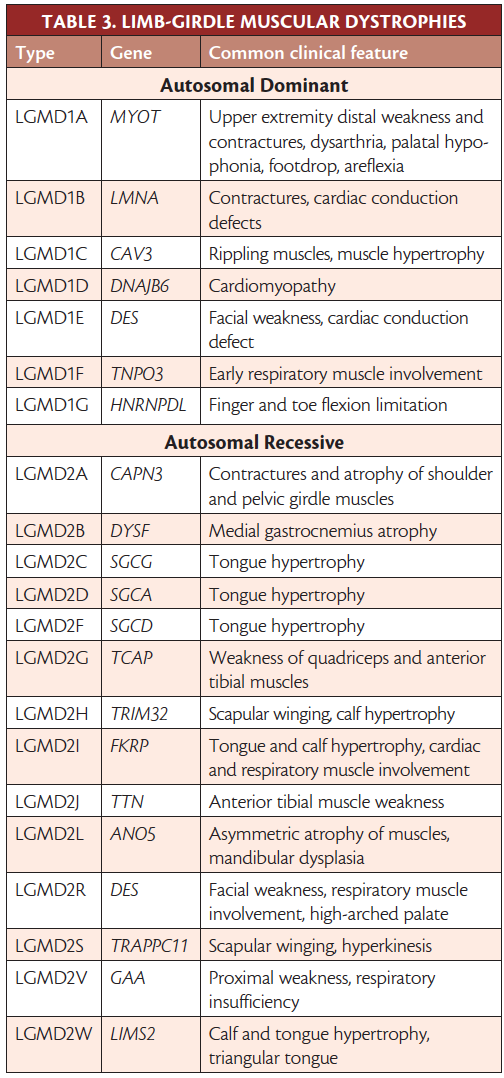
The LGMDs are heterogeneous, progressive, genetic disorders, characterized by shoulder and pelvic girdle muscle weakness, with incidence of 1 to 6 out of 100,000. Inheritance can be autosomal dominant (type 1), or recessive (type 2), or X-linked. Diagnosis is based on phenotype and genetic testing (Table 3),6 which should be done before ancillary testing (eg, biopsy, imaging, or electrodiagnostics).
1. Meola G. Clinical and genetic heterogeneity in myotonic dystrophies. Muscle Nerve. 2000;23(12);1789-1799.
2. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy; expansion of a trinucleotide (CTG) repeat at the 3’ end of transcript encoding protein kinase family member. Cell. 1992;68(4);799-808.
3. Fischbeck KH. The mechanism of myotonic dystrophy. Ann Neurol. 1994;35(3);255-256.
4. Fu Y-H, Friedman DL, Richards S, et al. Decreased expression of myotonin protein kinase messenger RNA and protein in adult for of myotonic dystrophy. Science. 1993;2(5102):60:235-238.
5. Fu Y-H, Pizzuti A, Fenwick R Jr, et al. An unstable triplet repeat in a gene related to myotonic dystrophy. Science. 1992;255(5049);1256-1258.
6. Harper PS, Harley HG, Reardon W, Shaw DJ. Anticipation in myotonic dystrophy: new light on an old problem. Am J Hum Genet. 1992;51(1):10-16.
7. Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3 prime untranslated region of the gene. Science. 1992;255:1253-1255.
8. Ptacek LJ, Johnson KJ, Griggs RC. Genetics and physiology of the myotonic disorders. New Engl J Med. 1993;328(7):482-489.
9. Shelbourne P, Davies J, Buxton J, et al. Direct diagnosis of myotonic dystrophy with a disease specific DNA marker. New Engl J Med. 1993;328(7):471-475.
10. Tian B, White RJ, Xia T, et al. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6(1):79-87.
11. Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of CIC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10(1):35-44.
12. Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic, and clinical spectrum. Neurology. 2003;60(4):657-664.
13. Schoser BG, Schneider-Gold C, Kress W, et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve. 2004a;29(2):275-281.
14. Vihola A, Bassez G, Meola G, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60(11):1854-1857.
15. Udd B, Meola G, Krake R, et al. Report of the 115th ENMC workshop: DM2/PROMM and other myotonic dystrophies. Neuromusc Disord. 2003;13;589-596.
16. George AL, Crackover MA, Abdulla JA, Hudson JA, Ebers GC. Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenital). Nat Genet. 1993;3:305-310.
17. Lorenz C, Meyer-Kleine C, Steinmeyer K, Koch MC, Jentsch TJ. Genomic organization of the human muscle chloride channel CIC-1 and analysis of novel mutations leading to Becker-type myotonia. Hum Mol Genet. 1994;3:941-946.
18. Wu FF, Ryan A, Devaney J, et al. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain. 2002;125:2392-2407.
19. Streib EW, Sun SF, Hanson M. Paramyotonia congenita: clinical and electrophysiologic studies. Electromyogr Clinical Neurophysiol. 1983;23(4):315-325.
20. Ricker K, Moxley RT 3rd, Heine R, Lehmann-Horn F. Myotonia fluctuans. A third type of muscle sodium channel disease. Arch Neurol. 1994;51(11):1095-1102..
21. Rüdel R, Ruppersberg JP, Spittelmeister W. Abnormalities of the fast sodium current in myotonic dystrophy, recessive generalized myotonia, and adynamia episodica. Muscle Nerve. 1989;12(4):281-287.
22. Bradley WG, Taylor R, Rice DR, et al. Progressive myopathy in hyperkalemic periodic paralysis. Arch Neurol. 1990;47:1013-1017.
23. Plaster NM, Tawil R, Trisani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s Syndrome. Cell. 2001;105(4):511-519.
24. Haller RG. Treatment of McArdle disease. Arch Neurol. 2000;57(7):923-924.
25. Hobson-Webb DD, Dearmey S, Kishnani PS. The clinical and electrodiagnostic characeristics of Pompe disease with post-enzyme replacement therapy findings. Clin Neurophysiol. 2011 Nov;122(11):2312-2317.
26. Tanopolsky MA. Metabolic myopathies. Continuum (Minneap Minn). 2016;22(6):1829-1851.
27. Statland JM, Tawil R. Facioscapulohumeral muscular dystrophy. Continuum (Minneap Minn). 2016;22(6):1916-1931.
YH, KP, and NK report no disclosures.



