Poor hDNA-Derived NGS Data May Provide Sufficient Phylogenetic Information of Potentially Extinct Taxa
 Catharina Clewing
Catharina Clewing Christian Kehlmaier
Christian Kehlmaier Björn Stelbrink
Björn Stelbrink Christian Albrecht1
Christian Albrecht1  Thomas Wilke
Thomas Wilke- 1Department of Animal Ecology and Systematics, Justus Liebig University Giessen, Giessen, Germany
- 2Museum of Zoology, Senckenberg Natural History Collections Dresden, Dresden, Germany
Museum material is an important source of metadata for past and recent biological events. With current sequencing technologies, it is possible to obtain historical DNA (hDNA) from older material and/or endangered species to answer taxonomic, systematic, and biogeographical questions. However, hDNA from museum collections is often highly degraded, making it difficult to assess relationships at or above the species level. We therefore studied two probably extinct gastropod species of the genus Laevicaspia, which were collected ∼140 years ago in the Caspian Sea, to map “standard” mitochondrial and nuclear markers and assess both the sequencing depth and the proportion of ambiguous sites as an indicator for the phylogenetic quality of the NGS data. Our study resulted in the first phylogenetically informative mitochondrial and nuclear markers for L. caspia. Assessment of both sequencing depth (mean coverage) and proportion of ambiguous sites suggests that our assembled consensus sequences are reliable for this species. In contrast, no informative gastropod-specific DNA was obtained for L. conus, likely due to a high degree of tissue digestion and contamination with non-gastropod DNA. Nevertheless, our results show that hDNA may in principle yield high-quality sequences for species-level phylogenetic analyses, which underlines the importance of museum collections as valuable archives of the biological past.
Introduction
Biological collections in museums represent archives of the recent and remote past, providing a variety of metadata that allow to address a wide range of research questions (e.g., Bakker et al., 2020; Miralles et al., 2020). In recent years, advances in molecular technology have enabled access to valuable genetic and genomic resources from both comparatively old ethanol- and formalin-fixed or dry materials (Bi et al., 2013; Hykin et al., 2015; Ruane and Austin, 2017; Derkarabetian et al., 2019; Kehlmaier et al., 2020; Card et al., 2021; Ernst et al., 2021; Orlando et al., 2021; Raxworthy and Smith, 2021). DNA from museum materials is often highly degraded (i.e., represented as ultrashort fragments) and sometimes cross-linked with proteins or other DNA fragments and thus difficult to access (see e.g., Card et al., 2021; Orlando et al., 2021; Raxworthy and Smith, 2021). Moreover, the corresponding DNA sequences may contain a high number of read errors, which usually makes population-level analyses infeasible. However, even small amounts of genetic (and genomic) information can still be valuable when placing individual species in a phylogenetic context (e.g., Guschanski et al., 2013; Fabre et al., 2014). This is of particular importance when the taxon of interest has gone extinct in the wild and/or its habitat is no longer accessible.
A prime example is the endemic Pontocaspian molluscan fauna that evolved in the Caspian Sea, the Black Sea, and the Aral Sea region. It has suffered from major anthropogenic disturbances since the mid-twentieth century and is facing a severe biodiversity crisis (Wesselingh et al., 2019). A large share of the c. 55–99 endemic species (see Wesselingh et al., 2019; Gogaladze et al., 2021) declined in abundance or completely vanished in the course of human activities in the last century, and have been replaced by invasive species. This affected both relatively large and highly abundant species such as the Caspian bivalves Dreissena caspia and D. elata, but also microgastropod species with restricted ranges such as Laevicaspia spp. (Hydrobiidae, Pyrgulinae). The latter genus comprises a total of 12 species, of which 10 are endemic to the Caspian Sea and 2 to the Black Sea (Wesselingh et al., 2019). However, with the exception of L. lincta from the Black Sea (Wilke et al., 2007), none of these species have been found alive recently and are thus only known from the fossil record and older museum materials (Gogaladze et al., 2021).
The lack of comparative genetic data not only complicates taxonomic decisions. More importantly, it makes the reconstruction of biogeographic patterns and evolutionary processes—such as the timing and causes of faunal separation between the Black Sea and Caspian Sea taxa—very difficult. Given the lack of recent material for these tiny species from the Caspian Sea, the question arises whether degraded historical DNA (hDNA; Raxworthy and Smith, 2021) from old museum collections is of sufficient quality to assess relationships at or above the species level. Mollusks might be particularly problematic as their soft bodies are typically rich in mucopolysaccharides, which hamper DNA isolation (Jaksch et al., 2016; Adema, 2021).
In this study, we therefore subjected two ∼140-year-old museum specimens of Laevicaspia from the Caspian Sea, L. caspia (Eichwald, 1838) and L. conus (Eichwald, 1838), to next-generation sequencing (NGS) protocols, which were developed for ancient and heavily degraded DNA. Specifically, we aimed to (i) map “standard” mitochondrial and nuclear markers from quality-filtered reads that are frequently used for taxonomic assignments and (ii) evaluate whether the quality of the NGS data is sufficient to establish reliable DNA barcode references and thus to provide robust phylogenetic information of potentially extinct taxa.
Materials and Methods
Materials
The ∼140-year-old specimens of Laevicaspia caspia and L. conus (Hydrobiidae, Pyrgulinae) were provided by the Zoological Institute of Russian Academy of Science (ZIN RAS), St. Petersburg, Russia (lot no. 4387/5 and 4614/4, respectively). Laevicaspia caspia was collected by O.A. Grimm in the Caspian Sea, ∼20 km off the eastern coast of Kazakhstan at a depth of ∼74 m (coordinates 43.28°N/51.05°E) on 9 July 1876. The individual of L. conus was collected by O.A. Grimm in the Caspian Sea, offshore near the city Baku at a depth of ∼11 m (geographical coordinates are not available) on 10 July 1874. In recent years, both specimens were stored in ethanol. However, it is not known in which fixative the individuals were originally preserved.
Genomic DNA was extracted from c. 3 mm3 of soft tissue using the GEN-IAL All-tissue DNA-Kit (GEN-IAL GmbH, Troisdorf, Germany) basic protocol for forensic material. The final DNA pellet was dissolved in 50 μL TE buffer. DNA concentration and average fragment length were measured with a Qubit Fluorometer High Sensitivity assay kit (Invitrogen, Carlsbad, CA, United States) and a TapeStation High Sensitivity D1000 assay kit (Agilent, Santa Clara, CA, United States), respectively (Supplementary Figures 1, 2). A final amount of 12.9 ng (L. caspia) and less than 0.2 ng (L. conus) of extracted DNA with average fragment lengths between 50 and 75 bp were converted into single-indexed, single-stranded Illumina sequencing libraries (see Gansauge and Meyer, 2013; Korlević et al., 2015), including the removal of uracil residues by uracil-DNA glycosylase (UDG) treatment. An Illumina MiSeq platform (Illumina, San Diego, CA, United States) housed at the Senckenberg Natural History Collections Dresden (Germany) was used for shotgun sequencing (75 bp paired-end reads), with each specimen being processed in its own private sequencing run.
Quality Control and Data Preparation
Raw reads were quality-checked and filtered using a previously established analytical pipeline (see Kehlmaier et al., 2017, 2019; Stelbrink et al., 2019). Adapters were trimmed with Skewer version 0.2.2 (Jiang et al., 2014), reads were merged (minimum length = 35 bp), filtered for quality (minimum Q-score = 20, corresponding to a base call accuracy of 99%), and duplicates were removed using BBMap version 37.241 (Bushnell, 2014). Per base sequence quality (i.e., base call accuracy) and read length distribution of trimmed (but unmerged) reads was analyzed and visualized using FastQC 0.11.9.2
Genomic Analysis
For the mitogenome assembly (see Table 1), the filtered reads (reduced readpool) were mapped against eight gastropod mitogenomes using Geneious Prime version 2021.1.1.3 Because no mitogenome is publicly available for the family Hydrobiidae, we chose the following representatives of the superfamily Truncatelloidea: (1) Bithynia leachii (Bithyniidae; GenBank acc. no. MT410857; N/A = locality unknown), (2) Caecum sp. (Caecidae; MT877093; Belize), (3) Oncomelania hupensis hupensis (Pomatiopsidae; NC_012899; China), (4) O. h. robertsoni (Pomatiopsidae; NC_013187; China), (5) Potamopyrgus antipodarum (Tateidae; MG979468; New Zealand), (6) Potamopyrgus estuarinus (Tateidae; GQ996415; N/A), (7) Stenothyra glabra (Stenothyridae; MN548735; China), and (8) Tricula hortensis (Pomatiopsidae; NC_013833; China). Geneious Prime settings used for the mitogenome mapping were: sensitivity = medium-low sensitivity/fast; 5 iterations; annotation similarity = 25%. Finally, the consensus sequence was generated using the default settings (threshold for highest quality = 60%; call Sanger heterozygotes > 50%).
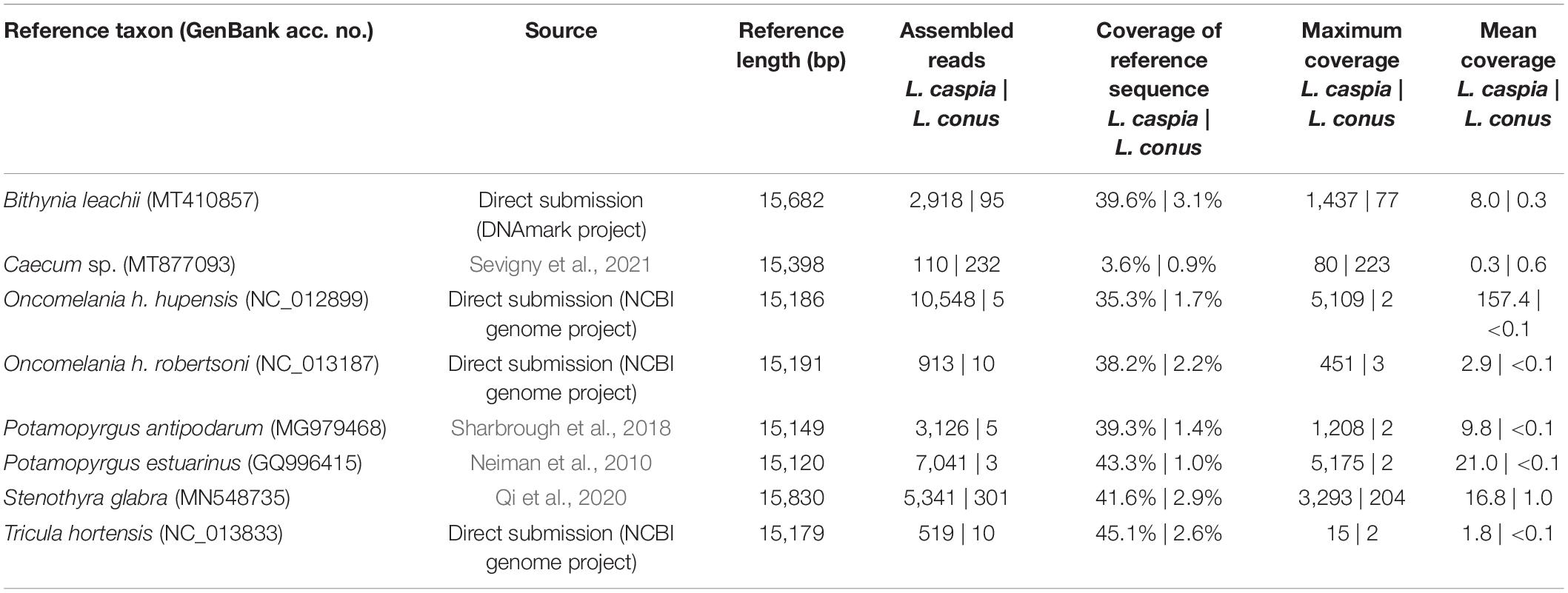
Table 1. Mitogenome mapping for L. caspia and L. conus.
In addition, single-gene mapping was performed (see Table 2) against standard genetic markers used for phylogenetic analyses (see phylogenies of truncatelloids of Wilke et al., 2013; Delicado et al., 2019; Layton et al., 2019). Overall, we focused on the following three mitochondrial and five nuclear gene fragments: (1) mitochondrial cytochrome c oxidase subunit I (COI), (2) mitochondrial small subunit ribosomal RNA (SSU rRNA, 12S), mitochondrial large subunit ribosomal RNA (LSU rRNA, 16S), (4) nuclear small subunit ribosomal RNA (SSU rRNA, 18S), (5) nuclear large subunit ribosomal RNA (LSU rRNA, 28S), (6) nuclear internal transcribed spacer 1 (ITS1), (7) nuclear internal transcribed spacer 2 (ITS2), and (8) nuclear histone 3 (H3). For the selection of gene fragments, we chose those seed reference sequences that were as closely related as possible, depending on the availability in GenBank (e.g., for COI, Laevicaspia lincta from the Azov Sea in Russia was selected; see Table 2). Settings for the single-gene fragment mapping in Geneious Prime were as follows: sensitivity = medium-low sensitivity/fast; 5 iterations. The consensus sequences were generated using the following settings: threshold for highest quality = 60%; call “N” if coverage < 2; call Sanger heterozygotes > 50%. Ambiguous sites (i.e., “N”) at the beginning and end of each sequence were removed afterward (see Table 2 for trimmed sequence lengths).
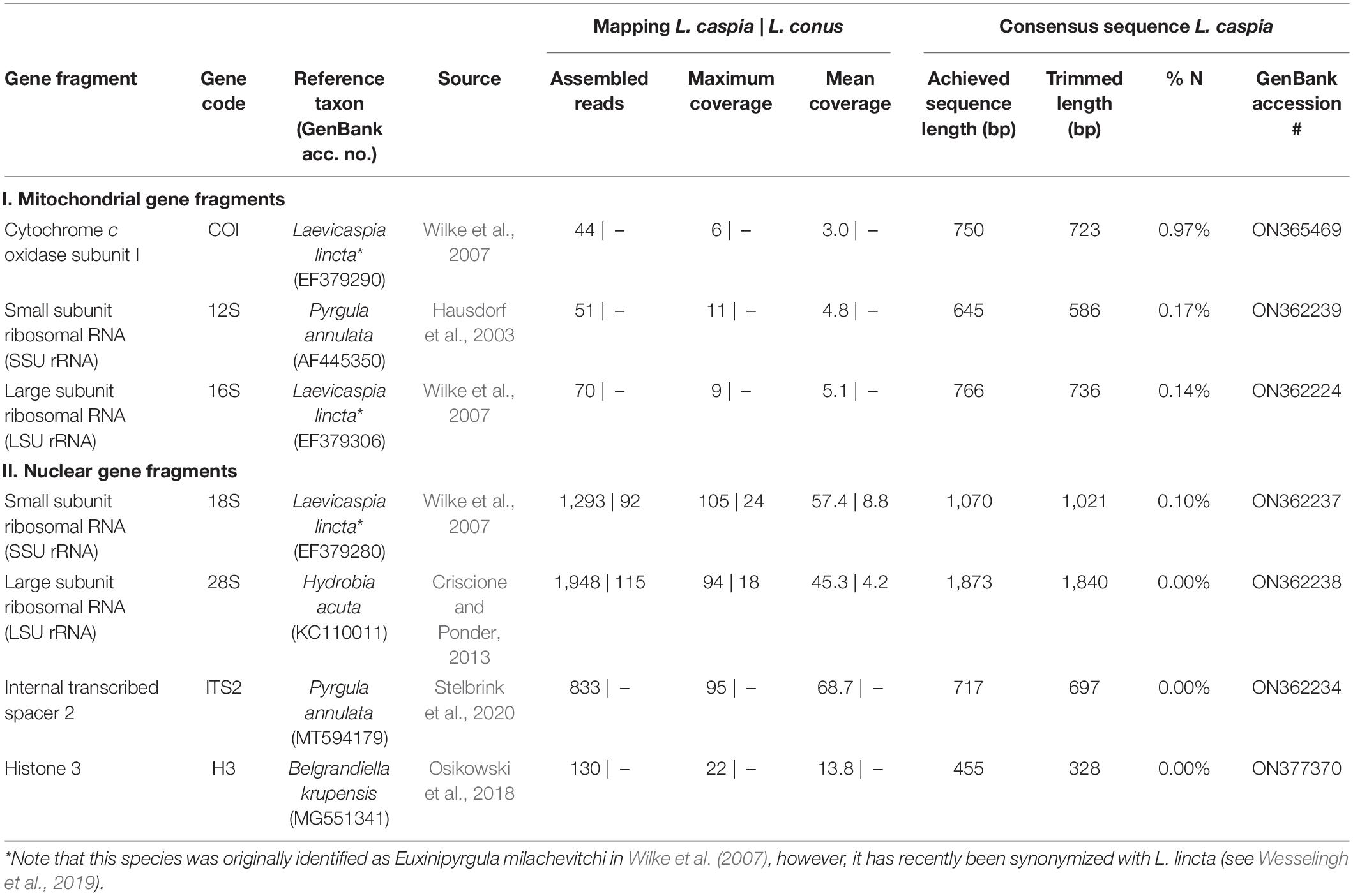
Table 2. Overview of achieved gene fragments for Laevicaspia caspia and L. conus (for the latter, only the mapping results are shown).
Phylogenetic Analysis
In order to place these two species in a phylogenetic context, we compiled a reduced multigene dataset (COI, 16S, and 18S) from Wilke et al. (2007). The dataset included Hydrobia acuta (Hydrobiinae; France; GenBank acc. no.: AF278808, AY222659, AF367680) and Pseudamnicola lucensis (Pseudamnicolinae; Italy; AF367651, AF478394, AF367687) as outgroup and the following taxa belonging to the Pyrgulinae: Dianella thiesseana (Greece; AY676127, AY676121, AY676125), Falsipyrgula pfeiferi (Turkey; EF379296, EF379312, EF379283), Laevicaspia lincta (=Euxinipyrgula milachevitchi; Russia; EF379290, EF379306, EF379280), Laevicaspia lincta (=Turricaspia sp.; Ukraine; EF379294, EF379310, EF379282), Laevicaspia lincta (=Micromelania lincta; Romania; EF379292, EF379308, EF379281), Ohridopyrgula macedonica (North Macedonia; EF379287, EF379302, EF379278), Pyrgula annulata (Italy; AY341258, AY676122, AY676124), and Xestopyrgula dybowskii (North Macedonia; EF379289, EF379304, EF379279). The 16S and 18S partitions were aligned with the MAFFT web service (Katoh and Toh, 2008; Katoh and Standley, 2013) with default settings, and best-fit substitution models for each partition were selected using jModelTest 2.1.4 (Darriba et al., 2012). Bayesian inference (BI) was performed as implemented in MrBayes 3.2.6 (Ronquist et al., 2012), with two independent MCMC searches running for 1,000,000 generations and sampling each 500th tree. A burn-in of 50% was applied a posteriori.
Results
Quality of Reads
A total of 37,339,378 (L. caspia) and 39,219,244 (L. conus) raw reads (read pairs) was generated in the two sequencing runs. The per base sequence quality was comparatively high for both untrimmed and trimmed reads. However, because the majority of trimmed reads was very short, i.e., ≤ 35 bp (c. 69.4% for L. caspia and 52.2% for L. conus; Figure 1), only c. 20.0% (L. caspia) and 45.1% (L. conus) of the read pairs could be joined in BBMap. After quality filtering, a total number of 6,036,414 (L. caspia) and 5,282,339 (L. conus) reads and thus only c. 16.2% (L. caspia) and 13.5% (L. conus) of the total reads sequenced could be used for subsequent analyses.
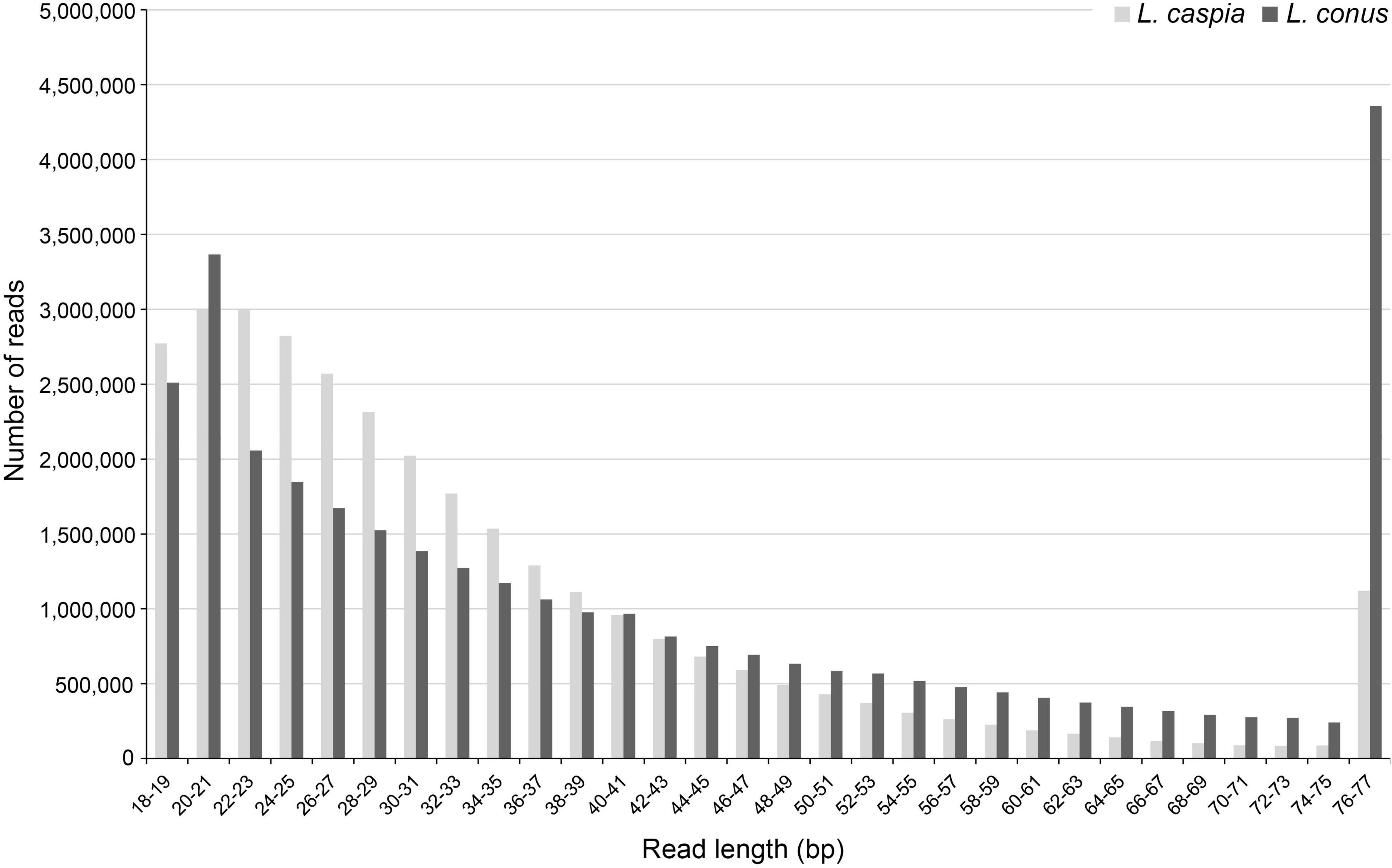
Figure 1. Fragment length distribution (in bp) of trimmed and quality-filtered reads (reduced readpool) of both Laevicaspia species.
Mitogenome Mapping
Eight truncatelloid mitogenomes were used to map the reduced readpool of L. caspia and L. conus. For L. caspia, the highest mean coverage (157.4) and second-highest maximum coverage (5,109) was obtained using the mitogenome data of Oncomelania hupensis hupensis (NC_012899; China) as seed reference (see Figure 2 and Table 1). Thereby, 10,548 reads from the reduced readpool could be assembled, covering 35.3% of the reference sequence and parts of the following five genes: COI (cytochrome c oxidase subunit 1; 84% similarity), 12S (small subunit rRNA), 18S (large subunit rRNA), ND2 (NADH-ubiquinone oxidoreductase chain 2), and ATP8 (ATP synthase protein 8). Neither the number nor the coverage of mapped tRNAs were examined here, although they were also found by the mapping algorithm. A similar number of genes was obtained when the reduced readpool was mapped against Tricula hortensis from China (NC_013833; see Table 1). For the Oncomelania hupensis hupensis mapping, the high coverage was, however, mainly due to an overrepresentation of mapped reads against ND2 starting at position 15,016. When this 778 bp-long fragment was removed, maximum and mean coverages were considerably lower (32 and 1.5, respectively; Figure 2). The lowest mean coverage (0.3), as well as coverage of the reference sequence (3.6%), was obtained with the mitogenome data of Caecum sp. (MT877093; Belize). For all other selected reference mitogenomes, mean and maximum coverage ranged from 1.8–21.0 to 15–5,175, respectively. This was sometimes a result of overrepresented mapped genes such as 16S, cyt b, and ND2. The coverage of these reference sequences was between 38.2 and 45.1% (for details see Table 1). For L. conus, considerably fewer reads (5–301) were mapped against all mitogenomes selected (Table 1). We therefore did not analyze these results in detail.
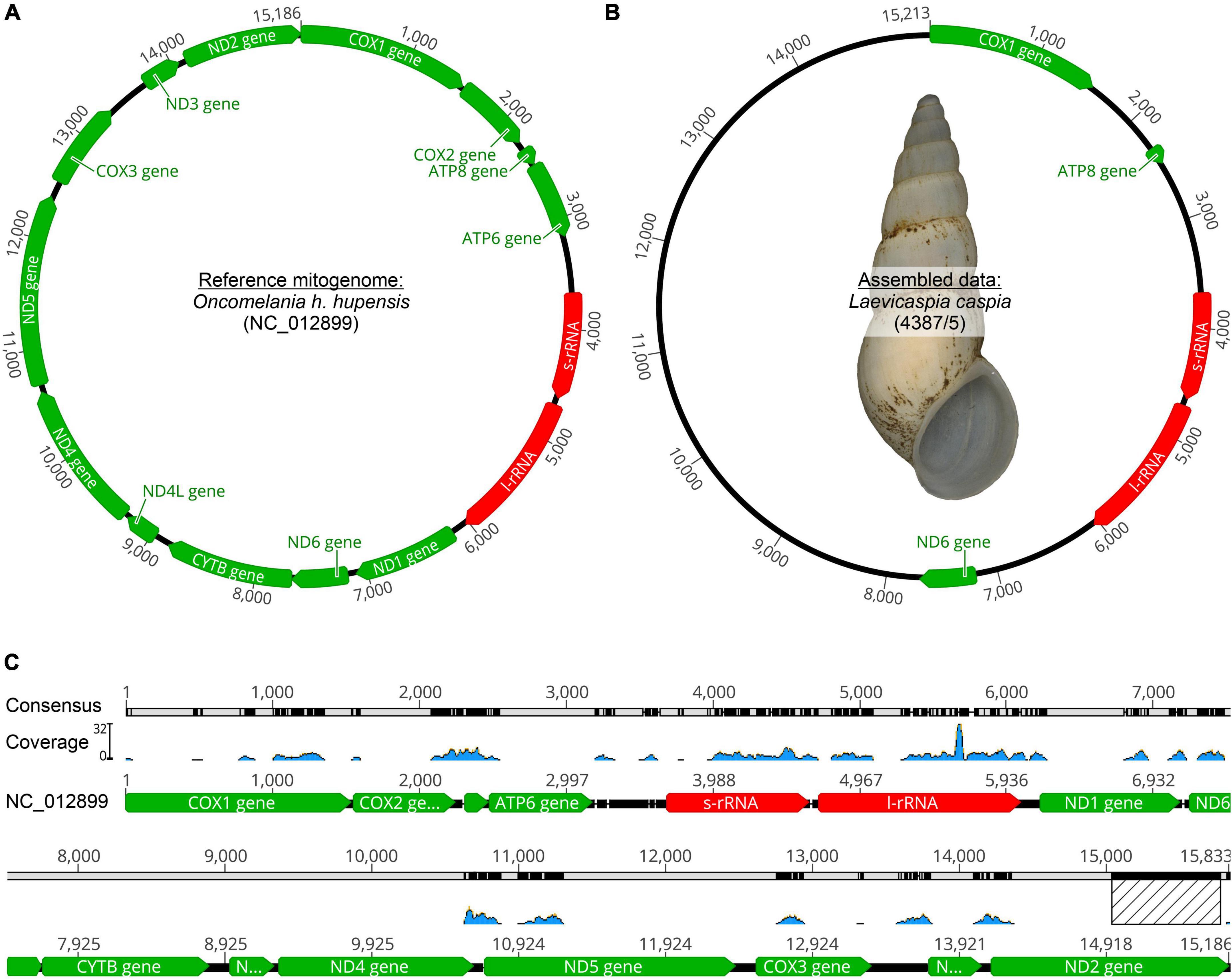
Figure 2. Mitogenome mapping using Geneious Prime (version 2021.1.1). (A) Seed reference mitogenome (O. h. hupensis: GenBank acc. no. NC_012899) where the highest coverage could be achieved, (B) Assembled data for Laevicaspia caspia (10,548 out of 6,036,414 reads; maximum coverage = 5,109; mean coverage = 157.4, see also Table 1) including a shell image of the sequenced individual (shell height = c. 12.4 mm), (C) Illustration of mitogenome coverage. Note that the overrepresented 778 bp fragment of ND2 (marked with a hatched rectangle) has been removed (see text for details).
Single-Gene Mapping
All selected “standard” genetic markers used for molecular phylogenies of truncatelloids could be successfully mapped using the reduced readpool of L. caspia (see Table 2). Mean and maximum coverage of the three mitochondrial markers (COI, 12S, and 16S) ranged from 3.0–5.1 to 6–11, respectively. Mean and maximum coverage of the four nuclear gene fragments (18S, 28S, ITS2, and H3) was considerably higher with values ranging from 13.8–68.7 to 22–105, respectively. The proportion of ambiguous sites (“N”) in the trimmed consensus sequence was used as an additional quality measure of the respective gene fragment. Thereby, the mitochondrial markers showed a generally higher N-content (0.14–0.97%) compared to the nuclear markers (0.00–0.10%), with COI having the highest (0.97%) and 28S, ITS2, and H3 having the lowest values (0.00%).
In contrast, the single-gene mapping was not successful for L. conus, similar to the mitogenome mapping (see above). Accordingly, only 18S and 28S could be mapped, though with a very low number of assembled reads (92 and 115, respectively; see Table 2). We therefore did not analyze these mapping results further. However, we applied a megablast search (as implemented in Geneious Prime; settings: nr/nt, maximum hits = 1) to the reduced readpool for fragments >100 bp (N = 365,445). Accordingly, 28,143 hits were found, of which 10,283 had a query coverage of 100%, i.e., a fragment length of 100 bp. In total, 1,490 unique organisms were found that mainly belong to bacteria (Supplementary Figure 3).
Phylogenetic Analysis
Due to the different mapping success, only sequence information from L. caspia could be used in the phylogenetic analyses. Accordingly, L. caspia from the Caspian Sea represents a genetically distinct lineage and forms a highly supported (Bayesian posterior probability, BPP = 1.00) clade within the Pyrgulinae, together with Falsipyrgula pfeiferi from Lake Egirdir (Turkey) and three individuals of Laevicaspia lincta sampled from different localities in the Black Sea basin (see Supplementary Figure 4).
Discussion
Leveraging genomic resources from historical museum material is a promising tool for addressing research topics related to the fields of biodiversity, conservation, taxonomy, and systematics, particularly for species that are rare or even extinct. Depending on age, tissue amount, and condition of the museum material, and the quality of generated sequences, complete mitogenomes and various nuclear loci of interest may, in principle, be assembled from raw sequencing data (e.g., Raxworthy and Smith, 2021). However, such analyses might be problematic in mollusks due to their high mucopolysaccharide content (Jaksch et al., 2016; Adema, 2021). Here, we used ∼140-year-old hydrobiid microgastropod specimens of Laevicaspia caspia and L. conus to map “standard” mitochondrial and nuclear markers for taxonomic assignments. We further assessed both the sequencing depth (mean coverage) as well as the proportion of ambiguous sites as an indicator of the phylogenetic quality of the NGS data.
The main problem in generating genomic information for both Laevicaspia species was probably not the DNA isolation and sequencing itself, but the preservation condition of the source tissue. Despite the overall high per base sequence quality, the reduced readpool was dominated by a large share of short DNA fragments and thus a low number of merged reads. Therefore, it was not possible to assemble a complete or near-complete mitogenome, although a high-quality mitogenome was previously generated for another ∼80-year-old freshwater gastropod specimen using the same laboratory pipeline in the same laboratory (see Stelbrink et al., 2019). However, applying our mitogenome and single-gene mapping approach, we were able to assemble taxonomically and phylogenetically informative mitochondrial and nuclear markers such as COI, 16S, and 18S (see e.g., Wilke et al., 2007), at least for L. caspia (Supplementary Figure 4). In contrast, virtually none of our mapping strategies were successful for L. conus. It is very likely that the specimen of this species was preserved under such poor conditions that the already small amount of tissue was too heavily digested and thus fragmented and further contaminated with non-gastropod DNA during decomposition (e.g., Raxworthy and Smith, 2021).
For assessing the reliability of the consensus sequences in L. caspia, we compared both the mean coverage as well as the proportion of ambiguous sites (see Table 2). The coverage of the mitochondrial target fragments was by an order of magnitude lower compared to the nuclear genes of interest and also considerably lower than the mean coverage of the previously published near-complete mitogenome of the paludomid gastropod Pseudocleopatra dartevellei (Stelbrink et al., 2019). We assume that this is related to the conditions under which the material was preserved. However, it would require a larger sequencing approach with several (fresh and old) samples to make a reliable statement. Similarly, given the higher mean coverage, the proportion of ambiguous sites was negligible for the nuclear fragments (<0.1%), whereas this ratio was higher, yet very low, for the mitochondrial markers (<1%). Overall, both factors—the moderate to high mean coverage together with the low amount of ambiguous sites—indicate that our assembled consensus sequences are reliable and can be used for taxonomic and phylogenetic purposes at the species level.
In summary, our pipeline using a set of single-gene and mitogenome seed reference sequences allowed us to map several phylogenetically relevant markers for L. caspia. These loci enabled us to provide the first DNA barcode sequences of this genus for the Caspian Sea. This will allow researchers to calculate genetic distances to other relatives, and to infer the phylogenetic position of this probably extinct species within the Pyrgulinae. Importantly, despite the relatively poor quality of our data, we here present information about an endangered ecosystem (e.g., Prange et al., 2020), whose endemic fauna is under increasing human pressure (e.g., Wesselingh et al., 2019).
Data Availability Statement
The data presented in the study are deposited in the NCBI GenBank repository, accession numbers ON362224, ON362234, ON362237, ON362238, ON362239, ON365469, and ON377370.
Author Contributions
CC analyzed the data, created the figures, and wrote the first draft of the manuscript. CK performed lab work and performed preliminary analyses. BS helped analyzing the data. CA and TW conceived the study. All authors contributed to drafting and reviewing the manuscript.
Funding
This study was funded by the German Research Foundation (DFG, Grant Nos. WI 1902-14 and WI 1902-17).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Vitaliy Anistratenko (I.I. Schmalhausen Institute of Zoology, Kiev, Ukraine), Dmitry M. Palatov (Zoological Institute of Russian Academy of Sciences, St. Petersburg, Russia), and Maxim V. Vinarski (Saint-Petersburg State University, St. Petersburg, Russia) for providing the Laevicaspia material. We thank two reviewers for their constructive comments on a previous version of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.907889/full#supplementary-material
Footnotes
- ^ https://sourceforge.net/projects/bbmap
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- ^ https://www.geneious.com
References
Adema, C. M. (2021). Sticky problems: extraction of nucleic acids from molluscs. Philos. Trans. R. Soc. Lond. B 376, 20200162. doi: 10.1098/rstb.2020.0162
Bakker, F. T., Antonelli, A., Clarke, J., Cook, J. A., Edwards, S. V., Faurby, S., et al. (2020). The Global Museum: natural history collections and the future of evolutionary biology and public education. PeerJ 8:e8225. doi: 10.7717/peerj.8225
Bi, K., Linderoth, T., Vanderpool, D., Good, J. M., Nielsen, R., and Moritz, C. (2013). Unlocking the vault: next-generation museum population genomics. Mol. Ecol. 22, 6018–6032. doi: 10.1111/mec.12516
Bushnell, B. (2014). BBMap: A Fast, Accurate, Splice-Aware Aligner. Berkeley, CA: Ernest Orlando Lawrence Berkeley National Laboratory.
Card, D. C., Shapiro, B., Giribet, G., Moritz, C., and Edwards, S. V. (2021). Museum genomics. Annu. Rev. Genet. 55, 633–659. doi: 10.1146/annurev-genet-071719-020506
Criscione, F., and Ponder, W. F. (2013). A phylogenetic analysis of rissooidean and cingulopsoidean families (Gastropoda: Caenogastropoda). Mol. Phylogenet. Evol. 66, 1075–1082. doi: 10.1016/j.ympev.2012.11.026
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Delicado, D., Arconada, B., Aguado, A., and Ramos, M. A. (2019). Multilocus phylogeny, species delimitation and biogeography of Iberian valvatiform springsnails (Caenogastropoda: Hydrobiidae), with the description of a new genus. Zool. J. Linn. Soc. 186, 892–914. doi: 10.1093/zoolinnean/zly093
Derkarabetian, S., Benavides, L. R., and Giribet, G. (2019). Sequence capture phylogenomics of historical ethanol-preserved museum specimens: unlocking the rest of the vault. Mol. Ecol. 19, 1531–1544. doi: 10.1111/1755-0998.13072
Ernst, R., Kehlmaier, C., Baptista, N. L., Pinto, P. V., Branquima, M. F., Dewynter, M., et al. (2021). Filling the gaps: the mitogenomes of Afrotropical egg-guarding frogs based on historical type material and a re-assessment of the nomenclatural status of Alexteroon Perret, 1988 (Hyperoliidae). Zool. Anz. 293, 215–224. doi: 10.1016/j.jcz.2021.06.002
Fabre, P.-H., Vilstrup, J. T., Raghavan, M., Der Sarkissian, C., Willerslev, E., Douzery, E. J. P., et al. (2014). Rodents of the Caribbean: origin and diversification of hutias unravelled by next-generation museomics. Biol. Lett. 10:20140266. doi: 10.1098/rsbl.2014.0266
Gansauge, M. T., and Meyer, M. (2013). Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 8, 737–748. doi: 10.1038/nprot.2013.038
Gogaladze, A., Son, M. O., Lattuada, M., Anistratenko, V. V., Syomin, V. L., Pavel, A. B., et al. (2021). Decline of unique Pontocaspian biodiversity in the Black Sea Basin: a review. Ecol. Evol. 11, 12923–12947. doi: 10.1002/ece3.8022
Guschanski, K., Krause, J., Sawyer, S., Valente, L. M., Bailey, S., Finstermeier, K., et al. (2013). Next-generation museomics disentangles one of the largest primate radiations. Syst. Biol. 62, 539–554. doi: 10.1093/sysbio/syt018
Hausdorf, B., Röpstorf, P., and Riedel, F. (2003). Relationships and origin of endemic Lake Baikal gastropods (Caenogastropoda: Rissooidea) based on mitochondrial DNA sequences. Mol. Phylogenet. Evol. 26, 435–443. doi: 10.1016/s1055-7903(02)00365-2
Hykin, S. M., Bi, K., and McGuire, J. A. (2015). Fixing formalin: a method to recover genomic-scale DNA sequence data from formalin-fixed museum specimens using high-throughput sequencing. PLoS One 10:e0141579. doi: 10.1371/journal.pone.0141579
Jaksch, K., Eschner, A., von Rintelen, T., and Haring, E. (2016). DNA analysis of molluscs from a museum wet collection: a comparison of different extraction methods. BMC Res. Notes 9:348. doi: 10.1186/s13104-016-2147-7
Jiang, H., Lei, R., Ding, S. W., and Zhu, S. (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182. doi: 10.1186/1471-2105-15-182
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katoh, K., and Toh, H. (2008). Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9, 286–298. doi: 10.1093/bib/bbn013
Kehlmaier, C., Barlow, A., Hastings, A. K., Vamberger, M., Paijmans, J. L. A., Steadman, D. W., et al. (2017). Tropical ancient DNA reveals relationships of the extinct bahamian giant tortoise Chelonoidis alburyorum. Proc. R. Soc. Lond. B 284:20162235. doi: 10.1098/rspb.2016.2235
Kehlmaier, C., Graciá, E., Campbell, P. D., Hofmeyr, M. D., Schweiger, S., Martínez-Silvestre, A., et al. (2019). Ancient mitogenomics clarifies radiation of extinct Mascarene giant tortoises (Cylindraspis spp.). Sci. Rep. 9:17487. doi: 10.1038/s41598-019-54019-y
Kehlmaier, C., Zinenko, O., and Fritz, U. (2020). The enigmatic Crimean green lizard (Lacerta viridis magnifica) is extinct but not valid: mitogenomics of a 120-year-old museum specimen reveals historical introduction. J. Zool. Syst. Evol. Res. 58, 303–307. doi: 10.1111/jzs.12345
Korlević, P., Gerber, T., Gansauge, M., Hajdinjak, M., Nagel, S., Aximu-Petri, A., et al. (2015). Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. Biotechniques 59, 87–93. doi: 10.2144/000114320
Layton, K. K. S., Middelfart, P. U., Tatarnic, N. J., and Wilson, N. G. (2019). Erecting a new family for Spirostyliferina, a truncatelloidean microgastropod, and further insights into truncatelloidean phylogeny. Zool. Scr. 48, 727–744. doi: 10.1111/zsc.12374
Miralles, A., Bruy, T., Wolcott, K., Scherz, M. D., Begerow, D., Beszteri, B., et al. (2020). Repositories for taxonomic data: where we are and what is missing. Syst. Biol. 69, 1231–1253. doi: 10.1093/sysbio/syaa026
Neiman, M., Hehman, G., Miller, J. T., Logsdon, J. M. Jr., and Taylor, D. R. (2010). Accelerated mutation accumulation in asexual lineages of a freshwater snail. Mol. Biol. Evol. 27, 954–963. doi: 10.1093/molbev/msp300
Orlando, L., Allaby, R., Skoglund, P., Der Sarkissian, C., Stockhammer, P. W., Ávila-Arcos, M. C., et al. (2021). Ancient DNA analysis. Nat. Rev. Methods Prim. 1:14. doi: 10.1038/s43586-020-00011-0
Osikowski, A., Hofman, S., Rysiewska, A., Sket, B., Prevorènik, S., and Falniowski, A. (2018). A case of biodiversity overestimation in the Balkan Belgrandiella A. J. Wagner, 1927 (Caenogastropoda: Hydrobiidae): molecular divergence not paralleled by high morphological variation. J. Nat. Hist. 52, 323–344. doi: 10.1080/00222933.2018.1424959
Prange, M., Wilke, T., and Wesselingh, F. P. (2020). The other side of sea level change. Commun. Earth Environ. 1:69. doi: 10.1038/s43247-020-00075-6
Qi, L., Kong, L., and Li, Q. (2020). Redescription of Stenothyra glabra A. Adam, 1861 (Truncatelloidea, Stenothyridae), with the first complete mitochondrial genome in the family Stenothyridae. Zookeys 991, 69–83. doi: 10.3897/zookeys.991.51408
Raxworthy, C. J., and Smith, B. T. (2021). Mining museums for historical DNA: advances and challenges in museomics. Trends Ecol. Evol. 36, 1049–1060. doi: 10.1016/j.tree.2021.07.009
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Ruane, S., and Austin, C. C. (2017). Phylogenomics using formalin-fixed and 100+ year-old intractable natural history specimens. Mol. Ecol. Resour. 17, 1003–1008. doi: 10.1111/1755-0998.12655
Sevigny, J., Leasi, F., Simpson, S., Di Domenico, M., Jörger, K. M., Norenburg, J. L., et al. (2021). Target enrichment of metazoan mitochondrial DNA with hybridization capture probes. Ecol. Indic. 121:106973. doi: 10.1016/j.ecolind.2020.106973
Sharbrough, J., Luse, M., Boore, J. L., Logsdon, J. M. Jr., and Neiman, M. (2018). Radical amino acid mutations persist longer in the absence of sex. Evolution 72, 808–824. doi: 10.1111/evo.13465
Stelbrink, B., Kehlmaier, C., Wilke, T., and Albrecht, C. (2019). The near-complete mitogenome of the critically endangered Pseudocleopatra dartevellei (Caenogastropoda: Paludomidae) from the Congo River assembled from historical museum material. Mitochondrial DNA B Resour. 4, 3229–3231. doi: 10.1080/23802359.2019.1669081
Stelbrink, B., Wilke, T., and Albrecht, C. (2020). Ecological opportunity enabled invertebrate radiations in ancient Lake Ohrid. J. Great Lakes Res. 46, 1156–1161. doi: 10.1016/j.jglr.2020.06.012
Wesselingh, F. P., Neubauer, T. A., Anistratenko, V. V., Vinarski, M. V., Yanina, T., ter Poorten, J. J., et al. (2019). Mollusc species from the Pontocaspian region – an expert opinion list. Zookeys 827, 31–124. doi: 10.3897/zookeys.827.31365
Wilke, T., Albrecht, C., Anistratenko, V. V., Sahin, S. K., and Yildirim, Z. (2007). Testing biogeographical hypotheses in space and time: faunal relationships of the putative ancient Lake Egirdir in Asia Minor. J. Biogeogr. 34, 1807–1821. doi: 10.1111/j.1365-2699.2007.01727.x
Keywords: historical DNA, museomics, Gastropoda, Caspian Sea, mapping, mitochondrial makers, nuclear markers
Citation: Clewing C, Kehlmaier C, Stelbrink B, Albrecht C and Wilke T (2022) Poor hDNA-Derived NGS Data May Provide Sufficient Phylogenetic Information of Potentially Extinct Taxa. Front. Ecol. Evol. 10:907889. doi: 10.3389/fevo.2022.907889
Received: 30 March 2022; Accepted: 04 May 2022;
Published: 23 May 2022.
Edited by:
Jonathan J. Fong, Lingnan University, ChinaReviewed by:
Parin Jirapatrasilp, Chulalongkorn University, ThailandLeila Belén Guzmán, Instituto de Biología Subtropical (CONICET-UNaM), Argentina
Copyright © 2022 Clewing, Kehlmaier, Stelbrink, Albrecht and Wilke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catharina Clewing, catharina.clewing@allzool.bio.uni-giessen.de
†Present address: Björn Stelbrink, Museum für Naturkunde – Leibniz Institute for Evolution and Biodiversity Science, Berlin, Germany
‡These authors share first authorship