 Xianzhen Zhou
Xianzhen Zhou Dan Yu
Dan Yu Zhimin Cao*
Zhimin Cao*- College of Forestry, Northwest A&F University, Yangling, China
Convergent evolution between distantly related taxa often mirrors adaptation to similar environments. Rust fungi and anther smuts, which belong to different classes in Pucciniomycotina, have independently evolved a phytoparasitic lifestyle, representing an example of convergent evolution in the fungal kingdom. To investigate their adaptations and the genetic bases underlying their phytoparasitic lifestyles, we performed genome-wide convergence analysis of amino acid substitutions, evolutionary rates, and gene gains and losses. Convergent substitutions were detected in ATPeV0D and RP-S27Ae, two genes important for the generation of turgor pressure and ribosomal biosynthesis, respectively. A total of 51 positively selected genes were identified, including eight genes associated with translation and three genes related to the secretion pathway. In addition, rust fungi and anther smuts contained more proteins associated with oligopeptide transporters and vacuolar proteases than did other fungi. For rust fungi and anther smuts, these forms of convergence suggest four adaptive mechanisms for a phytoparasitic lifestyle: 1) reducing the metabolic demand for hyphal growth and penetration at the pre-penetration stage, 2) maintaining the efficiency of protein synthesis during colonization, 3) ensuring the normal secretion of rapidly evolving secreted proteins, and 4) improving the capacity for oligopeptide metabolism. Our results are the first to shed light on the genetic convergence mechanisms and molecular adaptation underlying phytoparasitic lifestyles in fungi.
Introduction
As heterotrophs, many fungi are parasitic, where they develop a feeding relationship with other organisms (Zelmer, 1998). To obtain nourishment from hosts, parasitic fungi also need to penetrate the outer defenses of hosts and evade the immune response, often having harmful consequences for the hosts (Ewald, 1983). The spectrum of hosts ranges from plants and animals to fungi (Viterbo and Horwitz, 2010; Sharon and Shlezinger, 2013; Köhler et al., 2015), which has been implicated as one of the reasons for the high species diversity of fungi. Most parasitic fungi are parasites of plants and can cause severe damage to a wide range of agricultural crops, tree species used for wood production, and herbaceous plants with ornamental value (Pinon and Frey, 2005; Fontaine et al., 2013; Gramacho et al., 2013). To efficiently control phytoparasitic fungi, research on the genetic mechanisms underlying the phytoparasitic lifestyle is necessary.
Comparative genomics improved our understanding of how phytoparasitic lifestyles evolved. Phytoparasitic fungi possess very plastic and dynamic genomes because of the presence of many transposable elements (TEs) (Möller and Stukenbrock, 2017). This genomic environment has given a competitive advantage to phytoparasitic fungi that need to adapt constantly to the plant immune system (Raffaele and Kamoun, 2012). For phytoparasitic fungi, the expanded genes mainly encode lytic enzymes and putative transporters, while contracted gene families are involved in digesting plant cell walls (Powell et al., 2008). Transcriptomics technology has also helped reveal how phytoparasitic fungi change their gene expression upon host colonization. In most transcriptomics studies, a large proportion of genes were upregulated upon host colonization relative to ribosome-associated proteins, effectors, secreted enzymes, and membrane transporters. These responses involve forming infection structures, avoiding recognition, manipulating host defense, and absorbing nutrients from the host plant (van der Does and Rep, 2017). Although some molecular mechanisms underlying the phytoparasitic lifestyle have been revealed by comparative genomics and transcriptomics technology, how fungi adapt to a phytoparasitic lifestyle remains unclear.
Convergent evolution refers to the process in which species that are not closely related independently evolve similar traits for adaptation to similar selective pressures (Christin et al., 2010; Ghiselin, 2016). Phenotypic convergence often results from genetic convergence at multiple levels. Most studies fulfill the predominant criterion for inferring convergent molecular evolution, which is that the amino acid position of a protein changes from a different ancestral amino acid to the same descendant amino acid along independent evolutionary lineages (Zhang and Kumar, 1997). Examples include echolocation in bats and toothed whales (Parker et al., 2013), the bamboo diet of giant and red pandas (Hu et al., 2017), and the ability to live in marginal environments in mangroves (Xu et al., 2017). According to a more relaxed definition, convergence could also mean copy number or evolutionary rate changes in the same genes (Denoeud et al., 2014; Maria et al., 2016). Overall, convergence analyses provide more opportunities to reveal genetic mechanisms of adaptive evolution.
There is a great diversity of lifestyles among fungi in Pucciniomycotina, including phytoparasites, mycoparasites, entomoparasites, and saprobes (Aime et al., 2014). Two distantly related fungi, rust fungi in the order Pucciniales and anther smut fungi from Microbotryales, are the primary representatives of phytoparasitic fungi in Pucciniomycotina, causing a serious threat to agriculture and forestry worldwide (Aime et al., 2017; Begerow et al., 2017). In contrast, almost all the remaining members of Pucciniomycotina are nonphytoparasitic fungi (Aime et al., 2014). Phytoparasitic lifestyles have evolved independently in rust fungi and anther smuts, making them an ideal model of convergent evolution. To identify the genetic mechanism underlying convergence between these two distantly related phytoparasitic fungi, we performed analyses with three different perspectives: 1) the genes containing convergent amino acid substitutions between rust fungi and anther smuts, 2) the genes subjected to positive selection during the evolutionary process of rust fungi and anther smuts, and 3) convergent gene gain and loss in rust fungi and anther smuts. Our findings may help elucidate the adaptive mechanism associated with phytoparasitism in rust fungi and anther smuts. These findings might also facilitate the development of novel antifungal strategies for rust fungi and anther smuts.
Materials and Methods
Classification of Selected Fungi
The flow chart presented in Supplementary Figure S1 illustrates the main steps in our study. A total of 19 fungi were selected, including six rust fungi, three anther smuts, seven nonphytoparasitic fungi in Pucciniomycotina, and three nonbiotrophic fungi in Ustilaginomycotina. Among them, Austropuccinia psidii APSI1 (Aps), Cronartium quercuum f. sp. fusiforme G11 version 1.0 (Cqf) (Pendleton et al., 2014), Hemileia vastatrix XXXIII (Hvs), Melampsora larici-populina 98AG31 version 1.0 (Mlp) (Duplessis et al., 2011), Puccinia striiformis f. sp. tritici PST-130 (Pst) (Cantu et al., 2011), and Uromyces viciae-fabae I2 (Uvf) were selected as analytical data for representative rust fungi. Microbotryum intermedium 1389 BM 12 12 (Min) (Branco et al., 2017), Microbotryum lychnidis-dioicae p1A1 Lamole (Mld) (Perlin et al., 2015), and Microbotryum silenes-dioicae 1303 (Msd) were selected as phytoparasitic fungi for convergence analyses. The following nonphytoparasitic fungi were included for comparison: Cystobasidiopsis lactophilus JCM 7595 (Cla), Cystobasidium minutum MCA 4210 version 1.0 (Cmi) (Camiolo et al., 2019), Erythrobasidium yunnanense JCM 10687 (Eyu), Glaciozyma antarctica PI12 (Gan), Leucosporidiella creatinivora 62-1032 version 1.0 (Lcr), (Mondo et al., 2017), Rhodotorula graminis strain WP1 version 1.1 (Rgr) (Firrincieli et al., 2015), and Sporobolomyces roseus SR19 (Sro). Finally, Acaromyces ingoldii MCA 4198 (Ain) (Kijpornyongpan et al., 2018), Malassezia globosa CBS 7966 (Mgl) (Xu et al., 2007), and Pseudozyma antarctica T-34 (Pan) (Morita et al., 2013), which belong to three different classes in Ustilaginomycotina, were selected as the outgroup taxa.
The data of 11 selected fungi (Cqf, Mlp, Pst, Min, Mld, Cmi, Lcr, Rgr, Ain, Mgl, and Pan) are publicly available in the JGI MycoCosm portal (https://mycocosm.jgi.doe.gov/mycocosm/home). The protein and coding sequences (CDSs) of Msd were downloaded from the NCBI database. Genome assemblies without gene annotation for an additional seven fungi (Aps, Hvs, Uvf, Cla, Eyu, Gan, and Sro) publicly available in the NCBI database were downloaded. Then, protein-coding genes were predicted by BRAKER2 (Hoff et al., 2019). Detailed information for the selected fungi is available in Supplementary Table S1.
Building Orthologous Gene Groups and Phylogenomic Tree Construction
All amino acid sequences of the 19 fungi were analyzed to obtain orthologous groups by OrthoFinder with default parameters (Emms and Kelly, 2016). To determine the phylogenetic relationships between the selected fungi, we conducted a phylogenetic analysis using all the single-copy genes. Multiple sequence alignments of single-copy orthologous genes were performed using MAFFT (Katoh and Standley, 2013), and the gaps were removed using trimAl (Capella-Gutiérrez et al., 2009). Next, the genes were concatenated to generate a supergene sequence. The best-fitting model for phylogenetic analysis was estimated using ModelTest-NG (Darriba et al., 2020). With the input of these 19 supergene sequences, a phylogenetic tree was constructed using RAxML-NG software with 1,000 bootstrap replicates (Kozlov et al., 2019).
Convergence Analysis
Only single-copy genes were utilized to identify convergent amino acid sites. First, ancestral states were reconstructed using CodeML. We set node “a” as the ancestral state of rust fungi, node “b” as the most recent common ancestor (MRCA) of rust fungi and other nonphytoparasitic fungi, node “c” as the ancestral state of anther smuts, and node “d” as the MRCA of anther smuts and other nonphytoparasitic fungi (Figure 1). We identified convergent amino acid sites between rust fungi and anther smuts with the following rules: 1) the amino acid residues of node a and node c were identical, 2) amino acid changes were inferred to have occurred between node a and node b, and 3) amino acid changes were inferred to have occurred between node c and node d. Noteworthily, detection of false and random convergence events may result from limited taxon sampling.
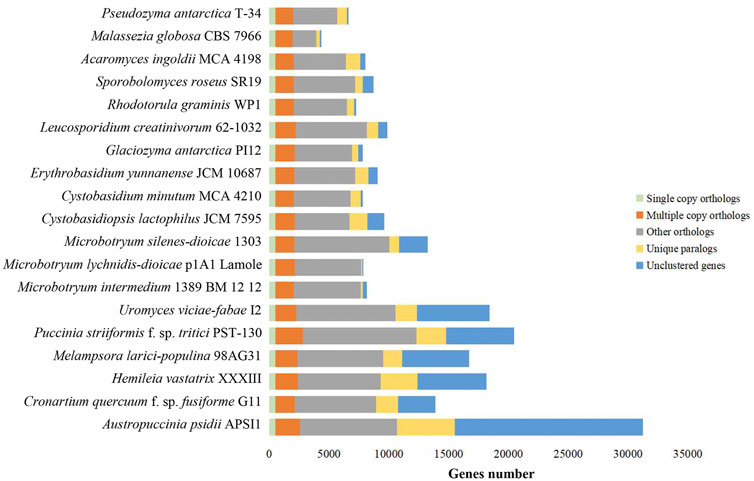
FIGURE 1. Orthologous groups for 19 selected fungi. Bars are subdivided to represent five different types of orthologs’ relationships. “Single_copy_orthologs” indicates one to one gene among all selected fungi. “Multiple_copy_orthologs” indicates other conserved genes present in multiple copies among all selected fungi. “Other_orthologs” indicates those genes present in at least two fungi, but not all fungi. “Unique_paralogs” indicates species-specific duplicated genes. “Unclustered_genes” indicates no homologous relationship between one and any other taxa.
Considering that conserved sites permit a more accurate inference of the ancestral state to avoid false convergence (Xu et al., 2017), the convergent amino acid sites with high support were selected with the following rules: 1) the amino acid residues of all rust fungi and anther smuts were identical, 2) there was a significant difference in the usage of amino acids between phytoparasitic and nonphytoparasitic fungi (p values less than 0.0005, calculated by Fisher’s exact test), and 3) more than five nonphytoparasitic fungi in Pucciniomycotina and more than two nonphytoparasitic fungi in Ustilaginomycotina tended to use the same amino acid. In general, the ancestral states of node a and node c should be more accurate when the first rule is met. The second and third rules were used to improve the accuracy of the ancestral states of node b and node d.
To filter out noise resulting from chance amino acid substitutions, we then performed a Poisson test to verify whether the observed number of highly supported convergent sites of each gene was significantly greater than the number expected with random substitution under the JTT-fgene model, following the method of Zou and Zhang (2015) (conv_cal, https://github.com/ztzou/conv_cal). The Benjamini–Hochberg method was used to correct multiple testing (q < 0.05).
Testing for Positive Selection
The aligned protein-coding sequences and the species tree obtained from the phylogenetic analysis were used to detect positive selection using the branch-site model in the CodeML program of PAML (Yang, 1997). For every single-copy gene, the branch-site model (model = 2 and NSsites = 2) of codon evolution was used. To identify genes under adaptive evolution related to phytoparasitism, the ancestral branch of rust fungi and the ancestral branch of anther smuts were designated as foreground branches, and the other branches were designated as background branches. We compared the model (which allowed sites to be under positive selection; fix_omega = 0) with the null model (in which sites may evolve neutrally or under purifying selection; fix_omega = 1 and omega = 1) with likelihood ratio tests (LRTs) performed by the CodeML program in PAML. The LRT statistics were evaluated by calculating the 2ΔLnL between the abovementioned models. The significance of LRT statistics was determined by a χ2 distribution, and p values were adjusted by following the Benjamini–Hochberg procedure. The alternative model was considered to be significantly better than the null model when q < 0.05.
Analyses of Gene Gain and Gene Loss
MCMCTree analysis was applied to estimate divergence times among the 19 fungi (Yang, 1997). According to the TimeTree database (http://www.timetree.org/), ancestral Basidiomycota fungi diverged into Ustilaginomycotina and Pucciniomycotina 489 million years ago (MYA), while the MRCA of Puccinia and Melampsora diverged 114.5 MYA (Kumar et al., 2017). Using these two fossil calibration points, we estimated the divergence times of the 19 selected fungi based on all single-copy genes. After acquiring this ultrametric tree, orthologous gene group expansion and contraction were detected by CAFE v5.0 (Mendes et al., 2020). Only the multicopy gene group that contained at least one protein in no fewer than eight species was chosen for analysis. Then, the p-value was calculated for each orthologous group, and orthologous groups with p values less than 0.05 were considered to have accelerated rates of expansion and contraction. The groups that had specifically expanded and contracted in rust fungi and anther smuts were considered candidate gene groups. We also performed a manual check to identify genes specifically missing or present in selected rust fungi and anther smuts.
Results
Orthologous Groups, Single-Copy Genes, and the Phylogeny
To study whether convergent evolution occurs in rust fungi and anther smuts, we selected 19 basidiomycetous fungi, including 16 fungi in Pucciniomycotina (six rust fungi, three anther smuts, and seven nonphytoparasitic fungi) and three nonphytoparasitic fungi in Ustilaginomycotina, for evolutionary analysis. We first identified all the orthologous gene groups in these fungi. In total, 19,479 orthologous gene groups were identified, including 530 single-copy orthologous gene groups and 1,276 multi-copy orthologous gene groups across all selected fungi (Supplementary Tables S2, S3). Consistent with the findings of previous studies (Cuomo et al., 2017), it is notable that rust fungi have very different gene contents from other selected fungi, harboring a larger number of unique paralogs (2,592, on average) and unclustered genes (7,004, on average) (Figure 2; Supplementary Table S2).
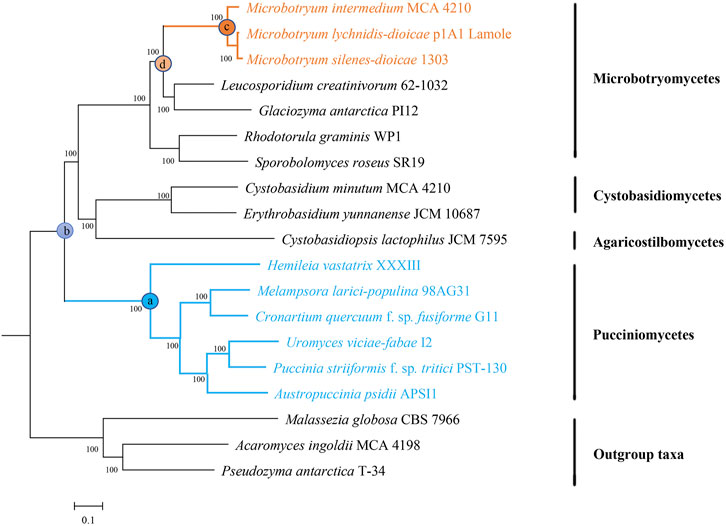
FIGURE 2. Genome-wide phylogenetic tree of 19 selected fungi. All single-copy genes were used to infer the phylogenomic tree topology using RAxML-NG. Numbers near the nodes are bootstrap support values. Anther smuts and rust fungi are indicated in orange and blue, respectively.
To identify the genomic signatures of convergent evolution between rust fungi and anther smuts, a maximum likelihood phylogeny was reconstructed on the basis of all the proteins encoded by the single-copy genes. The constructed genome-wide phylogenetic tree showed reasonable phylogenetic relationships. Within Pucciniomycotina, three anther smuts were clustered together with other Microbotryomycetes fungi. Compared with other selected Pucciniomycotina fungi, six rust fungi have occupied a basal position (Figure 1).
Convergent Signatures Between Rust Fungi and Anther Smuts
All 530 single-copy genes were used for genomic convergence analyses. Four hundred forty-four genes with 2,150 convergent amino acid substitutions between rust fungi and anther smuts were identified based on our initial criteria. Because genomic signatures of molecular convergence are generally not strong (Zou and Zhang, 2015), we surmised that random and false convergence events occurred frequently. Seventy-six genes with 84 highly supported sites that fit further criteria were chosen for the Poisson test. Overall, the Poisson test revealed that only two genes (OG0002989 and OG0003182) exhibited stronger signatures of convergent amino acid substitution than expected by chance (q < 0.05) (Supplementary Table S4).
These two convergent genes encode V-type H+-transporting ATPase subunit d (ATPeV0D) and ubiquitin-small subunit ribosomal protein S27Ae (RP-S27Ae). For ATPeV0D, two convergent substitutions occurred at site 17 (valine in nonphytoparasitic fungi to isoleucine in phytoparasitic fungi) and site 92 (lysine in nonphytoparasitic fungi to threonine in phytoparasitic fungi). These two sites are located in helix 1 and helix 5, respectively. Helix 1 is required for ATPase subunit assembly, and helix 5 contributes to the translocation of protons (Owegi et al., 2006). For RP-S27Ae, one convergent substitution occurred at site 57 (serine in nonphytoparasitic fungi to alanine in phytoparasitic fungi). This site is a phosphorylation site in the ubiquitin domain that regulates ubiquitin homeostasis (Figure 3) (Lee et al., 2017).
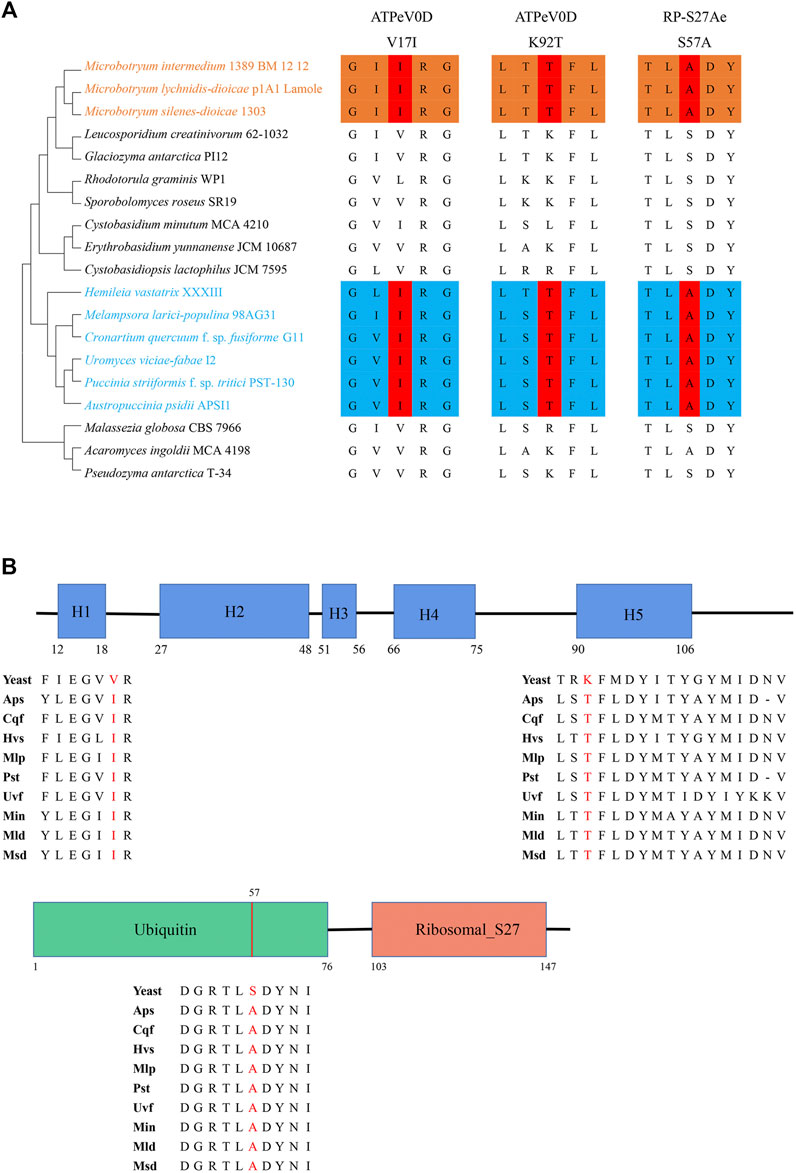
FIGURE 3. Two convergent genes. (A) Convergent amino acid substitutions in ATPeV0D and RP-S27Ae among the 19 fungi. The number above the convergent site denotes the amino acid position. (B) Structural domains of ATPeV0D and RP-S27Ae and the locations of three convergent amino acid sites. The structural domain of ATPeV0D was annotated based on the VMA6 protein in Saccharomyces cerevisiae, and that of RP-S27Ae was annotated based on the RPS31 protein in S. cerevisiae.
Positive Selection Signals in Rust Fungi and Anther Smuts
Across all 530 gene orthologues, we found convergent signals of positive selection in 51 genes during the evolution of rust fungi and anther smuts (Supplementary Table S5). To further characterize the functions of positively selected genes, we performed functional annotation with the Kyoto Encyclopaedia of Genes and Genomes (KEGG) database. In total, 43 of the 51 genes had KEGG annotations. Among them, 25 genes were associated with 27 pathways (Table 1). The genes were associated with very diverse pathways, including four main categories: metabolism, genetic information processing, environmental information processing, and cellular processes. There were eight genes in translation pathways (pathway ids: 00970, 03008, 03010, 03013, and 03015), which was the most abundant category. In addition, we found three positively selected genes associated with secretory pathways. They included a protein transport protein (Sec24, OG0003606), calnexin (CANX, OG0003436), and dolichyl-phosphate-mannose-protein mannosyltransferase (POMT, OG0002993). However, genes associated with other functions were relatively small in number.

TABLE 1. KEGG pathways of positively selected genes.
Expansion and Contraction of Orthologous Gene Groups in Rust Fungi and Anther Smuts
To identify convergent gene gain or loss in rust fungi and anther smuts, an analysis was performed in CAFÉ. First, a clock-calibrated phylogenetic tree was constructed based on the concatenation of all single-copy genes (Supplementary Figure S2). A total of 4,559 orthologous gene groups were used for CAFÉ analysis by filtering (Supplementary Table S6). There were a few orthologous gene groups that showed a convergent pattern of expansion or contraction between rust fungi and anther smuts. Only two gene groups were specifically expanded in the selected rust fungi and anther smuts. One orthologous group (OG0000061) was annotated as oligopeptide transporters (OPTs), and the other group (OG0000075) was associated with vacuolar protease (Table 2). However, there were no orthologous gene groups that showed a convergent pattern of contraction. Furthermore, no genes were specifically absent or present in rust fungi and anther smuts.
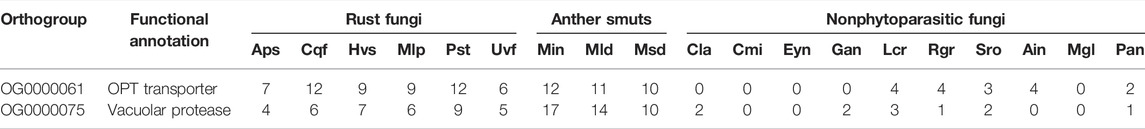
TABLE 2. Two expanded orthologous groups in rust fungi and anther smuts.
Discussion
By resolving the phylogeny of selected taxa, we clearly revealed that phytoparasitic lifestyles have evolved independently in rust fungi and anther smuts. Node b is the common ancestor of rust fungi and anther smuts. If rust fungi and anther smuts evolved a phytoparasitic lifestyle at the same time, all (or at least most) of the species arising from node b should be phytoparasitic, which is not consistent with the data (Figure 1). The phylogeny reconstructed in this study does not show the same topology as that in some previous studies in which Agaricostilbomycetes and Cystobasidiomycetes were sister to both Pucciniomycetes and Microbotryomycetes (Schell et al., 2011; Wang et al., 2015; Zhao et al., 2017). This is probably caused by the difference in datasets employed. Our results were inferred from the concatenation of single-copy genes, while previous studies were performed using one or several specific genes (ITS, RPB1, RPB2, TEF1, and CYTB). Phylogenetic studies based on one or several genes will probably be insufficient, with different phylogenetic studies producing highly contradictory results (Aime et al., 2006; Schell et al., 2011; Wang et al., 2015; Zhao et al., 2017). By comparison, the compilation of all single-copy genes will improve phylogenetic reconstructions due to increased statistical power (Salichos and Rokas, 2013). Furthermore, there are extreme differences in the life cycles, trophic modes, and genome contents of rust fungi and other fungi (Lorrain et al., 2019; Bakkeren and Szabo, 2020). In general, the more ancient the divergence is, the more diversity it accumulates (O'Meara, 2012). Thus, it is reasonable to place rust fungi as the earliest diverging lineage of the selected fungi.
For phytoparasitic fungi, external nutrient sources are likely to be scarce until successful penetration (Solomon et al., 2003; Divon and Fluhr, 2007). Using mechanical pressure to support hyphal elongation and penetration seems to be an ideal way to adapt to nutrient stress. Vacuoles undergo dramatic expansions during colonization in rust fungi and anther smuts (Chen et al., 2013; Kawamoto et al., 2017), which can produce turgor pressure to lessen the metabolic demands for cytoplasm biosynthesis (Richards et al., 2010). Vacuolar-type H+-ATPase (V-ATPase) is responsible for pumping protons into the vacuole, generating membrane potential for solute transport, which is important in the generation of turgor pressure (Smart et al., 1998). Consistently, signatures of convergence related to V-ATPase could be observed. ATPeV0D, which belongs to the V0 subunit of the V-ATPase, plays a role in the coupling of proton transport and ATP hydrolysis (Smith et al., 2005). In ATPeV0D, helix 1 and helix 5 are important domains required for V1Vo assembly and the translocation of protons, respectively (Owegi et al., 2006). The convergence that occurred in these two domains may enhance efficiency in the generation of turgor pressure, reducing energy consumption for hyphal growth and penetration.
The colonization process of phytoparasitic fungi is complex and continuous and requires support from protein synthesis. For this reason, ribosome-associated proteins could play an important role in colonization processes (Wang et al., 2019). We surmised that the sustainability and high efficiency of protein synthesis may facilitate colonization. RP-S27AE is a fusion protein composed of N-terminal ubiquitin and C-terminal ribosomal proteins. The ubiquitin component has a chaperone function, which aids in the correct folding and synthesis of ribosomal proteins (Lacombe et al., 2009). It has been proven that the Ser57Ala mutation occurring in the ubiquitin component can improve ubiquitin homeostasis in yeast (Lee et al., 2017). Hence, convergence may facilitate the sustainability of ribosomal biosynthesis because higher homeostasis could mean a longer working time. The positively selected genes were consistent with the results observed for convergent substitutions. In addition, there were eight genes involved in translation under positive selection in rust fungi and anther smuts. Nonsynonymous mutations occurring in these genes may help phytoparasitic fungi enhance their protein production, reflecting their adaptive evolution.
In rust fungi and anther smuts, secreted proteins are vital in interactions with host plants, and they have been proven to be under selection pressure to undergo nonsynonymous amino acid changes to avoid detection (Aguileta et al., 2010; Hacquard et al., 2012; Sperschneider et al., 2014). However, nonsynonymous amino acid changes may cause protein structural changes that impair folding and modification. To ensure the normal secretion of rapidly evolving secreted proteins, it is not surprising that genes related to the secretion pathway are under positive selection. We found that three genes essential for secretion were under positive selection in rust fungi and anther smuts. Among them, SEC24 promotes the formation of transport vesicles from the endoplasmic reticulum (ER), CANX promotes the accurate folding of glycoproteins entering the secretory pathway, and POMT is responsible for attaching mannose to serine and threonine residues of secreted proteins, modulating their location and function (Gentzsch and Tanner, 1996; Williams, 2006; Wendeler et al., 2007). Taken together, these findings suggest that positive selection on genes results in efficient transport, accurate folding, and precise modification of secreted proteins and thus has a favorable effect on colonization.
The convergent expansion of genes involved in oligopeptide uptake and metabolism could also reflect the adaptive evolution of a phytoparasitic lifestyle. OPTs are believed to translocate oligopeptides from the extracellular environment into the cytosol (Lubkowitz, 2011). Oligopeptides, an important nitrogen source for fungi, are abundant in plants (Becerra-Rodríguez et al., 2020). To adapt to this nutritional environment, OPTs were likely expanded in rust fungi and anther smuts. Moreover, some OPTs have been demonstrated to be significantly upregulated during infection by rust fungi and anther smuts (Duplessis et al., 2011; Toh et al., 2017). The expansion and high expression level of OPTs may improve the capacity for oligopeptide uptake in rust fungi and anther smuts, which benefits fungal growth and colonization. Moreover, the expansion of vacuolar proteases could support the above hypothesis because larger numbers of vacuolar proteases may be better for the degradation and recycling of oligopeptides.
Conclusion
This study is the first to show common adaptive mechanisms underlying phytoparasitic lifestyles in rust fungi and anther smuts. Signatures of convergence were identified based on convergent substitutions, positive selection, and gene gains or losses. A total of two convergent genes, 51 positively selected genes, and two expanded orthogroups were identified. These convergence events suggest four adaptive mechanisms underlying a phytoparasitic lifestyle: 1) reducing the metabolic demand for hyphal growth and penetration at the pre-penetration stage, 2) maintaining the efficiency of protein synthesis during colonization, 3) ensuring the normal secretion of rapidly evolving secreted proteins, and 4) improving the capacity for oligopeptide metabolism. However, existing methods mainly focus on strict single-copy genes, which account for a small portion of all genes in the selected fungi. For this reason, a small amount of evidence for convergence was found in our study. With the development of new methods, we expect that more evidence for convergence between rust fungi and anther smuts will be discovered in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
ZC planned and designed the research. XZ and DY performed the research, and XZ wrote the manuscript. All the authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This research was funded by the National Natural Science Foundation of China (Project No. 31870630, 31670650).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Prof. Yibo Hu (Institute of Zoology, Chinese Academy of Sciences), Prof. Zhengting Zou (Institute of Zoology, Chinese Academy of Sciences), Prof. Qi Wu (Institute of Microbiology, Chinese Academy of Sciences), and Dr. Qinhu Wang (Northwest A&F University) for providing suggestions for this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2022.863617/full#supplementary-material
Supplementary Figure 1 | Flow chart describing the main steps of convergence analyses.
Supplementary Figure 2 | The divergence times of 19 selected fungi.
Supplementary Table 1 | Detailed information on the selected fungi.
Supplementary Table 2 | General statistics about orthologous gene groups.
Supplementary Table 3 | The ID of genes for each orthogroup.
Supplementary Table 4 | Convergent analyses in rust fungi and anther smuts.
Supplementary Table 5 | Selective pressure analyses in rust fungi and anther smuts.
Supplementary Table 6 | The number of genes for each orthogroup.
Supplementary File 1 | The results of genome annotation, FASTA format sequences for RAxML-NG, FASTA format sequences for conv_cal, and PML format sequences for CodeML.
References
Aguileta, G., Lengelle, J., Marthey, S., Chiapello, H., Rodolphe, F., Gendrault, A., et al. (2010). Finding Candidate Genes under Positive Selection in Non-model Species: Examples of Genes Involved in Host Specialization in Pathogens. Mol. Ecol. 19 (2), 292–306. doi:10.1111/j.1365-294X.2009.04454.x
Aime, M. C., Matheny, P. B., Henk, D. A., Frieders, E. M., Nilsson, R. H., Piepenbring, M., et al. (2006). An Overview of the Higher Level Classification of Pucciniomycotina Based on Combined Analyses of Nuclear Large and Small Subunit rDNA Sequences. Mycologia 98 (6), 896–905. doi:10.3852/mycologia.98.6.896
Aime, M. C., McTaggart, A. R., Mondo, S. J., and Duplessis, S. (2017). “Phylogenetics and Phylogenomics of Rust Fungi,” in Phylogenetics and Phylogenomics of Rust FungiAdvances in Genetics. Editors J. P. Townsend,, and Z. Wang (Cambridge, Massachusetts: Academic Press), 267–307. doi:10.1016/bs.adgen.2017.09.011
Aime, M. C., Toome, M., and McLaughlin, D. J. (2014). “10 Pucciniomycotina,” in Systematics and Evolution: Part A. Editors D. J. McLaughlin,, and J. W. Spatafora (Berlin, Heidelberg: Springer Berlin Heidelberg), 271–294. doi:10.1007/978-3-642-55318-9_10
Bakkeren, G., and Szabo, L. J. (2020). Progress on Molecular Genetics and Manipulation of Rust Fungi. Phytopathology 110 (3), 532–543. doi:10.1094/phyto-07-19-0228-ia
Becerra-Rodríguez, C., Marsit, S., and Galeote, V. (2020). Diversity of Oligopeptide Transport in Yeast and its Impact on Adaptation to Winemaking Conditions. Front. Genet. 11 (602). doi:10.3389/fgene.2020.00602
Begerow, D., Kemler, M., Feige, A., and Yurkov, A. (2017). “Parasitism in Yeasts,” in Yeasts in Natural Ecosystems: Ecology. Editors P. Buzzini, M. A. Lachance, and A. Yurkov (Cham: Springer International Publishing), 179–210. doi:10.1007/978-3-319-61575-2_7
Branco, S., Badouin, H., Rodríguez de la Vega, R. C., Gouzy, J., Carpentier, F., Aguileta, G., et al. (2017). Evolutionary Strata on Young Mating-type Chromosomes Despite the Lack of Sexual Antagonism. Proc. Natl. Acad. Sci. USA 114 (27), 7067–7072. doi:10.1073/pnas.1701658114
Camiolo, S., Toome-Heller, M., Aime, M. C., Haridas, S., Grigoriev, I. V., Porceddu, A., et al. (2019). An Analysis of Codon Bias in Six Red Yeast Species. Yeast 36 (1), 53–64. doi:10.1002/yea.3359
Cantu, D., Govindarajulu, M., Kozik, A., Wang, M., Chen, X., Kojima, K. K., et al. (2011). Next Generation Sequencing Provides Rapid Access to the Genome of Puccinia Striiformis F. Sp. Tritici, the Causal Agent of Wheat Stripe Rust. PLoS One 6 (8), e24230. doi:10.1371/journal.pone.0024230
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). trimAl: a Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 25 (15), 1972–1973. doi:10.1093/bioinformatics/btp348
Chen, W., Wellings, C., Chen, X., Kang, Z., and Liu, T. (2014). Wheat Stripe (Yellow) Rust Caused byPuccinia Striiformisf. sp.Tritici. Mol. Plant Pathol. 15 (5), 433–446. doi:10.1111/mpp.12116
Christin, P.-A., Weinreich, D. M., and Besnard, G. (2010). Causes and Evolutionary Significance of Genetic Convergence. Trends Genet. 26 (9), 400–405. doi:10.1016/j.tig.2010.06.005
Cuomo, C. A., Bakkeren, G., Khalil, H. B., Panwar, V., Joly, D., Linning, R., et al. (2017). Comparative Analysis Highlights Variable Genome Content of Wheat Rusts and Divergence of the Mating Loci. G3-genes Genom Genet. 7 (2), 361–376. doi:10.1534/g3.116.032797
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 37 (1), 291–294. doi:10.1093/molbev/msz189
Denoeud, F., Carretero-Paulet, L., Dereeper, A., Droc, G., Guyot, R., Pietrella, M., et al. (2014). The Coffee Genome Provides Insight into the Convergent Evolution of Caffeine Biosynthesis. Science 345 (6201), 1181–1184. doi:10.1126/science.1255274
Divon, H. H., and Fluhr, R. (2007). Nutrition Acquisition Strategies during Fungal Infection of Plants. FEMS Microbiol. Lett. 266 (1), 65–74. doi:10.1111/j.1574-6968.2006.00504.x
Duplessis, S., Cuomo, C. A., Lin, Y.-C., Aerts, A., Tisserant, E., Veneault-Fourrey, C., et al. (2011). Obligate Biotrophy Features Unraveled by the Genomic Analysis of Rust Fungi. Proc. Natl. Acad. Sci. 108 (22), 9166–9171. doi:10.1073/pnas.1019315108
Emms, D. M., and Kelly, S. (2015). OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 16 (1), 1–14. doi:10.1186/s13059-015-0721-2
Ewald, P. W. (1983). Host-parasite Relations, Vectors, and the Evolution of Disease Severity. Annu. Rev. Ecol. Syst. 14 (1), 465–485. doi:10.1146/annurev.es.14.110183.002341
Firrincieli, A., Otillar, R., Salamov, A., Schmutz, J., Khan, Z., Redman, R. S., et al. (2015). Genome Sequence of the Plant Growth Promoting Endophytic Yeast Rhodotorula Graminis WP1. Front. Microbiol. 6, 978. doi:10.3389/fmicb.2015.00978
Fontaine, M. C., Gladieux, P., Hood, M. E., and Giraud, T. (2013). History of the Invasion of the Anther Smut Pathogen onSilene latifoliainNorthAmerica. New Phytol. 198 (3), 946–956. doi:10.1111/nph.12177
Gentzsch, M., and Tanner, W. (1996). The PMT Gene Family: Protein O-Glycosylation in Saccharomyces cerevisiae Is Vital. EMBO J. 15 (21), 5752–5759. doi:10.1002/j.1460-2075.1996.tb00961.x
Ghiselin, M. T. (2016). Homology, Convergence and Parallelism. Phil. Trans. R. Soc. B 371 (1685), 20150035. doi:10.1098/rstb.2015.0035
Gramacho, K., Miller, T., and Schmidt, R. (2013). Comparative Histopathology of Host Reaction Types in Slash Pine Resistant to Cronartium Quercuum F. Sp. Fusiforme. Forests 4 (2), 319–328. doi:10.3390/f4020319
Hacquard, S., Joly, D. L., Lin, Y.-C., Tisserant, E., Feau, N., Delaruelle, C., et al. (2012). A Comprehensive Analysis of Genes Encoding Small Secreted Proteins Identifies Candidate Effectors in Melampsora Larici-populina (poplar Leaf Rust). Mpmi 25 (3), 279–293. doi:10.1094/mpmi-09-11-0238
Hoff, K. J., Lomsadze, A., Borodovsky, M., and Stanke, M. (2019). “Whole-genome Annotation with BRAKER,” in Gene Prediction: Methods and Protocols. Editor M. Kollmar (New York, NY: Springer), 65–95. doi:10.1007/978-1-4939-9173-0_5
Hu, Y., Wu, Q., Ma, S., Ma, T., Shan, L., Wang, X., et al. (2017). Comparative Genomics Reveals Convergent Evolution between the Bamboo-Eating Giant and Red Pandas. Proc. Natl. Acad. Sci. USA 114 (5), 1081–1086. doi:10.1073/pnas.1613870114
Katoh, K., and Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30 (4), 772–780. doi:10.1093/molbev/mst010
Kawamoto, H., Hirata, A., and Kawano, S. (2017). Three-dimensional Ultrastructural Study of the Anther of Silene Latifolia Infected with Microbotryum Lychnidis-dioicae. PLoS One 12 (8), e0182686. doi:10.1371/journal.pone.0182686
Kijpornyongpan, T., Mondo, S. J., Barry, K., Sandor, L., Lee, J., Lipzen, A., et al. (2018). Broad Genomic Sampling Reveals a Smut Pathogenic Ancestry of the Fungal Clade Ustilaginomycotina. Mol. Biol. Evol. 35 (8), 1840–1854. doi:10.1093/molbev/msy072
Kohler, J. R., Casadevall, A., and Perfect, J. (2015). The Spectrum of Fungi that Infects Humans. Cold Spring Harbor Perspect. Med. 5 (1), a019273. doi:10.1101/cshperspect.a019273
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 35 (21), 4453–4455. doi:10.1093/bioinformatics/btz305
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 34 (7), 1812–1819. doi:10.1093/molbev/msx116
Lacombe, T., García-Gómez, J. J., De la Cruz, J., Roser, D., Hurt, E., Linder, P., et al. (2009). Linear Ubiquitin Fusion to Rps31 and its Subsequent Cleavage Are Required for the Efficient Production and Functional Integrity of 40S Ribosomal Subunits. Mol. Microbiol. 72 (1), 69–84. doi:10.1111/j.1365-2958.2009.06622.x
Lee, S., Tumolo, J. M., Ehlinger, A. C., Jernigan, K. K., Qualls-Histed, S. J., Hsu, P.-C., et al. (2017). Ubiquitin Turnover and Endocytic Trafficking in Yeast Are Regulated by Ser57 Phosphorylation of Ubiquitin. Elife 6, e29176. doi:10.7554/eLife.29176
Lorrain, C., Gonçalves dos Santos, K. C., Germain, H., Hecker, A., and Duplessis, S. (2019). Advances in Understanding Obligate Biotrophy in Rust Fungi. New Phytol. 222 (3), 1190–1206. doi:10.1111/nph.15641
Lubkowitz, M. (2011). The Oligopeptide Transporters: a Small Gene Family with a Diverse Group of Substrates and Functions? Mol. Plant 4 (3), 407–415. doi:10.1093/mp/ssr004
Maria, C., Robinson, J. D., and Clark, N. L. (2016). Hundreds of Genes Experienced Convergent Shifts in Selective Pressure in marinemammals. Mol. Biol. Evol. 33 (9), 2182–2192. doi:10.1093/molbev/msw112
Mendes, F. K., Vanderpool, D., Fulton, B., and Hahn, M. W. (2020). CAFE 5 Models Variation in Evolutionary Rates Among Gene Families. Bioinformatics 36 (22-23), 5516–5518. doi:10.1093/bioinformatics/btaa1022
Möller, M., and Stukenbrock, E. H. (2017). Evolution and Genome Architecture in Fungal Plant Pathogens. Nat. Rev. Microbiol. 15 (12), 756–771. doi:10.1038/nrmicro.2017.76
Mondo, S. J., Dannebaum, R. O., Kuo, R. C., Louie, K. B., Bewick, A. J., Labutti, K., et al. (2017). Widespread Adenine N6-Methylation of Active Genes in Fungi. Nat. Genet. 49 (6), 964–968. doi:10.1038/ng.3859
Morita, T., Koike, H., Koyama, Y., Hagiwara, H., Ito, E., Fukuoka, T., et al. (2013). Genome Sequence of the Basidiomycetous Yeast Pseudozyma antarctica T-34, a Producer of the Glycolipid Biosurfactants Mannosylerythritol Lipids. Genome Announc 1 (2), e0006413. doi:10.1128/genomeA.00064-13
O'Meara, B. C. (2012). Evolutionary Inferences from Phylogenies: a Review of Methods. Annu. Rev. Ecol. Evol. Syst. 43 (1), 267–285. doi:10.1146/annurev-ecolsys-110411-160331
Owegi, M. A., Pappas, D. L., Finch, M. W., Bilbo, S. A., Resendiz, C. A., Jacquemin, L. J., et al. (2006). Identification of a Domain in the Vo Subunit D that Is Critical for Coupling of the Yeast Vacuolar Proton-Translocating ATPase. J. Biol. Chem. 281 (40), 30001–30014. doi:10.1074/jbc.M605006200
Parker, J., Tsagkogeorga, G., Cotton, J. A., Liu, Y., Provero, P., Stupka, E., et al. (2013). Genome-wide Signatures of Convergent Evolution in Echolocating Mammals. Nature 502 (7470), 228–231. doi:10.1038/nature12511
Pendleton, A. L., Smith, K. E., Feau, N., Martin, F. M., Grigoriev, I. V., Hamelin, R., et al. (2014). Duplications and Losses in Gene Families of Rust Pathogens Highlight Putative Effectors. Front. Plant Sci. 5, 299. doi:10.3389/fpls.2014.00299
Perlin, M. H., Amselem, J., Fontanillas, E., Toh, S. S., Chen, Z., Goldberg, J., et al. (2015). Sex and Parasites: Genomic and Transcriptomic Analysis of Microbotryum Lychnidis-dioicae, the Biotrophic and Plant-Castrating Anther Smut Fungus. BMC Genomics 16 (1), 461. doi:10.1186/s12864-015-1660-8
Pinon, J., and Frey, P. (2005). “Interactions between poplar Clones and Melampsora Populations and Their Implications for Breeding for Durable Resistance,” in Rust Diseases of Willow and Poplar. Editors M. H. Pei, and A. R. McCracken (Wallingford, United Kingdom: CABI), 139–154. doi:10.1079/9780851999999.0139
Powell, A. J., Conant, G. C., Brown, D. E., Carbone, I., and Dean, R. A. (2008). Altered Patterns of Gene Duplication and Differential Gene Gain and Loss in Fungal Pathogens. BMC Genomics 9, 147. doi:10.1186/1471-2164-9-147
Raffaele, S., and Kamoun, S. (2012). Genome Evolution in Filamentous Plant Pathogens: Why Bigger Can Be Better. Nat. Rev. Microbiol. 10 (6), 417–430. doi:10.1038/nrmicro2790
Richards, A., Veses, V., and Gow, N. (2010). Vacuole Dynamics in Fungi. Fungal Biol. Rev. 24 (3–4), 93–105. doi:10.1016/j.fbr.2010.04.002
Salichos, L., and Rokas, A. (2013). Inferring Ancient Divergences Requires Genes with strong Phylogenetic Signals. Nature 497 (7449), 327–331. doi:10.1038/nature12130
Schell, W. A., Lee, A. G., and Aime, M. C. (2011). A New Lineage in Pucciniomycotina: Class Tritirachiomycetes, Order Tritirachiales, Family Tritirachiaceae. Mycologia 103 (6), 1331–1340. doi:10.3852/10-333
Sharon, A., and Shlezinger, N. (2013). Fungi Infecting Plants and Animals: Killers, Non-killers, and Cell Death. Plos Pathog. 9 (8), e1003517. doi:10.1371/journal.ppat.1003517
Smart, L. B., Vojdani, F., Maeshima, M., and Wilkins, T. A. (1998). Genes Involved in Osmoregulation during Turgor-Driven Cell Expansion of Developing Cotton Fibers Are Differentially Regulated1. Plant Physiol. 116 (4), 1539–1549. doi:10.1104/pp.116.4.1539
Smith, A. N., Jouret, F., Bord, S., Borthwick, K. J., Al-Lamki, R. S., Wagner, C. A., et al. (2005). Vacuolar H+-ATPase D2 Subunit: Molecular Characterization, Developmental Regulation, and Localization to Specialized Proton Pumps in Kidney and Bone. Jasn 16 (5), 1245–1256. doi:10.1681/ASN.2004090761
Solomon, P. S., Tan, K.-C., and Oliver, R. P. (2003). The Nutrient Supply of Pathogenic Fungi; a fertile Field for Study. Mol. Plant Pathol. 4 (3), 203–210. doi:10.1046/j.1364-3703.2003.00161.x
Sperschneider, J., Ying, H., Dodds, P. N., Gardiner, D. M., Upadhyaya, N. M., Singh, K. B., et al. (2014). Diversifying Selection in the Wheat Stem Rust Fungus Acts Predominantly on Pathogen-Associated Gene Families and Reveals Candidate Effectors. Front. Plant Sci. 5, 372. doi:10.3389/fpls.2014.00372
Toh, S. S., Chen, Z., Schultz, D. J., Cuomo, C. A., and Perlin, M. H. (2017). Transcriptional Analysis of Mating and Pre-infection Stages of the Anther Smut, Microbotryum Lychnidis-dioicae. Microbiology 163 (3), 410–420. doi:10.1099/mic.0.000421
van der Does, H. C., and Rep, M. (2017). Adaptation to the Host Environment by Plant-Pathogenic Fungi. Annu. Rev. Phytopathol. 55 (1), 427–450. doi:10.1146/annurev-phyto-080516-035551
Viterbo, A., and Horwitz, B. A. (2010). “Mycoparasitism,” in Cellular and Molecular Biology of Filamentous Fungi. Editors K. A. Borkovich,, and D. J. Ebbole (Washington: ASM Press), 676–693.
Wang, B., Song, N., Tang, C., Ma, J., Wang, N., Sun, Y., et al. (2019). PsRPs26, a 40S Ribosomal Protein Subunit, Regulates the Growth and Pathogenicity of Puccinia Striiformis F. Sp. Tritici. Front. Microbiol. 10, 968. doi:10.3389/fmicb.2019.00968
Wang, Q.-M., Groenewald, M., Takashima, M., Theelen, B., Han, P.-J., Liu, X.-Z., et al. (2015). Phylogeny of Yeasts and Related Filamentous Fungi within Pucciniomycotina Determined from Multigene Sequence Analyses. Stud. Mycol. 81, 27–53. doi:10.1016/j.simyco.2015.08.002
Wendeler, M. W., Paccaud, J. P., and Hauri, H. P. (2007). Role of Sec24 Isoforms in Selective export of Membrane Proteins from the Endoplasmic Reticulum. EMBO Rep. 8 (3), 258–264. doi:10.1038/sj.embor.7400893
Williams, D. B. (2006). Beyond Lectins: the Calnexin/calreticulin Chaperone System of the Endoplasmic Reticulum. J. Cel Sci. 119 (4), 615–623. doi:10.1242/jcs.02856
Xu, J., Saunders, C. W., Hu, P., Grant, R. A., Boekhout, T., Kuramae, E. E., et al. (2007). Dandruff-associated Malassezia Genomes Reveal Convergent and Divergent Virulence Traits Shared with Plant and Human Fungal Pathogens. Proc. Natl. Acad. Sci. 104 (47), 18730–18735. doi:10.1073/pnas.0706756104
Xu, S., He, Z., Guo, Z., Zhang, Z., Wyckoff, G. J., Greenberg, A., et al. (2017). Genome-wide Convergence during Evolution of Mangroves from Woody Plants. Mol. Biol. Evol. 34 (4), msw277–1015. doi:10.1093/molbev/msw277
Yang, Z. (1997). PAML: a Program Package for Phylogenetic Analysis by Maximum Likelihood. Bioinformatics 13 (5), 555–556. doi:10.1093/bioinformatics/13.5.555
Zelmer, D. A. (1998). An Evolutionary Definition of Parasitism. Int. J. Parasitol. 28 (3), 531–533. doi:10.1016/S0020-7519(97)00199-9
Zhang, J., and Kumar, S. (1997). Detection of Convergent and Parallel Evolution at the Amino Acid Sequence Level. Mol. Biol. Evol. 14 (5), 527–536. doi:10.1093/oxfordjournals.molbev.a025789
Zhao, R.-L., Li, G.-J., Sánchez-Ramírez, S., Stata, M., Yang, Z.-L., Wu, G., et al. (2017). A Six-Gene Phylogenetic Overview of Basidiomycota and Allied Phyla with Estimated Divergence Times of Higher Taxa and a Phyloproteomics Perspective. Fungal Divers. 84 (1), 43–74. doi:10.1007/s13225-017-0381-5
Keywords: rust fungi, anther smuts, convergent evolution, amino acid substitution, positive selection, gene gain or loss
Citation: Zhou X, Yu D and Cao Z (2022) Convergence Analysis of Rust Fungi and Anther Smuts Reveals Their Common Molecular Adaptation to a Phytoparasitic Lifestyle. Front. Genet. 13:863617. doi: 10.3389/fgene.2022.863617
Received: 27 January 2022; Accepted: 25 February 2022;
Published: 08 April 2022.
Edited by:
Xiaoming Song, North China University of Science and Technology, ChinaReviewed by:
Danyu Shen, Nanjing Agricultural University, ChinaChengming Tian, Beijing Forestry University, China
Copyright © 2022 Zhou, Yu and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Cao, zmcao@nwsuaf.edu.cn
†These authors have contributed equally to this work