
C’est l’histoire d’un nourrisson de cinq semaines admis à l’hôpital car cela fait deux heures qu’il n’arrête pas de pleurer. Il est inconsolable et irritable. L’examen clinique révèle une déviation anormale des yeux vers le bas lors des pleurs qui sont, par ailleurs, atypiques.
Le nourrisson présente une encéphalopathie infantile. On estime qu’il existe environ 1 500 maladies génétiques pouvant être responsables de cette pathologie qui se traduit très souvent par le même tableau clinique mais dont les traitements varient grandement selon la cause.
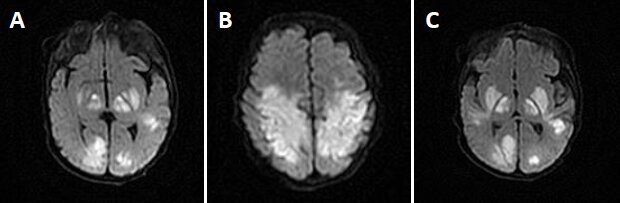
Il est 22h49 lorsque les parents du nourrisson se présentent aux urgences d’un hôpital californien. Quelques minutes plus tard, à 23h18, l’imagerie par résonance magnétique (IRM) du cerveau du nourrisson montre de multiples anomalies, à savoir la présence de plusieurs grandes zones hypodenses bilatérales. L’enfant est aussitôt transféré en soins intensifs néonataux.
Le lendemain matin, à 9h30, les médecins interrogent les parents sur l’histoire médicale de la famille. Ils apprennent alors que, dix ans auparavant, les parents du bébé, qui sont par ailleurs cousins germains, ont eu un enfant qui avait présenté les mêmes symptômes neurologiques. Ce premier enfant est décédé à l’âge de onze mois, sans qu’à l’époque un diagnostic ne soit établi, et ce malgré les nombreux examens complémentaires réalisés.
Séquençage génomique
Trois heures plus tard, à 12h30, des spécialistes en séquençage génomique, en génétique médicale et en néonatologie discutent ce cas clinique. Une heure et demie plus tard, ils obtiennent le consentement des parents pour que soit réalisé un séquençage rapide de l’ADN de leur fils à partir d’un échantillon de sang. Il est alors 14h.
A 15h52, la prise de sang a lieu. Il est 17h01 quand le tube arrive au centre de génomique. La préparation de l’échantillon sanguin en vue du séquençage débute à 17h50. Le séquençage de l’ADN débute à 19h23.
Le lendemain matin, il est 6h30 lorsque le séquençage prend fin et que l’analyse bioinformatique commence. Les premiers résultats sont téléchargés à 7h24 dans un système de diagnostic moléculaire par intelligence artificielle. Dix minutes plus tard, l’analyse est terminée.
Il s’est donc écoulé 14 heures et 33 minutes entre l’arrivée du prélèvement sanguin au centre génomique et l’obtention d’un diagnostic provisoire. Celui-ci est confirmé au vu des signes cliniques 49 minutes plus tard.
Une maladie neurologique au pronostic redoutable
Le nourrisson présente une maladie génétique autosomique récessive, les parents étant tous deux porteurs d’une anomalie génétique. Celle-ci concerne le gène SLC19A3 qui code hTHTR2, « transporteur 2 de la thiamine », protéine située sur la membrane cellulaire*.
La pathologie neurologique pédiatrique dont souffre ce nourrisson est caractérisée par un dysfonctionnement du métabolisme de la thiamine. Elle est désignée par le sigle THMD2 (pour thiamine metabolism dysfunction syndrome 2).
La thiamine (ou vitamine B1) est une vitamine d’origine uniquement alimentaire car l’organisme ne peut pas la synthétiser. Elle est surtout présente dans les céréales (riz, son), les noix, les haricots secs, les pois, le soja et la viande (porc, volaille). En cas d’apport insuffisant, la carence apparaît en quelques semaines.
Il s’avère que ce nourrisson présente un syndrome THMD2 infantile précoce, dit « de type Leigh », qui se manifeste au cours des trois premiers mois de vie par une détérioration neurologique rapide, suivie plus tard en l’absence de traitement de la mort de l’enfant. Cette pathologie neurologique est sans doute à l’origine du décès d’un enfant dans cette famille. En effet, il existe des similitudes entre les signes cliniques et radiologiques des deux enfants. En outre, les parents, rappelons-le cousins germains, sont porteurs du même défaut génétique.
A l’hôpital, il est 8h11 lorsque le nourrisson, à son 43ème jour de vie, présente plusieurs crises d’épilepsie enregistrées par vidéo-électro-encéphalographie (examen consistant à enregistrer simultanément l’électroencéphalogramme et le comportement du patient).
Environ un quart d’heure plus tard, à 8h23, les spécialistes médicaux se réunissent à nouveau, cette fois pour décider de la prise en charge du petit malade.
Un traitement administré 37 heures après l’hospitalisation du petit patient
A 9h44, il est décidé de traiter le nourrisson par de la thiamine et de la biotine** (vitamine B8). A 12h13, les premières doses lui sont administrées par voie orale.
Ce traitement, parfaitement adapté à la pathologie neurologique du jeune patient, est ainsi débuté 37,5 heures après son admission à l’hôpital. Deux heures plus tard, les médecins lui administrent du phénobarbital (barbiturique utilisé pour contrôler les convulsions). Une seule crise épileptique, de quinze secondes, est enregistrée par la suite.
En fin d’après-midi, à 18h, l’irritabilité a disparu et l’enfant ne présente plus de crises d’épilepsie. Le nourrisson est calme et alerte. L’alimentation au biberon peut reprendre.
L’enfant quitte l’hôpital cinq jours après son admission. Aujourd’hui âgé de 7 mois, il se porte bien.
Publié le 3 juin 2021 dans l’hebdomadaire médical américain The New England Journal of Medicine, l’histoire se termine donc heureusement fort bien pour ce petit patient souffrant d’une maladie génétique mortelle en l’absence de traitement.
L’analyse rapide du génome, aide au diagnostic pédiatrique
Selon Mallory J. Owen, Stephen F. Kingsmore et leurs collègues du Rady’s Children’s de San Diego, ce cas clinique illustre comment l’analyse rapide du génome, en tant qu’aide au diagnostic d’un enfant hospitalisé en réanimation néonatale, a permis de diminuer la souffrance et d’améliorer considérablement le pronostic.
Procéder à l’analyse rapide du génome entier fait partie d’une démarche de « médecine de précision », également appelée médecine personnalisée. Son objectif est de proposer au patient un traitement adapté aux caractéristiques propres de sa maladie. Il a ainsi été possible d’identifier chez ce nourrisson une maladie génétique dès le lendemain de son admission à l’hôpital, ceci via le séquençage rapide de son génome, et d’obtenir des résultats facilitant la mise en route dans les meilleurs délais d’un traitement parfaitement adapté à la cause de sa maladie.
Marc Gozlan (Suivez-moi sur Twitter, Facebook, LinkedIn)
* Duplication (c.597dup) dans le gène SLC19A3, situé sur le bras long du chromosome 2 (frameshift variant: p.His200fs).
** L’efficacité de fortes doses de biotine dans cette maladie demeure énigmatique. La biotine n’étant pas un substrat de hTHTR2 (transporteur 2 de la thiamine), le mécanisme précis par lequel la biotine rétablit le phénotype clinique est inconnu. Il est probable que de fortes doses de biotine augmentent l’expression du gène SLC19A3, rétablissant ainsi une certaine fonction de la protéine mutée en augmentant son expression.
Pour en savoir plus :
Owen MJ, Niemi AK, Dimmock DP, et al. Rapid Sequencing-Based Diagnosis of Thiamine Metabolism Dysfunction Syndrome. N Engl J Med. 2021 Jun 3;384(22):2159-2161. doi: 10.1056/NEJMc2100365
Lee JS, Yoo T, Lee M, Lee Y, et al. Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clin Genet. 2020 Apr;97(4):586-594. doi: 10.1111/cge.13713
Debs R, Depienne C, Rastetter A, et al. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch Neurol. 2010 Jan;67(1):126-30. doi: 10.1001/archneurol.2009.293
Ozand PT, Gascon GG, Al Essa M, et al. Biotin-responsive basal ganglia disease: a novel entity. Brain. 1998 Jul;121 ( Pt 7):1267-79. doi: 10.1093/brain/121.7.1267
Sur le web :
Biotin-Thiamine-Responsive Basal Ganglia Disease (Maladie des ganglions de la base sensible à la biotine). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021. 2013 Nov 21 [updated 2020 Aug 20]