
Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals

Abstract
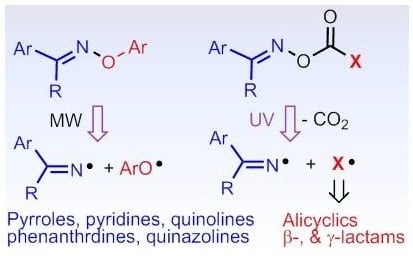
:
1. Introduction

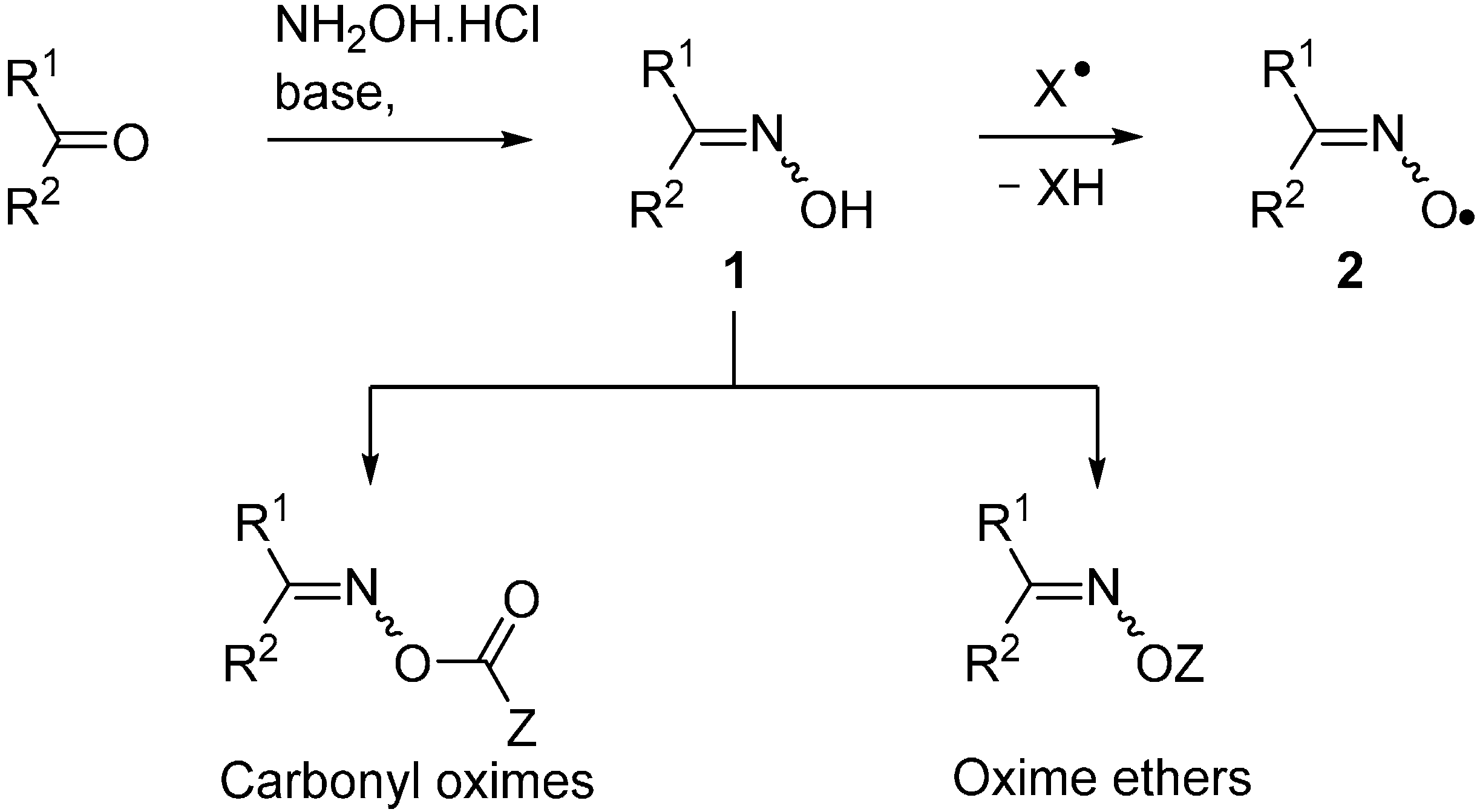
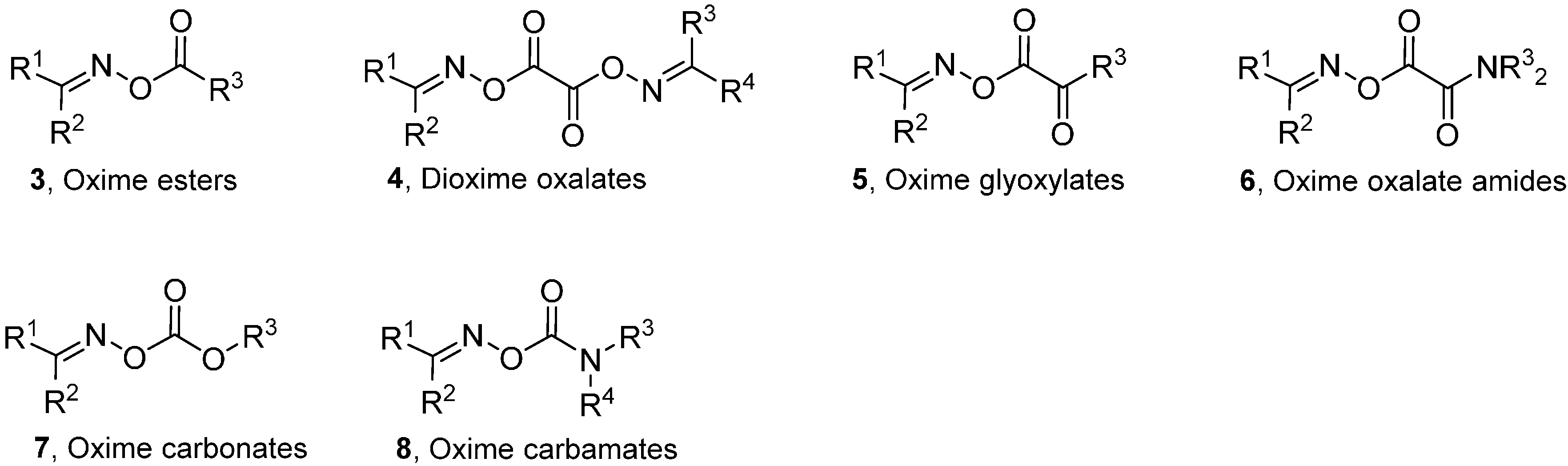
2. Oxime Esters and Related Carbonyl Oximes
2.1. General Features of Carbonyl Oxime Reactions

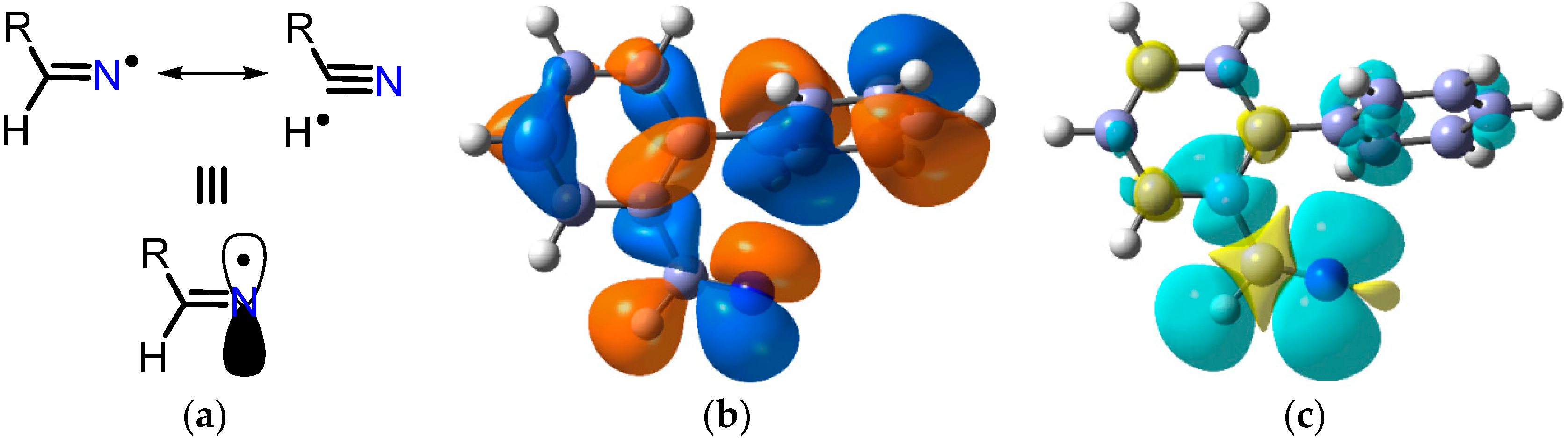
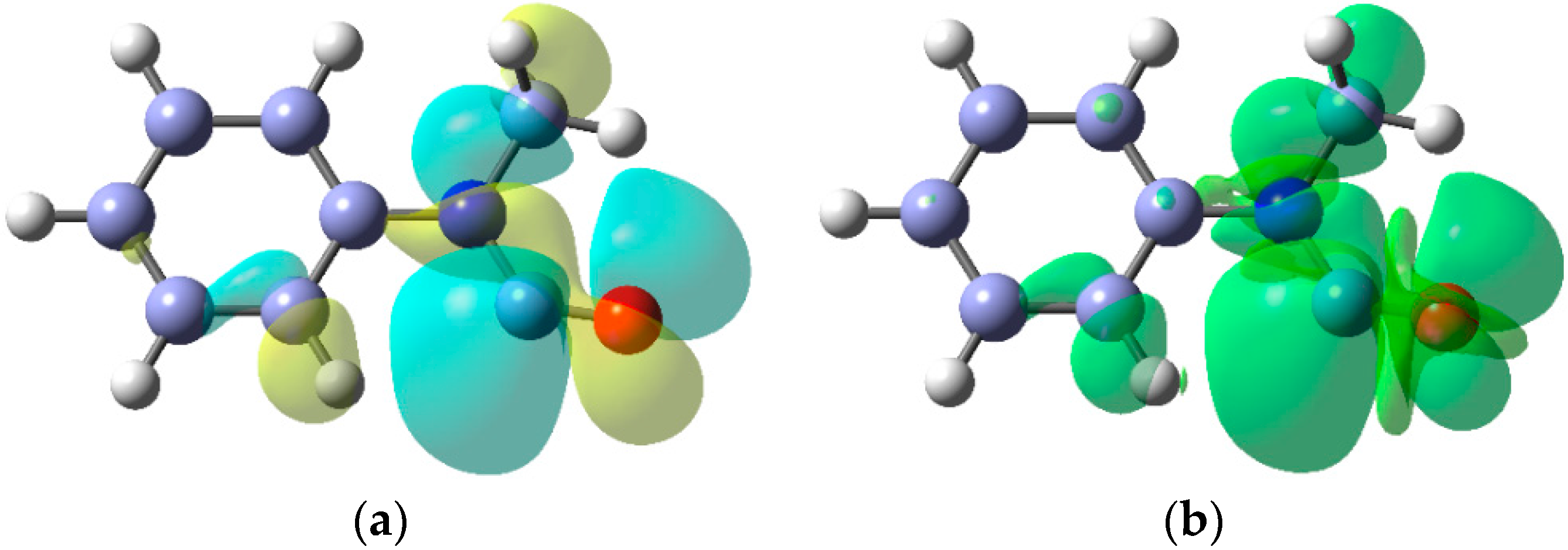
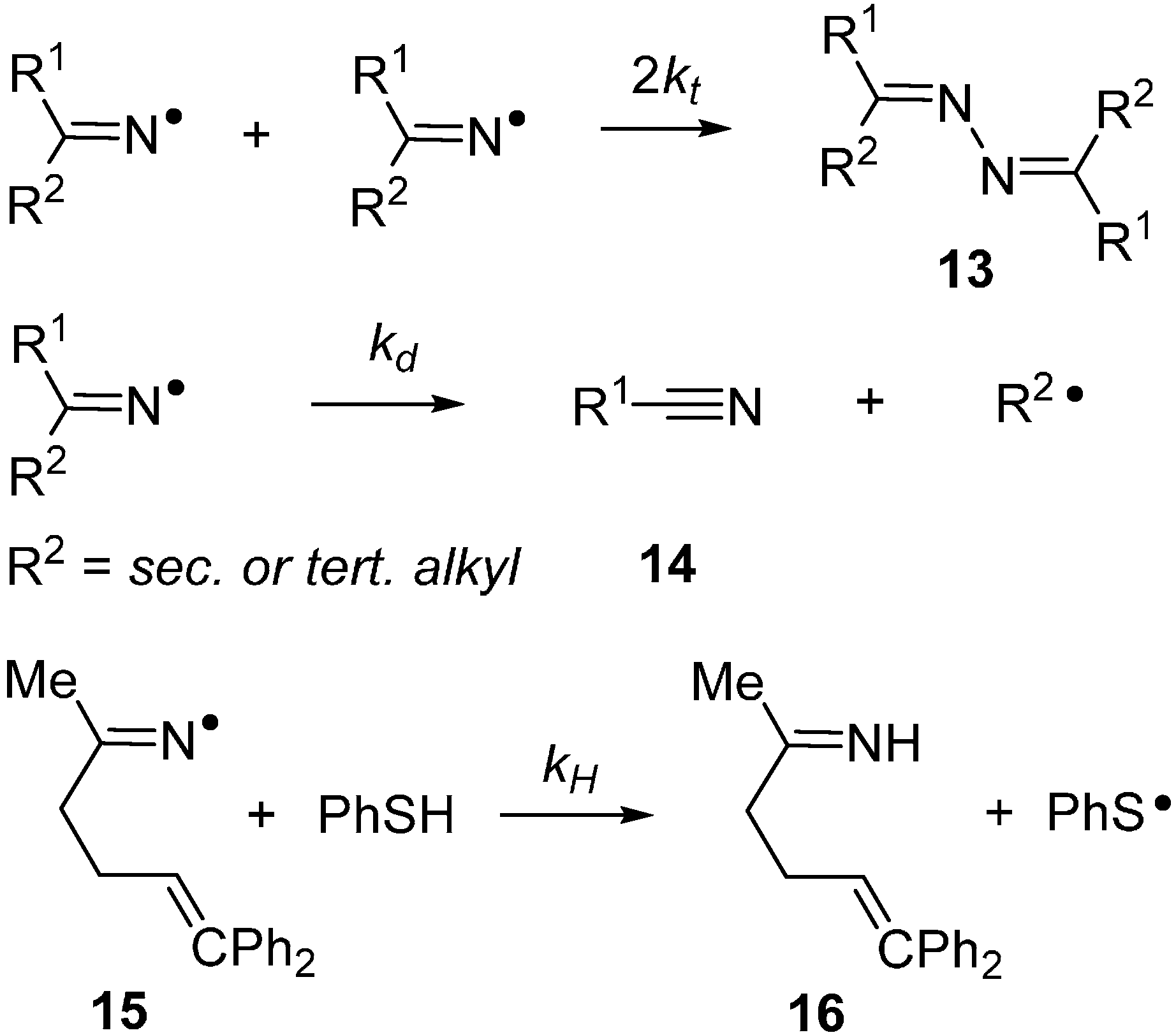
2.2. Iminyl Radical Structures and Transformations



| Radical | Solvent | T/K | g-Factor | a(14N) | a(Hβ) | a(Other) | Reference |
|---|---|---|---|---|---|---|---|
| H2C=N• | c-C3H6 | 223 | 2.0028 | 9.7 | 85.2(2H) | - | [19] |
| MeHC=N• | H2O | 300 | 2.0028 | 10.2 | 82.0 | 2.5(3H) | [16] |
| EtHC=N• | c-C3H6 | 220 | 2.0028 | 9.6 | 79.5 | 2.8(2H), 0.5(3H) | [20] |
| PhHC=N• | CCl4 | 270 | 2.0031 | 10.0 | 80.1 | 0.4(2H), 0.3(1H) | [17] |
| ArHC=N• (11) | PhBu-t | 300 | 2.0034 | 10.7 | 84.0 | - | [21] |
| Me2C=N• | c-C3H6 | 223 | 2.0029 | 9.6 | - | 1.4(6H) | [19] |
| PhMeC=N | PhBu-t | 308 | 2.0030 | 10.0 | - | 0.8(3H) | [23] |
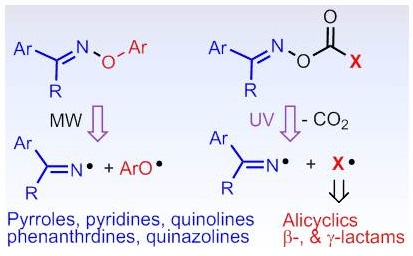
| BnMeC=N• (9) | PhBu-t | 240 | 2.0033 | 9.8 | - | 1.5(3H), 1.1(2H) | [tw] |
| Ph2C=N• | CCl4 | 308 | 2.0033 | 10.0 | - | 0.4(8H) | [24] |


 | |||
| Radical; R1, R2, R3 | mode | kc/s−1 (300 K) | Ec/kcal·mol−1 |
| 17 b | 5-exo | 10 × 103 | 9.2 |
| 18; H, H, H | 5-exo | 8.8 × 103 | 8.3 |
| 18; H, Me, H | 5-exo | 0.15 × 103 | 10.7 |
| 18; H, H, Et | 5-exo | 60 × 103 | 7.2 |
| 18; Me, H, H | 5-exo | 0.31 × 103 | 10.3 |
| 19 | 5-exo | 22 × 103 | 7.8 |
| 20 c | 6-endo | <5 × 103 | >9 |
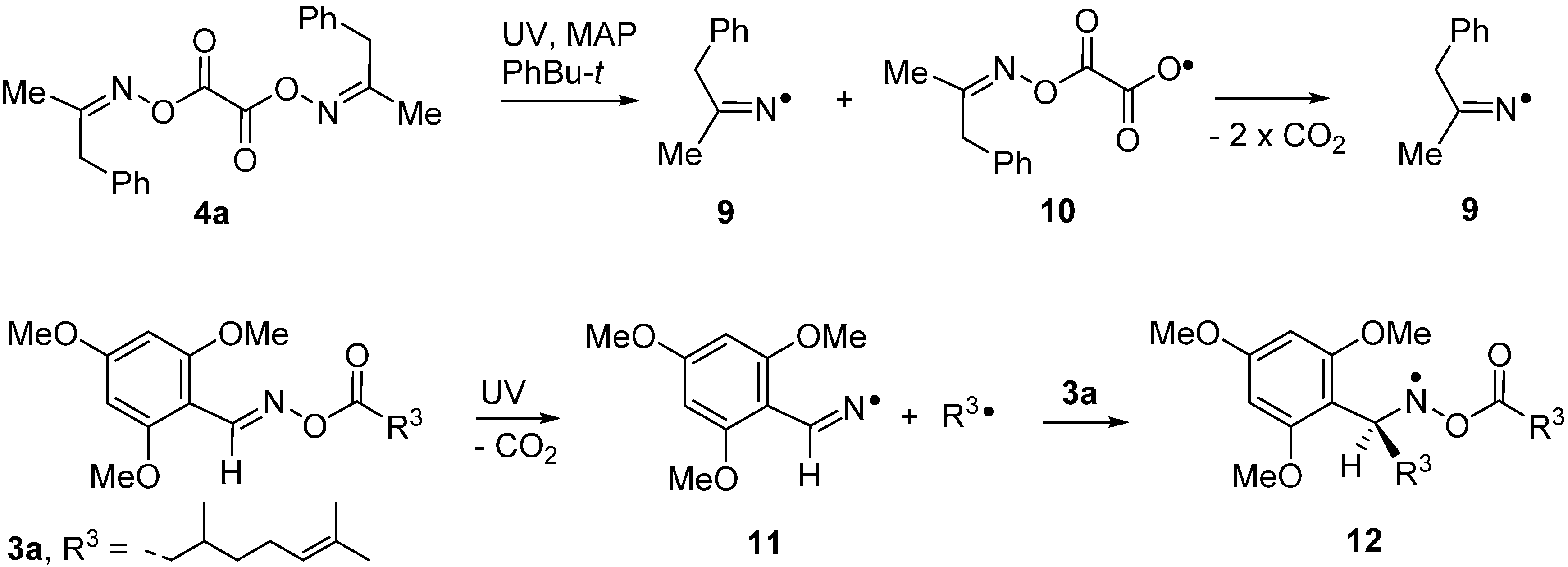
2.3. Radical Based Transformations of Oxime Esters and Dioxime Oxalates

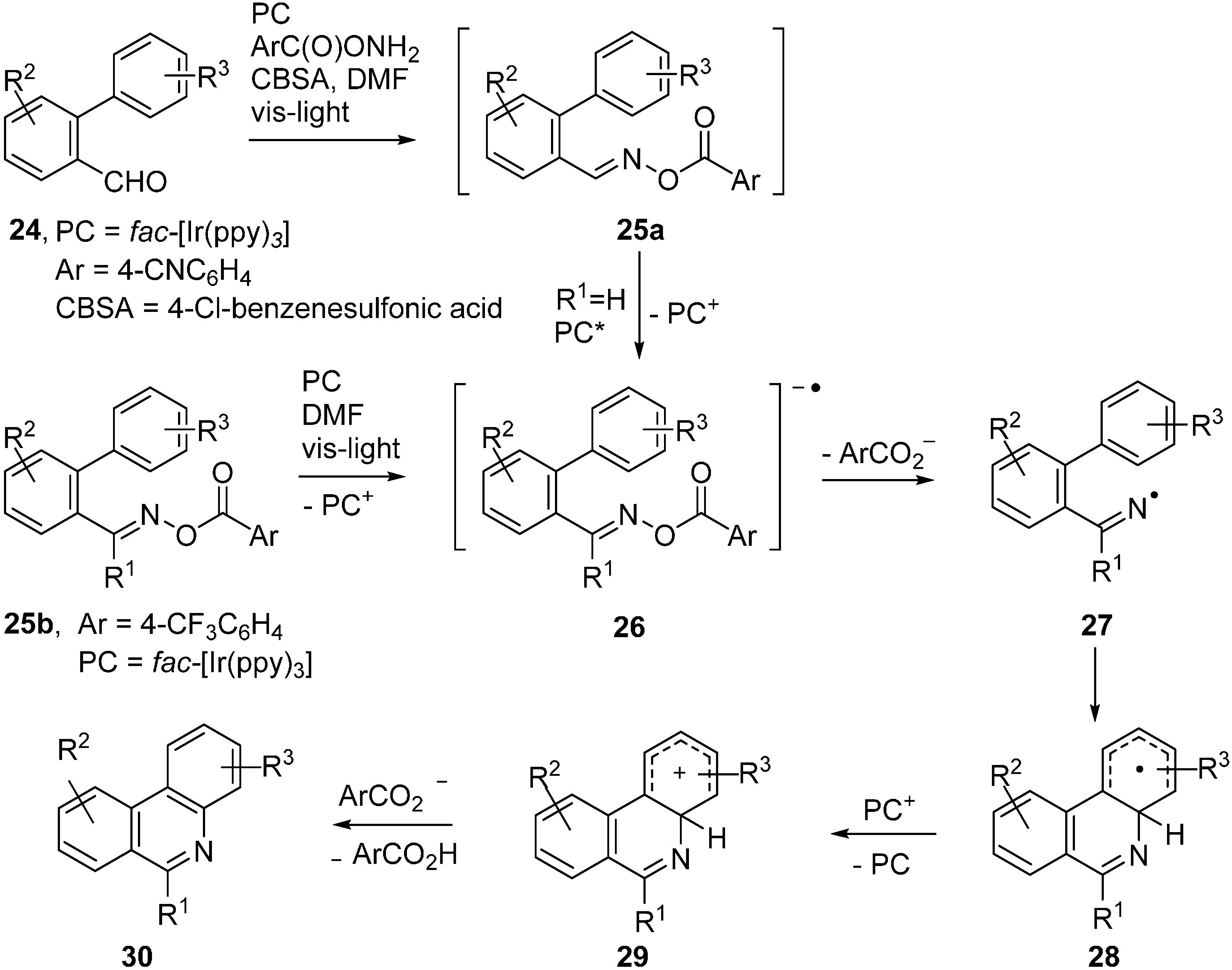
2.4. Oxime Esters and Photoredox Catalysis

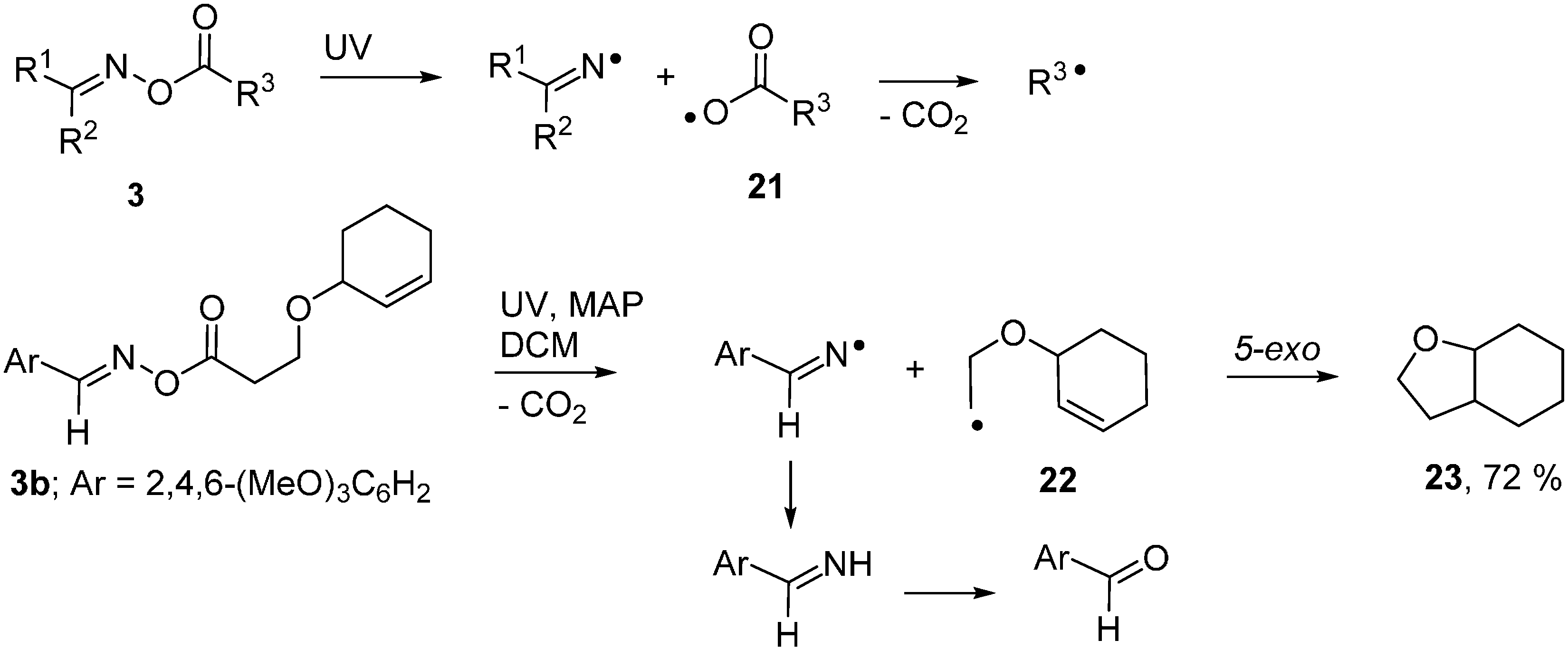
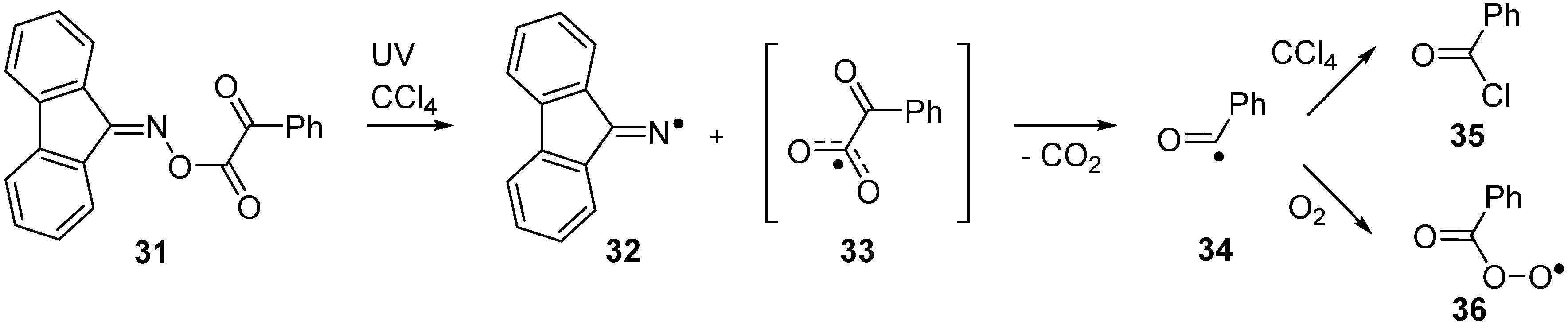
2.5. Photodissociation of Ketoxime Glyoxalates

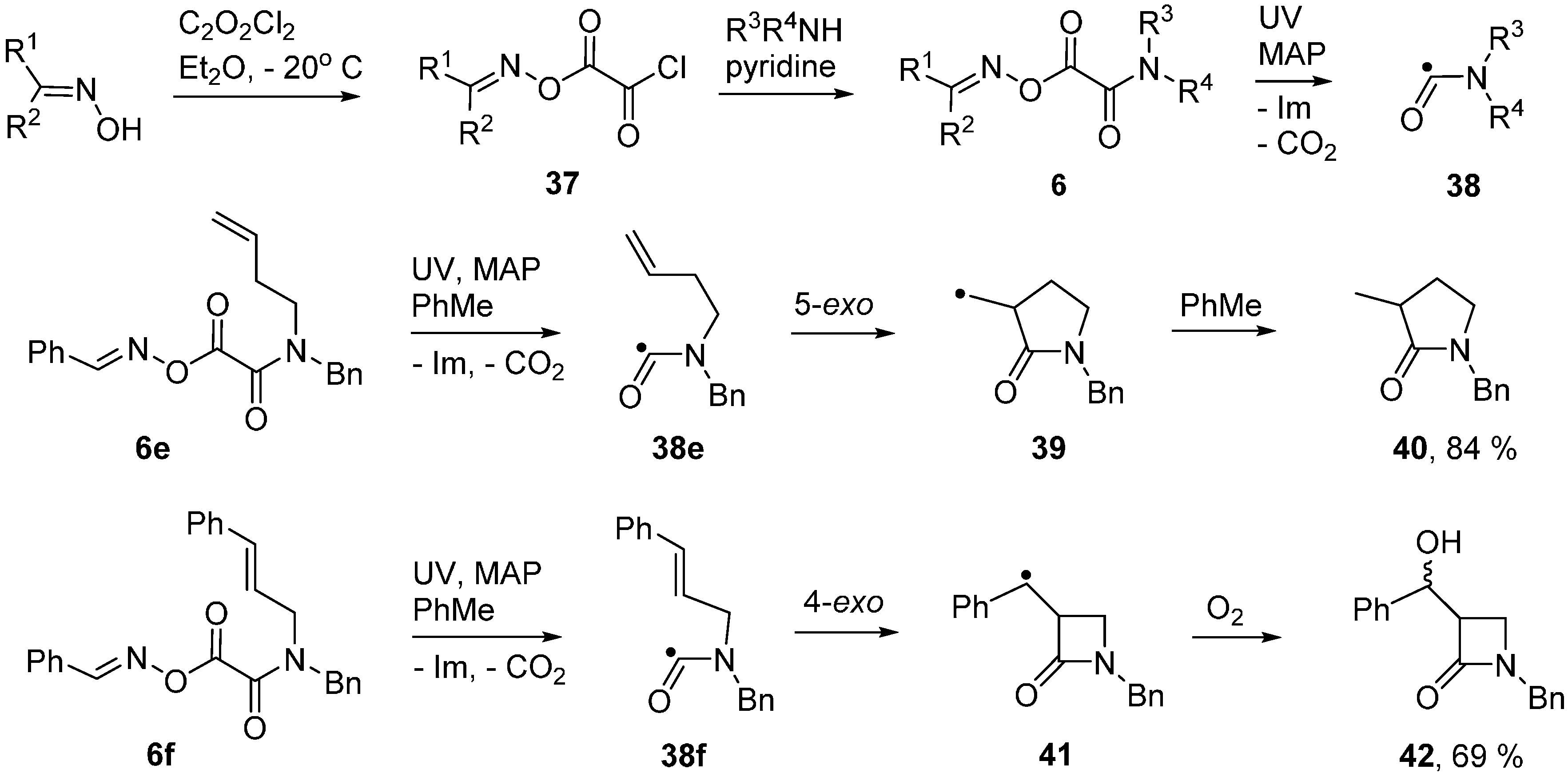
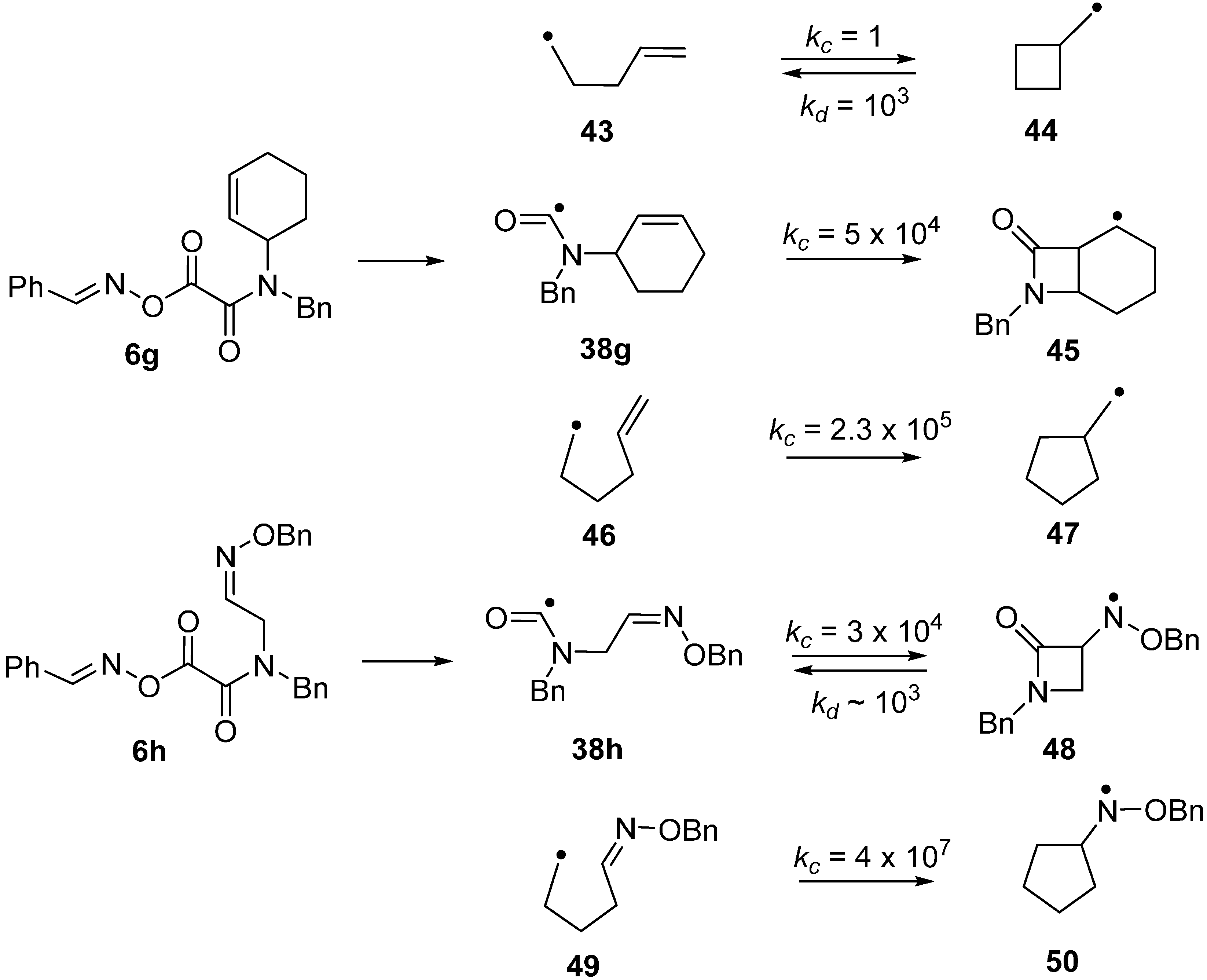
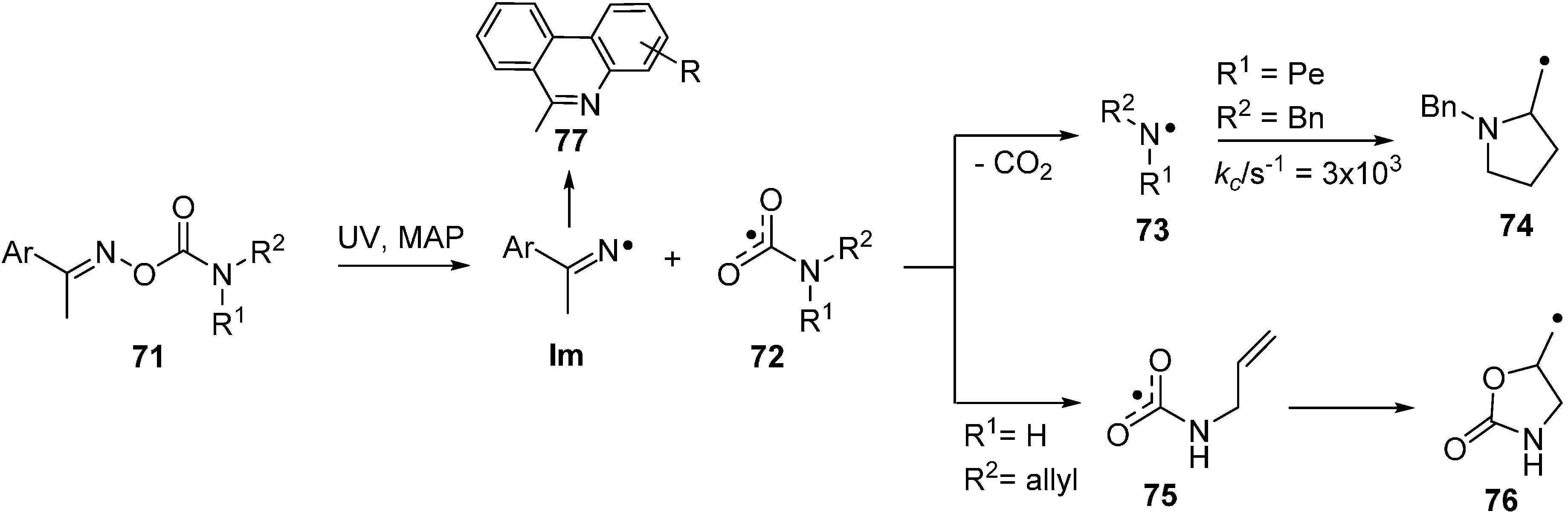
2.6. Carbamoyl Radicals from Oxime Oxalate Amides: Ring Closures to β- and γ-Lactams

 | ||||||
| Radical | Solvent | T/K | g-Factor | a(N) | a(Other) | Reference |
| Me(H)NC•(O) (38a, trans) | PhMe | 208 | 2.0015 | 24.0 | 0.9(NH), 0.9(3H) | [56] |
| Me(H)NC•(O) (38b, cis) | PhMe | 208 | 2.0015 | 21.2 | 25.1(NH) | [56] |
| but-4-enyl(Bn)NC•(O) (38e) | PhBu-t | 230 | 2.0017 | 21.7 | 0.8(1H) | [55] |
| but-2-enyl(Bn)NC•(O) | DTBP | 360 | 2.0019 | 22.1 | [57] | |
| n-Bu(Bn)NC•(O) | DTBP | 360 | 2.0019 | 21.9 | 0.9(4H) | [57] |
| 38c | PhBu-t | 220 | 2.0018 | 23.3 | [58] | |
| 38d | PhBu-t | 220 | 2.0018 | 21.0 | 1.6(1H) | [55] |


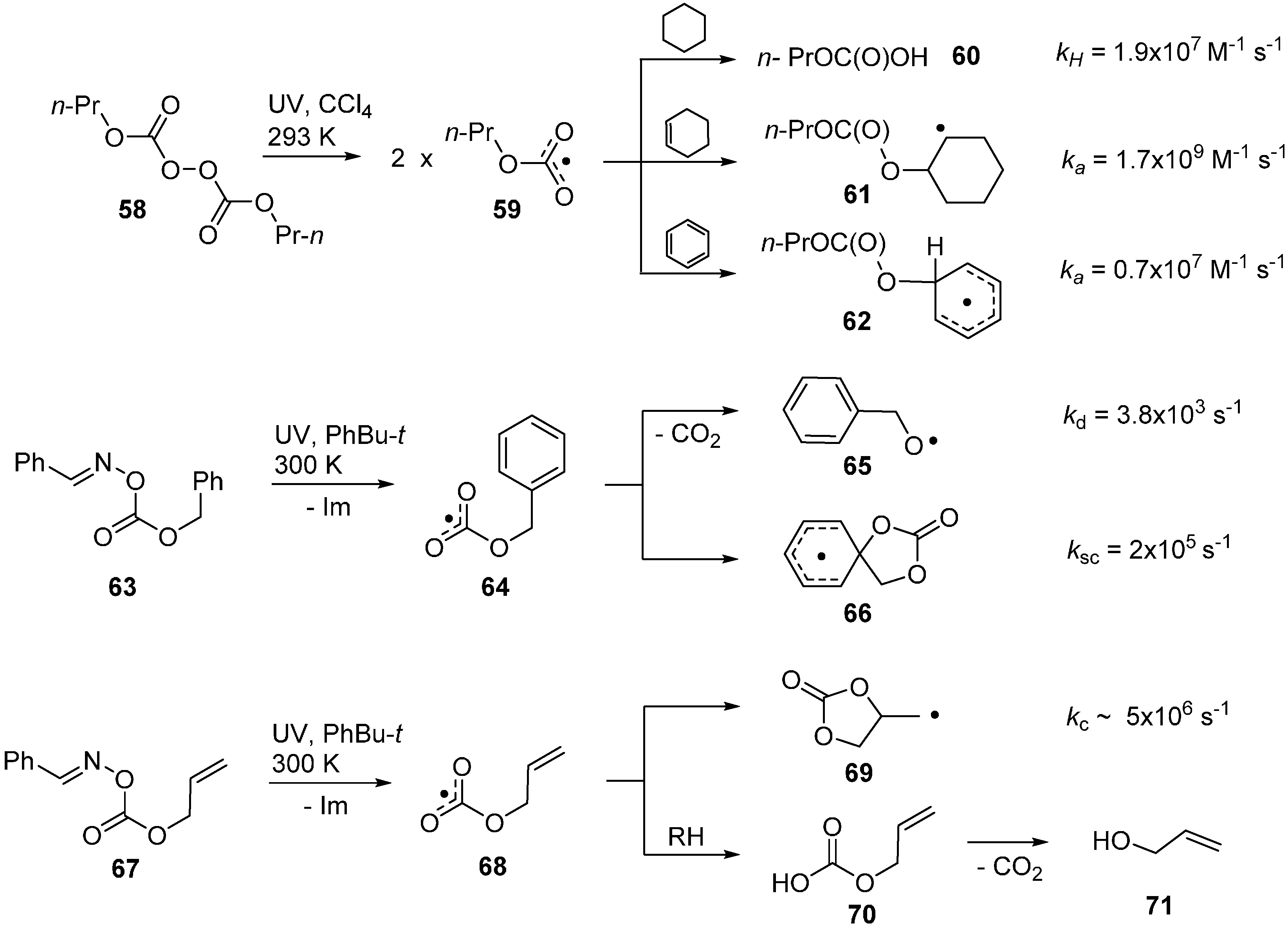
2.7. Dissociation of Oxime Carbonates and Generation of Alkoxycarbonyloxyl Radicals



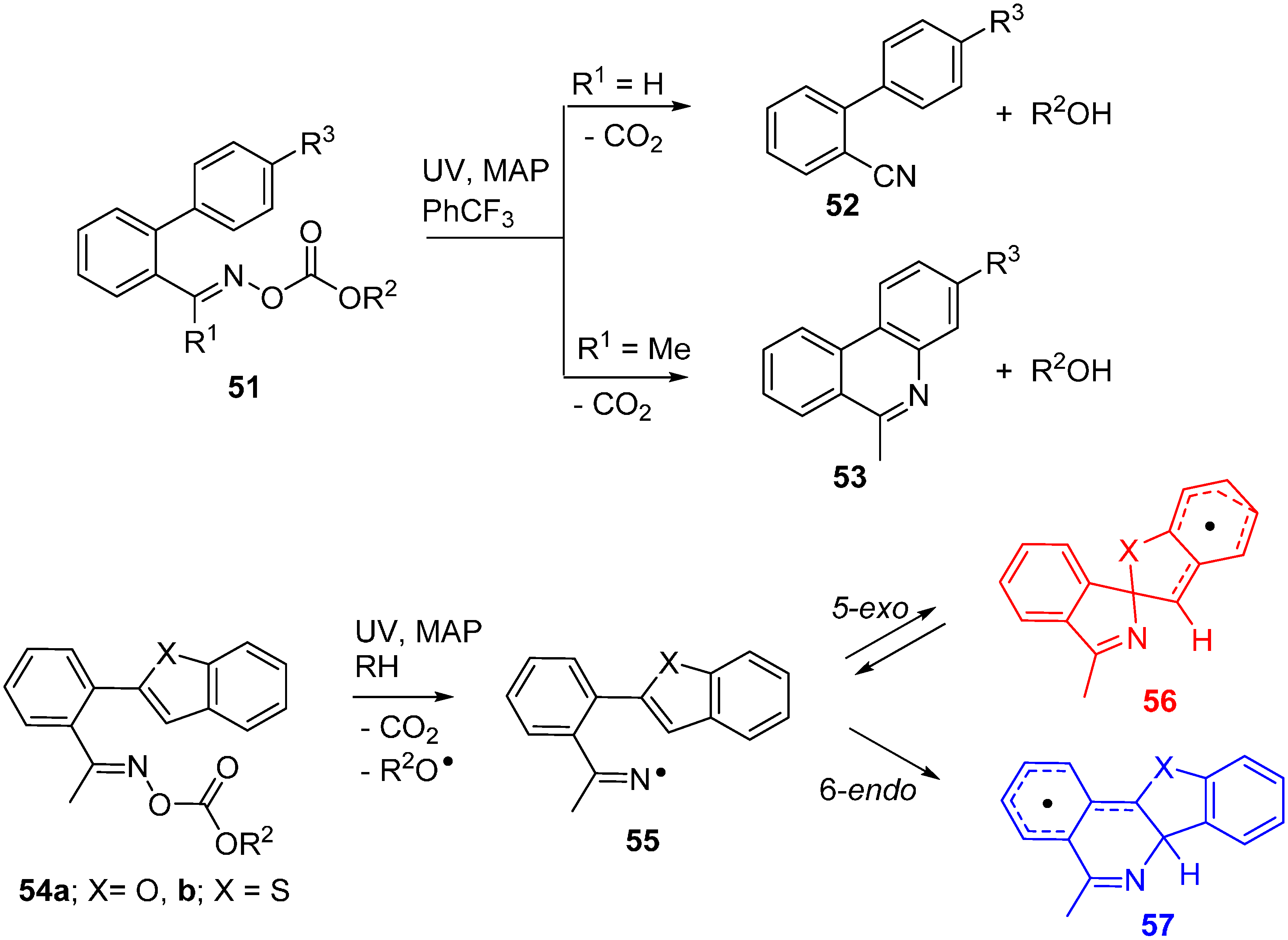
2.8. Oxime Carbamates: Precursors for Iminyl and Aminyl Radicals
| Radical | T/K | g-Factor | a(N)/G | a(Hβ)/G | a(Hβ)/G | Reference |
|---|---|---|---|---|---|---|
| Me2N• | 183 b | 2.0044 | 14.8 | 27.4 (6H) | [97,98] | |
| Et2N• | 210 | 2.0047 | 14.4 | 35.7 (2H) | 35.7(2H) | [95] |
| Bn2N• | 220 | 2.0046 | 14.3 | 37.1 (2H) | 37.1 (2H) | [95] |
| allyl2N• | 210 | 2.0048 | 14.6 | 36.0 (2H) | 36.0 (2H) | [95] |
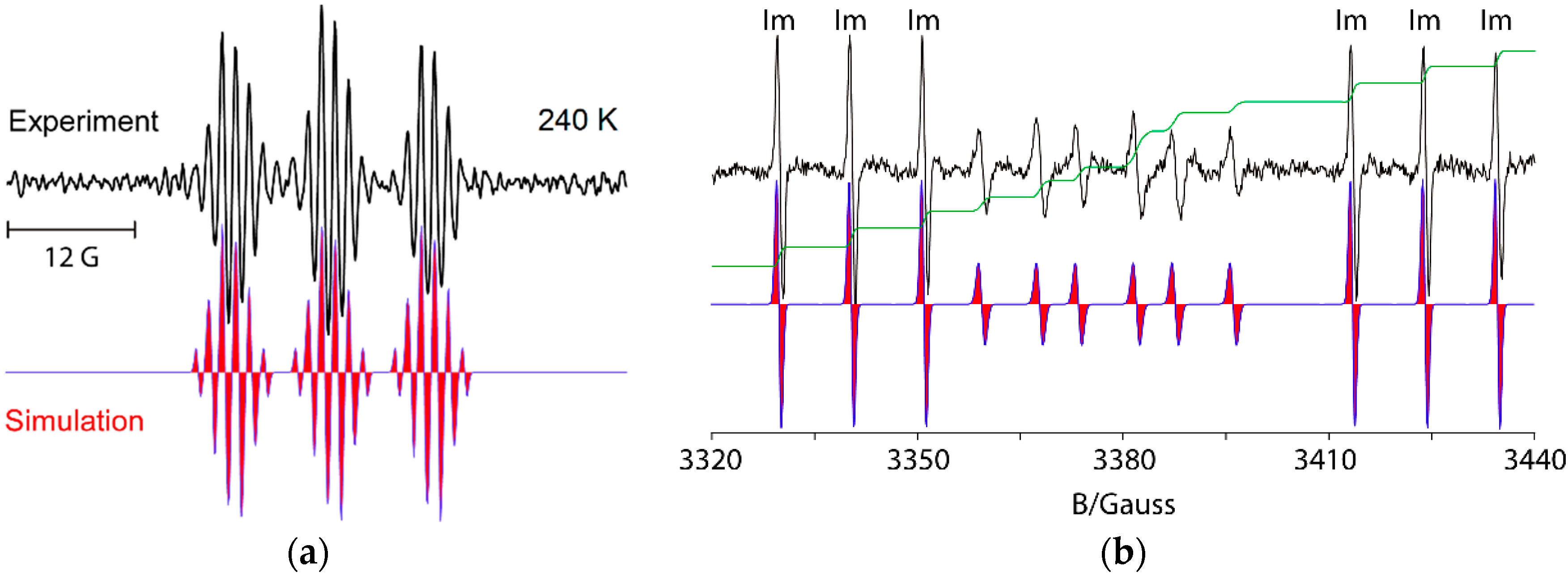
| BnN•Pe | 230 | 2.0048 | 14.2 | 36.9 (2H) | 35.4 (2H) | [95] |


3. Oxime Ethers in Radical-Mediated Reactions
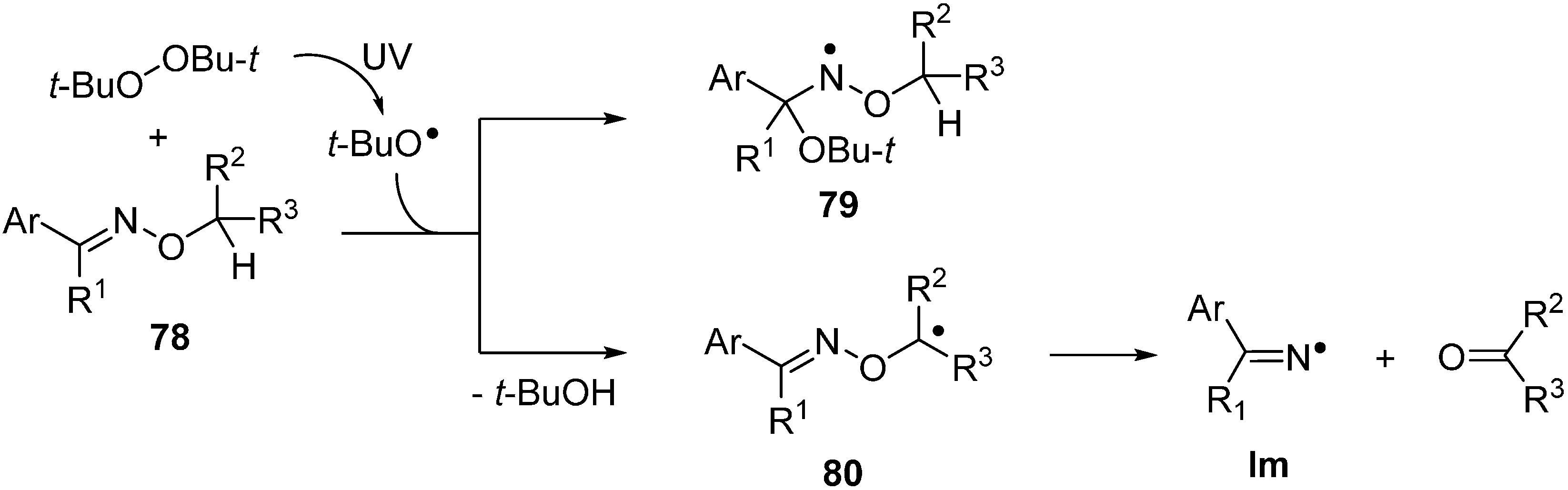
3.1. Homolytic Reactions of O-alkyl and O-aryl Oxime Ethers

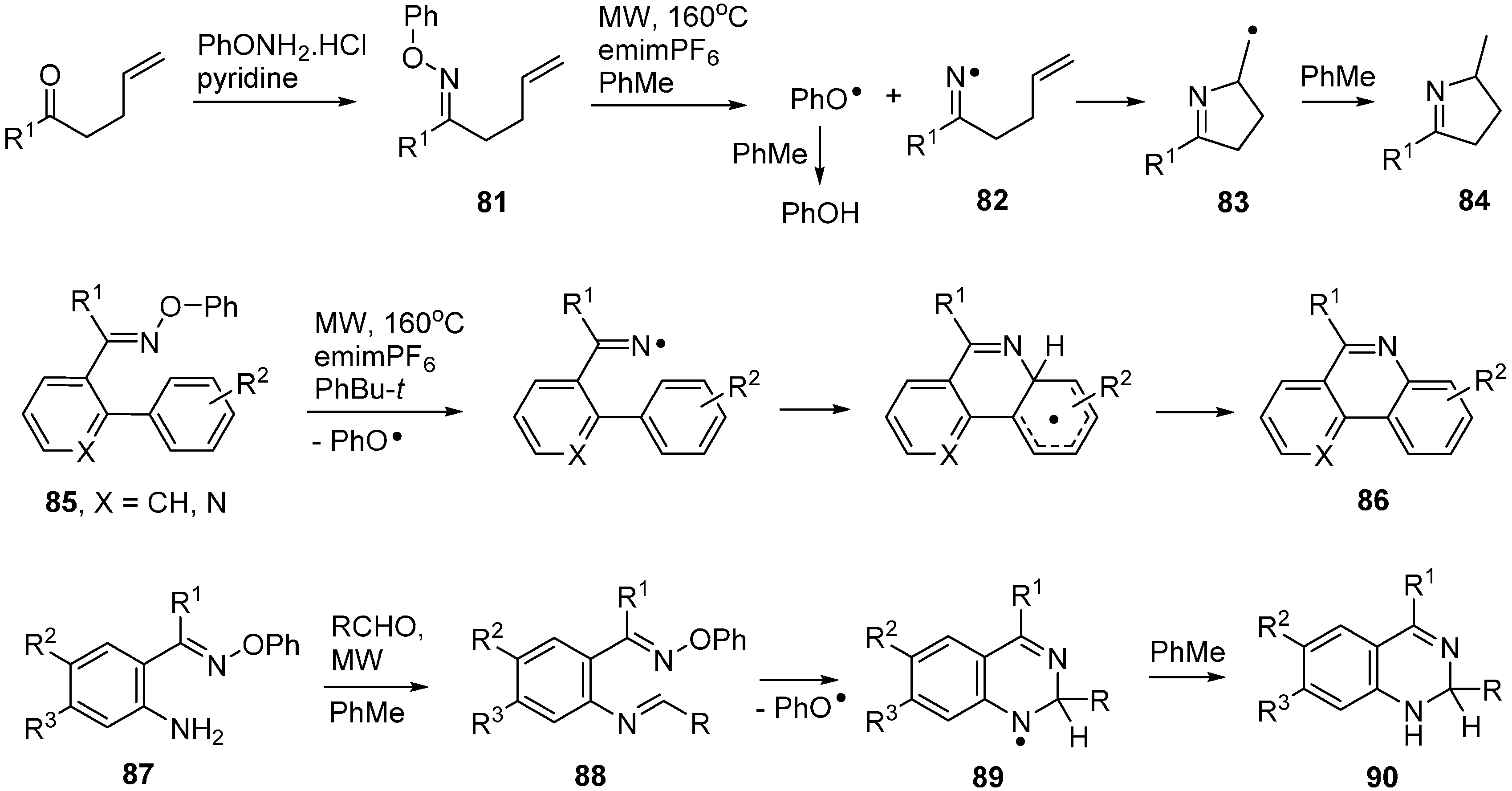
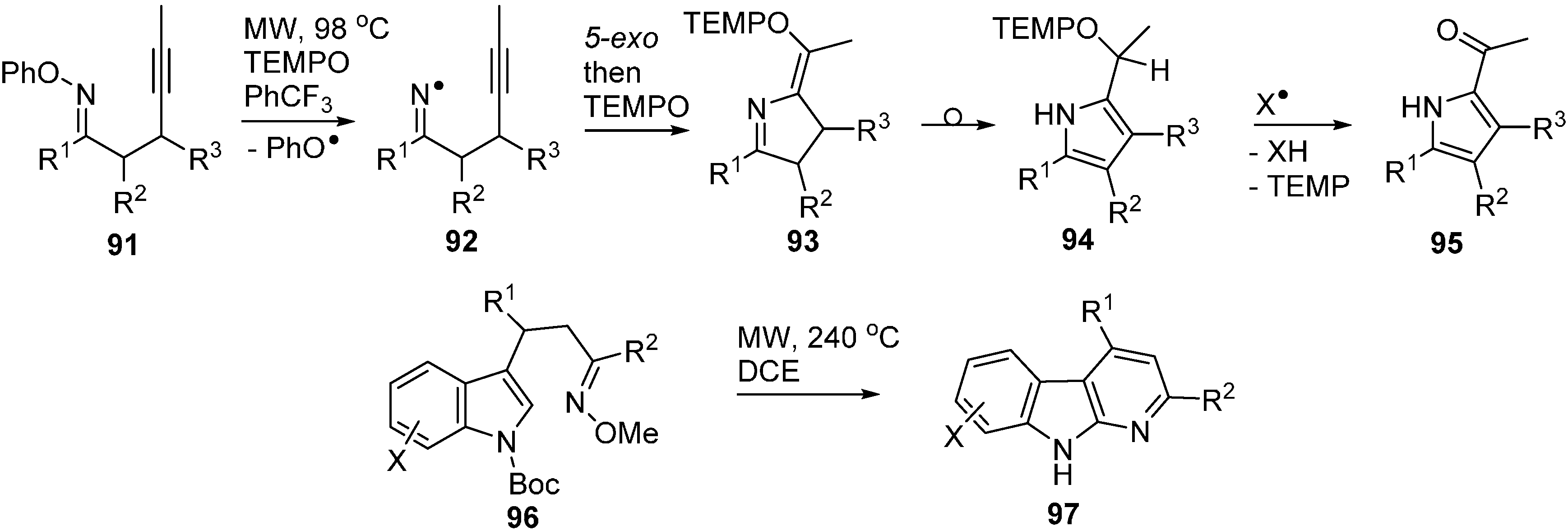
3.2. Conventional and Microwave Mediated Thermolyses of Oxime Ethers


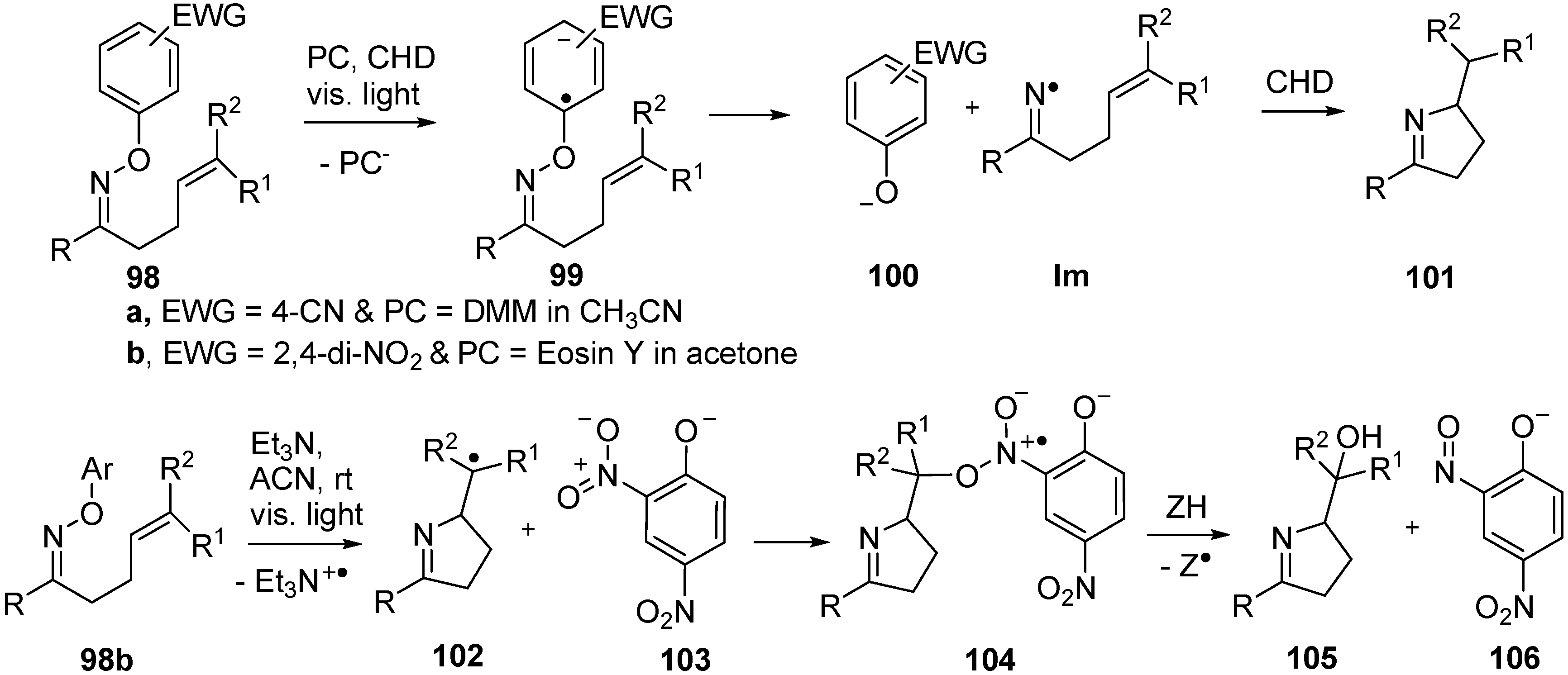
3.3. Oxime Ethers and Photoredox Catalysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
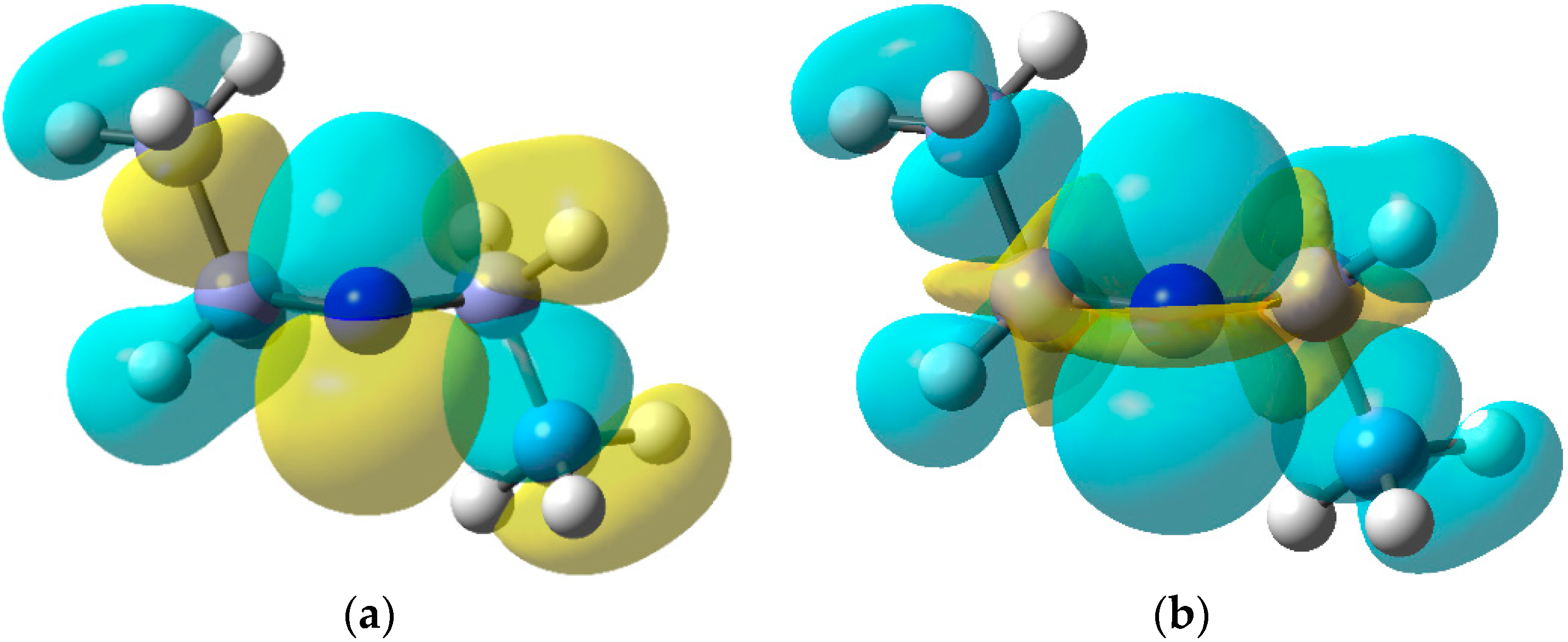{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Pratt, D.A.; Blake, J.A.; Mulder, P.; Walton, J.C.; Korth, H.-G.; Ingold, K.U. OH Bond dissociation enthalpies in oximes: Order restored. J. Am. Chem. Soc. 2004, 126, 10667–10675. [Google Scholar] [PubMed]
- Brokenshire, J.L.; Roberts, J.R.; Ingold, K.U. Kinetic applications of electron paramagnetic resonance spectroscopy. VII. Self-reactions of iminoxy radicals. J. Am. Chem. Soc. 1972, 94, 7040–7049. [Google Scholar] [CrossRef]
- Ingold, K.U. The only stable organic sigma radicals: Di-tert-alkyliminoxyls. In Stable Radicals; Hicks, R.G., Ed.; Wiley: Chichester, UK, 2010; pp. 231–244. [Google Scholar]
- Eisenhauer, B.M.; Wang, M.; Brown, R.E.; Labaziewicz, H.; Ngo, M.; Kettinger, K.W.; Mendenhall, G.D. Spectral and kinetic measurements on a series of persistent iminoxyl radicals. J. Phys. Org. Chem. 1997, 10, 737–746. [Google Scholar] [CrossRef]
- Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds; CRC Press: Boca Raton, FL, USA, 2003; pp. 225–228. [Google Scholar]
- Blake, J.A.; Pratt, D.A.; Lin, S.; Walton, J.C.; Mulder, P.; Ingold, K.U. Thermolyses of O-phenyl oxime ethers. A new source of iminyl radicals and a new source of aryloxyl radicals. J. Org. Chem. 2004, 69, 3112–3120. [Google Scholar] [CrossRef] [PubMed]
- Forrester, A.R.; Gill, M.; Sadd, J.S.; Thomson, R.H. Iminyls. Part 2. Intramolecular aromatic substitution by iminyls. A new route to phenanthridines and quinolines. J. Chem. Soc. Perkin Trans. 1979, 1, 612–615. [Google Scholar] [CrossRef]
- Forrester, A.R.; Napier, R.J.; Thomson, R.H. Iminyls. Part 7. Intramolecular hydrogen abstraction: Synthesis of heterocyclic analogs of α-tetralone. J. Chem. Soc. Perkin Trans. 1981, 1, 984–987. [Google Scholar] [CrossRef]
- Hasebe, M.; Kogawa, K.; Tsuchiya, T. Photochemical arylation by oxime esters in benzene and pyridine: Simple synthesis of biaryl compounds. Tetrahedron Lett. 1984, 25, 3887–3890. [Google Scholar] [CrossRef]
- Hasebe, M.; Tsuchiya, T. Photodecarboxylative chlorination of carboxylic acids via their benzophenone oxime esters. Tetrahedron Lett. 1988, 29, 6287–6290. [Google Scholar] [CrossRef]
- Boivin, J.; Callier-Dublanchet, A.-C.; Quiclet-Sire, B.; Schiano, A.-M.; Zard, S.Z. Iminyl, amidyl, and carbamyl radicals from O-benzoyl oximes and O-benzoyl hydroxamic acid derivatives. Tetrahedron 1995, 51, 6517–6528. [Google Scholar] [CrossRef]
- Boivin, J.; Fouquet, E.; Zard, S.Z. Iminyl radicals: Part I. Generation and intramolecular capture by an olefin. Tetrahedron 1994, 50, 1745–1756. [Google Scholar] [CrossRef]
- Boivin, J.; Fouquet, E.; Zard, S.Z. Iminyl radicals: Part II. Ring opening of cyclobutyl- and cyclopentyliminyl radicals. Tetrahedron 1994, 50, 1757–1768. [Google Scholar] [CrossRef]
- Zard, S.Z. Iminyl radicals. A fresh look at a forgotten species (and some of its relatives). Synlett 1996, 1148–1154. [Google Scholar] [CrossRef]
- Portela-Cubillo, F.; Surgenor, B.A.; Aitken, R.A.; Walton, J.C. Thermal rearrangement of indolyl oxime esters to pyridoindoles. J. Org. Chem. 2008, 73, 8124–8127. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Fessenden, R.W. Reaction of nitriles with hydrated electrons and hydrogen atoms in aqueous solution as studied by electron spin resonance. J. Phys. Chem. 1970, 74, 3362–3365. [Google Scholar] [CrossRef]
- Hudson, R.F.; Lawson, A.J.; Record, K.A.F. Conformation and stability of 1,1-diphenylmethyleneiminyl. J. Chem. Soc. Chem. Commun. 1974, 12, 488–489. [Google Scholar] [CrossRef]
- Griller, D.; Mendenhall, G.D.; van Hoof, W.; Ingold, K.U. Kinetic applications of electron paramagnetic resonance spectroscopy. XV. Iminyl radicals. J. Am. Chem. Soc. 1974, 96, 6068–6070. [Google Scholar] [CrossRef]
- Cooper, J.W.; Roberts, B.P.; Winter, J.N. Electron spin resonance study of iminyl and triazenyl radicals derived from organic azides. J. Chem. Soc. Chem. Commun. 1977, 320–321. [Google Scholar] [CrossRef]
- Roberts, B.P.; Winter, J.N. Electron spin resonance studies of radicals derived from organic azides. J. Chem. Soc. Perkin Trans. 1979, 2, 1353–1361. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Walton, J.C. Enhanced radical delivery from aldoxime esters for EPR and ring closure applications. Chem. Commun. 2000, 351–352. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Walton, J.C. Exploitation of aldoxime esters as radical precursors in preparative and EPR spectroscopic roles. J. Chem. Soc. Perkin Trans. 2000, 2, 2399–2409. [Google Scholar] [CrossRef]
- McBurney, R.T.; Harper, A.D.; Slawin, A.M.Z.; Walton, J.C. An all-purpose preparation of oxime carbonates and resultant insights into the chemistry of alkoxycarbonyloxyl radicals. Chem. Sci. 2012, 3, 3436–3444. [Google Scholar] [CrossRef]
- Brown, C.; Hudson, R.F.; Record, K.A.F. The reaction between oximes and sulfinyl chlorides: A ready, low-temperature radical rearrangement process. J. Chem. Soc. Perkin Trans. 1978, 2, 822–828. [Google Scholar] [CrossRef]
- Hudson, R.F.; Lawson, A.J.; Lucken, E.A.C. A free-radical intermediate in the thermal rearrangement of oxime thionocarbamates. J. Chem. Soc. Chem. Commun. 1971, 807–808. [Google Scholar] [CrossRef]
- Bowman, W.R.; Bridge, C.F.; Brookes, P. Radical cyclization onto nitriles. Tetrahedron Lett. 2000, 41, 8989–8994. [Google Scholar] [CrossRef]
- Le Tadic-Biadatti, M.-H.; Callier-Dublanchet, A.-C.; Horner, J.H.; Quiclet-Sire, B.; Zard, S.Z.; Newcomb, M. Absolute rate constants for iminyl radical reactions. J. Org. Chem. 1997, 62, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Mikami, T.; Narasaka, K. Photochemical transformation of γ,δ-unsaturated ketone O-(p-cyanophenyl)oximes to 3,4-dihydro-2H-pyrrole derivatives. Chem. Lett. 2000, 29, 338–339. [Google Scholar] [CrossRef]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. Microwave-assisted preparations of dihydropyrroles from alkenone O-phenyl oximes. Chem. Commun. 2007, 4041–4043. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. Microwave-assisted syntheses of N-heterocycles using alkenone-, alkynone- and aryl-carbonyl O-phenyl oximes: Formal synthesis of neocryptolepine. J. Org. Chem. 2008, 73, 5558–5565. [Google Scholar] [CrossRef] [PubMed]
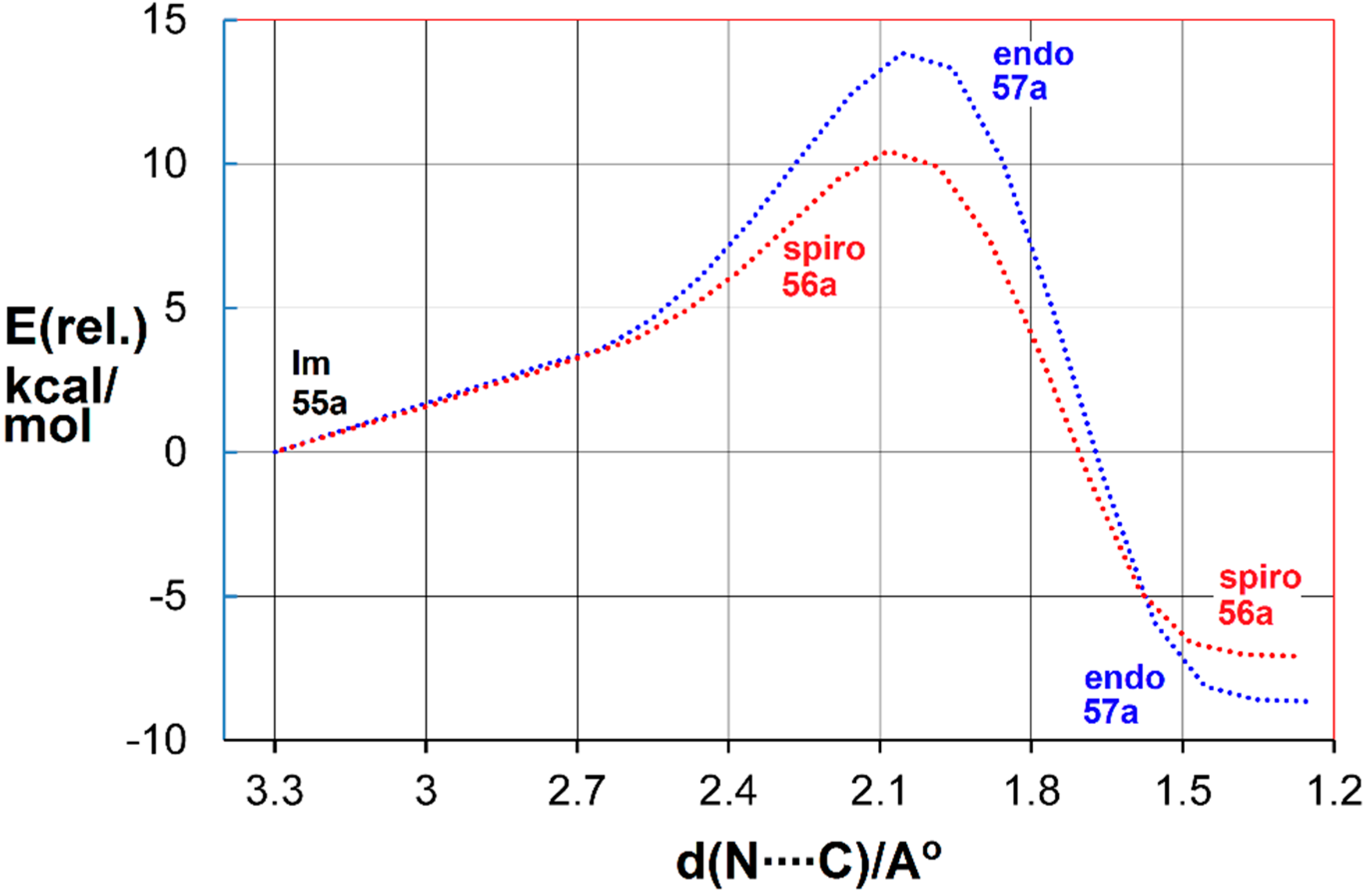
- Portela-Cubillo, F.; Alonso-Ruiz, R.; Sampedro, D.; Walton, J.C. 5-Exo-cyclizations of pentenyliminyl radicals: Inversion of the gem-dimethyl effect. J. Phys. Chem. A 2009, 113, 10005–10012. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, M. Synthetic Strategies & Applications. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; Wiley: New York, NY, USA, 2012; Volume 2. [Google Scholar]
- McBurney, R.T.; Walton, J.C. Interplay of ortho- with spiro-cyclisation during iminyl radical closures onto arenes and heteroarenes. Beilstein J. Org. Chem. 2013, 9, 1083–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, J.C. The importance of chain conformational mobility during 5-exo-cyclizations of C-, N- and O-centred radicals. Org. Biomol. Chem. 2014, 12, 7983–7992. [Google Scholar] [CrossRef] [PubMed]
- Skakovskii, E.D.; Stankevich, A.I.; Lamotkin, S.A.; Tychinskaya, L.Y.; Rykov, S.V. Thermolysis of methanolic solutions of acetyl propionyl peroxide. Russ. J. Gen. Chem. 2001, 71, 614–622. [Google Scholar] [CrossRef]
- Miyake, Y.; Takahashi, H.; Akai, N.; Shibuya, K.; Kawai, A. Structure and reactivity of radicals produced by photocleavage of oxime ester compounds studied by time-resolved electron paramagnetic resonance spectroscopy. Chem. Lett. 2014, 43, 1275–1277. [Google Scholar] [CrossRef]
- Alonso, R.; Campos, P.J.; Garcıa, B.; Rodrıguez, M.A. New light-induced iminyl radical cyclization reactions of acyloximes to isoquinolines. Org. Lett. 2006, 8, 3521–3523. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Campos, P.J.; Rodrıguez, M.A.; Sampedro, D. Photocyclization of iminyl radicals: Theoretical study and photochemical aspects. J. Org. Chem. 2008, 73, 2234–2239. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Caballero, A.; Campos, P.J.; Rodriguez, M.A. Photochemistry of acyloximes: Synthesis of heterocycles and natural products. Tetrahedron 2010, 66, 8828–8831. [Google Scholar] [CrossRef]
- Portela-Cubillo, F.; Scanlan, E.M.; Scott, J.S.; Walton, J.C. From dioxime oxalates to dihydropyrroles and phenanthridines via iminyl radicals. Chem. Commun. 2008, 4189–4191. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Lymer, J.; Scanlan, E.M.; Scott, J.S.; Walton, J.C. Dioxime oxalates; new iminyl radical precursors for syntheses of N-heterocycles. Tetrahedron 2008, 64, 11908–11916. [Google Scholar] [CrossRef]
- Hoffman, N. Photocatalysis with TiO2 applied to organic synthesis. Aust. J. Chem. 2015, 68, 1621–1639. [Google Scholar] [CrossRef]
- Manley, D.W.; Walton, J.C. Preparative semiconductor photoredox catalysis: An emerging theme in organic synthesis. Beilstein J. Org. Chem. 2015, 11, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Howe, R.F.; Rhydderch, S.; Walton, J.C. Unconventional titania photocatalysis: Direct deployment of carboxylic acids in alkylations and annulations. J. Am. Chem. Soc. 2012, 134, 13580–13583. [Google Scholar] [CrossRef] [PubMed]
- Manley, D.W.; McBurney, R.T.; Miller, P.; Walton, J.C.; Mills, A.; O’Rourke, C. Titania-promoted carboxylic acid alkylations of alkenes and cascade addition-cyclizations. J. Org. Chem. 2014, 79, 1386–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, T.P.; Ischay, M.A.; Du, J. Visible light photocatalysis as a greener approach to photochemical synthesis. Nat. Chem. 2010, 2, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Narayanam, J.M.R.; Stephenson, C.R.J. Visible light photoredox catalysis: Applications in organic synthesis. Chem. Soc. Rev. 2011, 40, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Xiao, W.-J. Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2012, 51, 6828–6838. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-Q.; Chen, J.-R.; Xiao, W.-J. Homogeneous visible-light photoredox catalysis. Angew. Chem. Int. Ed. 2013, 52, 11701–11703. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- An, X.-D.; Yu, S. Visible-light-promoted and one-pot synthesis of phenanthridines and quinolines from aldehydes and O-acyl hydroxylamine. Org. Lett. 2015, 17, 2692–2695. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; An, X.; Tong, K.; Zheng, T.; Zhang, Y.; Yu, S. Visible-light-promoted iminyl-radical formation from acyl oximes: A unified approach to pyridines, quinolines, and phenanthridines. Angew. Chem. Int. Ed. 2015, 54, 4055–4059. [Google Scholar]
- Kolano, C.; Bucher, G.; Wenk, H.H.; Jaeger, M.; Schade, O.; Sander, W. Photochemistry of 9-fluorenone oxime phenylglyoxylate: A combined TRIR, TREPR and ab initio study. J. Phys. Org. Chem. 2004, 17, 207–214. [Google Scholar] [CrossRef]
- Scanlan, E.M.; Walton, J.C. Preparation of oxime oxalate amides and their use in free-radical mediated syntheses of lactams. Chem. Commun. 2002, 2086–2087. [Google Scholar] [CrossRef]
- Scanlan, E.M.; Slawin, A.M.Z.; Walton, J.C. Preparation of β- and γ-lactams from carbamoyl radicals derived from oxime oxalate amides. Org. Biomol. Chem. 2004, 2, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, R.; Ingold, K.U. Cis-Alkylcarbamoyl radicals. The overlooked conformer. J. Am. Chem. Soc. 1981, 103, 7687–7689. [Google Scholar] [CrossRef]
- Bella, A.F.; Jackson, L.V.; Walton, J.C. A kinetic EPR study of the dissociation of 1-carbamoyl-1-methylcyclohexa-2,5-dienyl radicals: Release of aminoacyl radicals and their cyclisation. J. Chem. Soc. Perkin Trans. 2002, 2, 1839–1843. [Google Scholar]
- DiLabio, G.A.; Scanlan, E.M.; Walton, J.C. Kinetic and theoretical study of 4-exo ring closures of carbamoyl radicals onto C=C and C=N bonds. Org. Lett. 2005, 7, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Other acyl radicals [RC•(O)], diazenyls [RN=N•] and a few P-centred radicals share this characteristic
- Park, S.-U.; Varick, T.R.; Newcomb, M. Acceleration of the 4-exo radical cyclization to a synthetically useful rate. Cyclization of the 2,2-dimethyl-5-cyano-4-pentenyl radical. Tetrahedron Lett. 1990, 31, 2975–2978. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Moad, G. The kinetics and mechanism of ring opening of radicals containing the cyclobutylcarbinyl system. J. Chem. Soc. Perkin Trans. 1980, 2, 1083–1092. [Google Scholar] [CrossRef]
- Ingold, K.U.; Maillard, B.; Walton, J.C. The ring-opening reactions of cyclobutylmethyl and cyclobutenylmethyl radicals. J. Chem. Soc. Perkin Trans. 1981, 2, 970–974. [Google Scholar] [CrossRef]
- Gill, G.B.; Pattenden, G.; Reynolds, S.J. Cobalt-mediated reactions: Inter- and intramolecular additions of carbamoyl radical to alkenes in the synthesis of amides and lactams. J. Chem. Soc. Perkin Trans. 1994, 1, 369–378. [Google Scholar] [CrossRef]
- Bella, A.F.; Jackson, L.V.; Walton, J.C. Preparation of β-and γ-lactams via ring closures of unsaturated carbamoyl radicals derived from 1-carbamoyl-1-methylcyclohexa-2,5-dienes. Org. Biomol. Chem. 2004, 2, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Ryu, I.; Miyazato, H.; Kuriyama, H.; Tanaka, M.; Komatsu, M.; Sonoda, N. Broad-spectrum radical cyclizations boosted by polarity matching. carbonylative access to α-stannylmethylene lactams from azaenynes and CO. J. Am. Chem. Soc. 2003, 125, 5632–5633. [Google Scholar] [CrossRef] [PubMed]
- Fremont, S.L.; Belletire, J.L.; Ho, D.M. Free radical cyclizations leading to four-membered rings. I. Beta-lactam production using tributyltin hydride. Tetrahedron Lett. 1991, 32, 2335–2338. [Google Scholar] [CrossRef]
- Ishibashi, H.; Kameoka, C.; Kodama, K.; Ikeda, M. Asymmetric radical cyclization leading to β-lactams: stereoselective synthesis of chiral key intermediates for carbapenem antibiotics PS-5 and thienamycin. Tetrahedron 1996, 52, 489–502. [Google Scholar] [CrossRef]
- Cassayre, J.; Quiclet-Sire, B.; Saunier, J.-B.; Zard, S.Z. β- and γ-lactams by nickel powder mediated 4-exo or 5-endo radical cyclizations. A concise construction of the mesembrine skeleton. Tetrahedron 1998, 54, 1029–1040. [Google Scholar] [CrossRef]
- D’Annibale, A.; Pesce, A.; Resta, S.; Trogolo, C. Manganese(III)-promoted free radical cyclizations of enamides leading to β-lactams. Tetrahedron 1997, 53, 13129–13138. [Google Scholar] [CrossRef]
- Bryans, J.S.; Chessum, N.E.A.; Parsons, A.F.; Ghelfi, F. The synthesis of functionalized β- and γ-lactams by cyclization of enamides using copper(I) or ruthenium(II). Tetrahedron Lett. 2001, 42, 2901–2905. [Google Scholar] [CrossRef]
- Clark, A.J.; Peacock, J.L. An amidyl radical cyclization approach towards the synthesis of β-lactams. Tetrahedron Lett. 1998, 39, 1265–1268. [Google Scholar] [CrossRef]
- Walton, J.C. Unusual radical cyclisations. Top. Curr. Chem. 2006, 264, 163–200. [Google Scholar]
- Scanlan, E.M.; Walton, J.C. Radical 4-exo cyclizations onto O-alkyloxime acceptors: Towards the synthesis of penicillin-containing antibiotics. Helv. Chim. Acta 2006, 89, 2133–2143. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Easton, C.J.; Lawrence, T.; Serelis, A.K. Reactions of methyl-substituted 5-hexenyl and 4-pentenyl radicals. Aust. J. Chem. 1983, 36, 545–556. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Schiesser, C.H. A force-field study of alkenyl radical ring closure. Tetrahedron Lett. 1985, 26, 373–376. [Google Scholar] [CrossRef]
- Kim, S.; Joe, G.H.; Do, J.Y. Highly efficient intramolecular addition of aminyl radicals to carbonyl groups: A new ring expansion reaction leading to lactams. J. Am. Chem. Soc. 1993, 115, 3328–3329. [Google Scholar] [CrossRef]
- Walton, J.C. The oxime portmanteau motif: Released heteroradicals undergo incisive EPR interrogation and deliver diverse heterocycles. Acc. Chem. Res. 2014, 47, 1406–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBurney, R.T.; Slawin, A.M.Z.; Smart, L.A.; Yu, Y.; Walton, J.C. UV promoted phenanthridine syntheses from oxime carbonate derived iminyl radicals. Chem. Commun. 2011, 47, 7974–7976. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Edge, D.J.; Kochi, J.K. Electron spin resonance studies of carboxy radicals. Adducts to alkenes J. Am. Chem. Soc. 1973, 95, 2635–2643. [Google Scholar] [CrossRef]
- Chateauneuf, J.; Lusztyk, J.; Maillard, B.; Ingold, K.U. First spectroscopic and absolute kinetic studies on (alkoxycarbonyl)oxyl radicals and an unsuccessful attempt to observe carbamoyloxyl radicals. J. Am. Chem. Soc. 1988, 110, 6727–6731. [Google Scholar] [CrossRef]
- Bühl, M.; DaBell, P.; Manley, D.W.; McCaughan, R.P.; Walton, J.C. Bicarbonate and alkyl carbonate radicals: Their structural integrity and reactions with lipid components. J. Am. Chem. Soc. 2016. [Google Scholar] [CrossRef] [PubMed]
- McBurney, R.T.; Eisenschmidt, A.; Slawin, A.M.Z.; Walton, J.C. Rapid and selective spiro-cyclisations of O-centred radicals onto aromatic acceptors. Chem. Sci. 2013, 4, 2028–2035. [Google Scholar] [CrossRef]
- Kochi, J.K.; Gilliom, R.D. Decompositions of peroxides by metal salts. VII. Competition between intramolecular rearrangement of free radicals and oxidation by metal salts. J. Am. Chem. Soc. 1964, 86, 5251–5256. [Google Scholar] [CrossRef]
- Julia, M. Free radical cyclizations. XVII. Mechanistic studies. Pure Appl. Chem. 1974, 40, 553–567. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Ingold, K.U. Free-radical Rearrangements. In Rearrangements in Ground and Excited States; De Mayo, P., Ed.; Academic Press: New York, NY, USA, 1980; pp. 161–310. [Google Scholar]
- Hartung, J.; Gallou, F. Ring closure reactions of substituted 4-pentenyl-1-oxy radicals. The stereoselective synthesis of functionalized disubstituted tetrahydrofurans. J. Org. Chem. 1995, 60, 6706–6716. [Google Scholar] [CrossRef]
- Hartung, J.; Daniel, K.; Rummey, C.; Bringmann, G. On the stereoselectivity of 4-penten-1-oxyl radical 5-exo-trig cyclizations. Org. Biomol. Chem. 2006, 4, 4089–4100. [Google Scholar] [CrossRef] [PubMed]
- Pocker, Y.; Davison, B.L.; Deits, T.L. Decarboxylation of monosubstituted derivatives of carbonic acid. Comparative studies of water- and acid-catalyzed decarboxylation of sodium alkyl carbonates in water and water-d2. J. Am. Chem. Soc. 1978, 100, 3564–3567. [Google Scholar] [CrossRef]
- Reisenauer, H.P.; Wagner, J.P.; Schreiner, P.R. Gas-phase preparation of carbonic acid and its monomethyl ester. Angew. Chem. Int. Ed. 2014, 53, 11766–11771. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A.P.; Durden, J.A., Jr.; Sousa, A.A.; Weiden, M.H.J. Novel insecticidal oxathiolane and oxathiane oxime carbamates. J. Agric. Food Chem. 1987, 35, 106–114. [Google Scholar] [CrossRef]
- Patil, S.S.; Jadhav, S.D.; Deshmukh, M.B. Synthesis and antimicrobial activities of new oxime carbamates of 3-aryl-2-thioquinazolin-4(3H)-one. J. Chem. Sci. 2012, 124, 1043–1048. [Google Scholar] [CrossRef]
- Gattinoni, S.; de Simone, C.; Dallavalle, S.; Fezza, F.; Nannei, R.; Battista, N.; Minetti, P.; Quattrociocchi, G.; Caprioli, A.; Borsini, F.; et al. A new group of oxime carbamates as reversible inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2010, 20, 4406–4411. [Google Scholar] [CrossRef] [PubMed]
- Sit, S.Y.; Conway, C.M.; Xie, K.; Bertekap, R.; Bourin, C.; Burris, K.D. Oxime carbamate-discovery of a series of novel FAAH inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- McBurney, R.T.; Walton, J.C. Dissociation or cyclization: Options for a triad of radicals released from oxime carbamates. J. Am. Chem. Soc. 2013, 135, 7349–7354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelsen, S.F.; Landis, R.T. Diazenium cation-hydrazyl equilibrium. Three-electron-two-center π systems. J. Am. Chem. Soc. 1974, 96, 1788–1793. [Google Scholar] [CrossRef]
- Danen, W.C.; Rickard, R.C. Nitrogen-centered free radicals, IV. Electron spin resonance study of transient dialkylaminium radical cations. J. Am. Chem. Soc. 1972, 94, 3254–3256. [Google Scholar] [CrossRef]
- Danen, W.C.; Kensler, T.T. Electron spin resonance study of dialkylamino free radicals in solution. J. Am. Chem. Soc. 1970, 92, 5235–5237. [Google Scholar] [CrossRef]
- McCarroll, A.J.; Walton, J.C. Photolytic and radical induced decompositions of O-alkyl aldoxime ethers. J. Chem. Soc. Perkin Trans. 2000, 2, 1868–1875. [Google Scholar] [CrossRef]
- Tauh, P.; Fallis, A.G. Rate constants for 5-exo secondary alkyl radical cyclizations onto hydrazones and oxime ethers via intramolecular competition experiments. J. Org. Chem. 1999, 64, 6960–6968. [Google Scholar] [CrossRef]
- Blake, J.A.; Ingold, K.U.; Lin, S.; Mulder, P.; Pratt, D.A.; Sheeller, B.; Walton, J.C. Thermal decomposition of O-benzyl ketoximes; role of reverse radical disproportionation. Org. Biomol. Chem. 2004, 2, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. 2-(Aminoaryl)alkanone O-phenyl oximes: Versatile reagents for syntheses of quinazolines. Chem. Commun. 2008, 2935–2937. [Google Scholar] [CrossRef] [PubMed]
- Portela-Cubillo, F.; Scott, J.S.; Walton, J.C. Microwave-promoted syntheses of quinazolines and dihydroquinazolines from 2-aminoarylalkanone O-phenyl oximes. J. Org. Chem. 2009, 74, 4934–4942. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Jalan, A.; Kubosumi, A.R.; Castle, S.L. Microwave-promoted tin-free iminyl radical cyclization with TEMPO trapping: A practical synthesis of 2-acylpyrroles. Org. Lett. 2015, 17, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Markey, S.J.; Lewis, W.; Moody, C.J. A new route to α-carbolines based on 6π-electrocyclization of indole-3-alkenyl oximes. Org. Lett. 2013, 15, 6306–6308. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Booth, S.G.; Essafi, S.; Dryfe, R.A.W.; Leonori, D. Visible-light-mediated generation of nitrogen-centered radicals: Metal-free hydroimination and iminohydroxylation cyclization reactions. Angew. Chem. Int. Ed. 2015, 54, 14017–14021. [Google Scholar]
- Hofstra, J.L.; Grassbaugh, B.R.; Tran, Q.M.; Armada, N.R.; de Lijser, H.J.P. Catalytic oxidative cyclization of 2′-arylbenzaldehyde oxime ethers under photoinduced electron transfer conditions. J. Org. Chem. 2015, 80, 256–265. [Google Scholar] [CrossRef] [PubMed]
- McBurney, R.T.; Portela-Cubillo, F.; Walton, J.C. Microwave assisted radical organic syntheses. RSC Adv. 2012, 2, 1264–1274. [Google Scholar] [CrossRef]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walton, J.C. Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals. Molecules 2016, 21, 63. https://doi.org/10.3390/molecules21010063
Walton JC. Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals. Molecules. 2016; 21(1):63. https://doi.org/10.3390/molecules21010063
Chicago/Turabian StyleWalton, John C. 2016. "Functionalised Oximes: Emergent Precursors for Carbon-, Nitrogen- and Oxygen-Centred Radicals" Molecules 21, no. 1: 63. https://doi.org/10.3390/molecules21010063