
ACKR4 in Tumor Cells Regulates Dendritic Cell Migration to Tumor-Draining Lymph Nodes and T-Cell Priming

 and
and 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
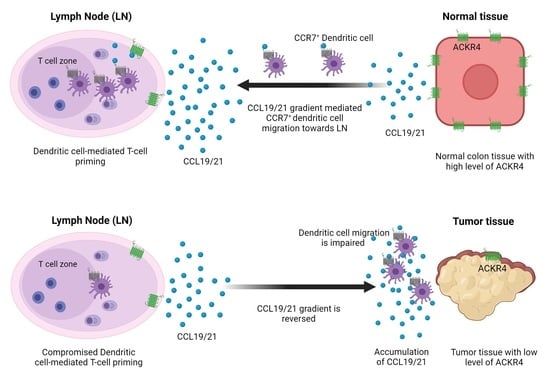
1. Introduction
2. Material and Methods
2.1. Cell Lines and Organoids
2.2. Immunofluorescence and Histology
2.3. Western Blotting of ACKR4
2.4. Cell Line Transfection and Transduction
2.5. Dendritic Cell Isolation
2.6. In Vivo DC Migration Assay
2.7. Flow Cytometry
2.8. In Vivo T-Cell Priming Assay
2.9. Mouse Subcutaneous Models
2.10. Mouse Treatment
2.11. Quantitative PCR (qPCR) Analysis
2.12. Enzyme-Linked Immunosorbent Assay (ELISA) for CCL21
2.13. Statistical Analysis
3. Results
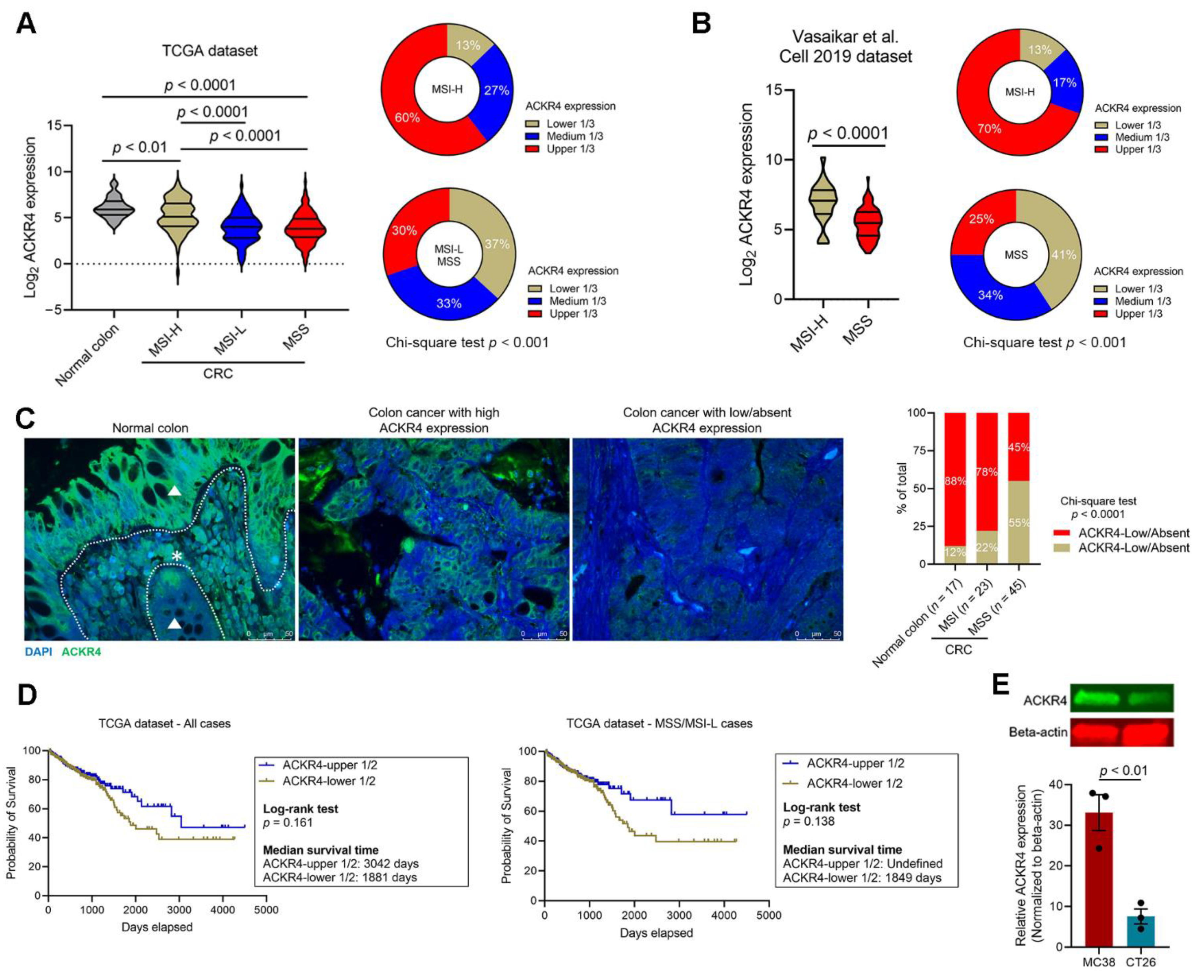
3.1. ACKR4 Is Downregulated in CRC Compared with Normal Colon
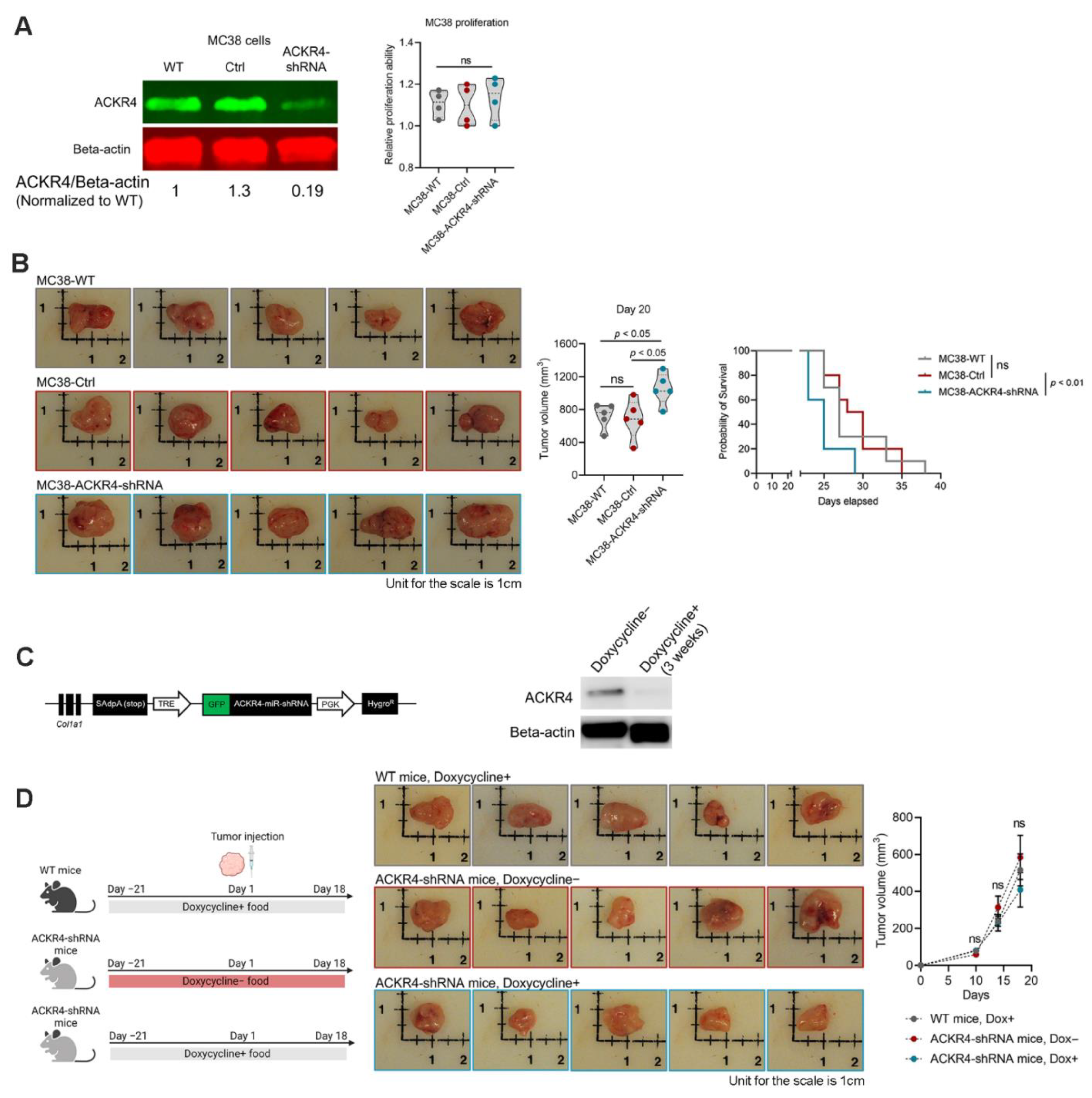
3.2. Knockdown of ACKR4 in Tumor Cells but Not the Host Tissues Accelerate Tumor Growth
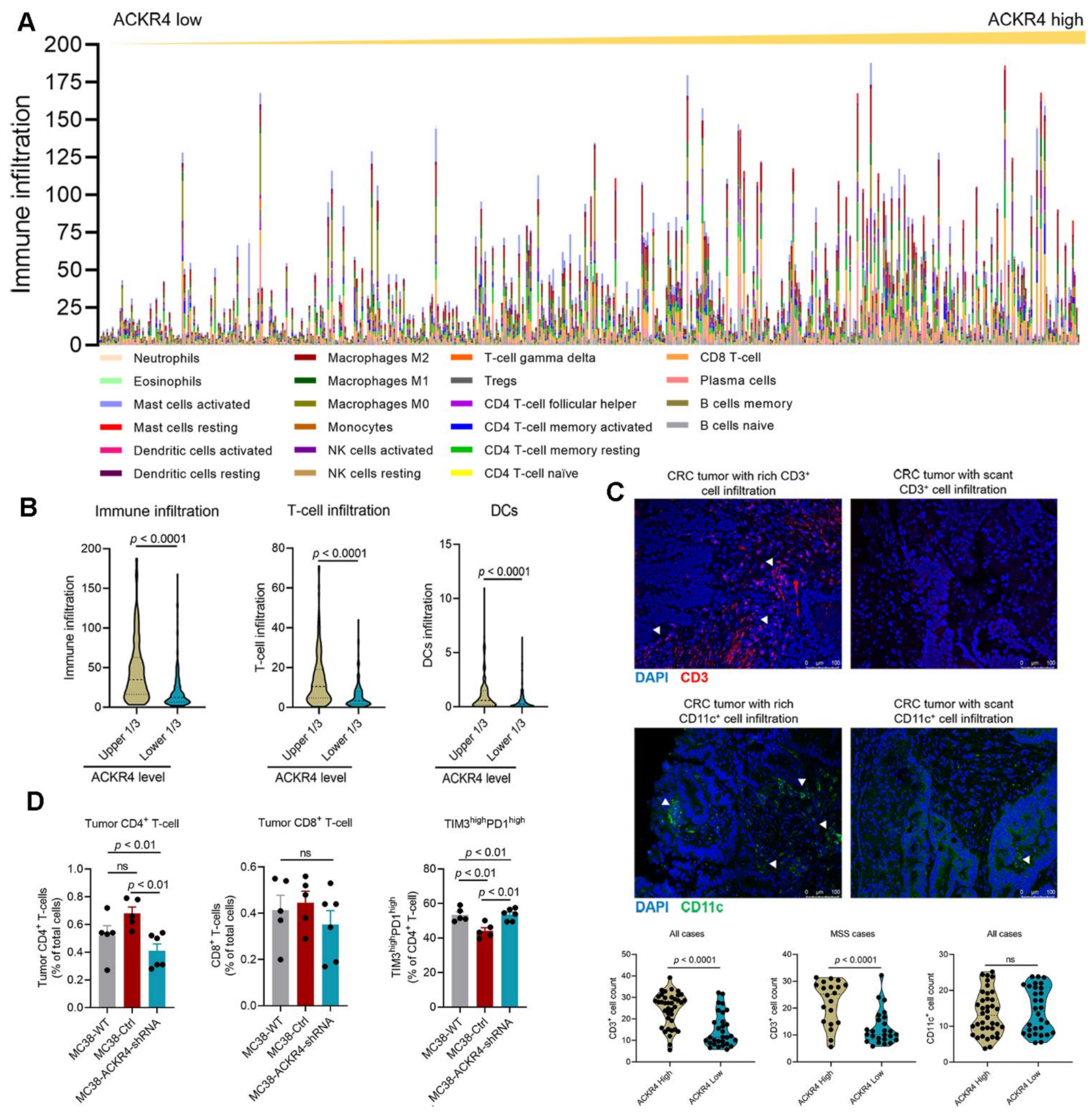
3.3. Loss of ACKR4 Reduces Tumor T-Cell Infiltration
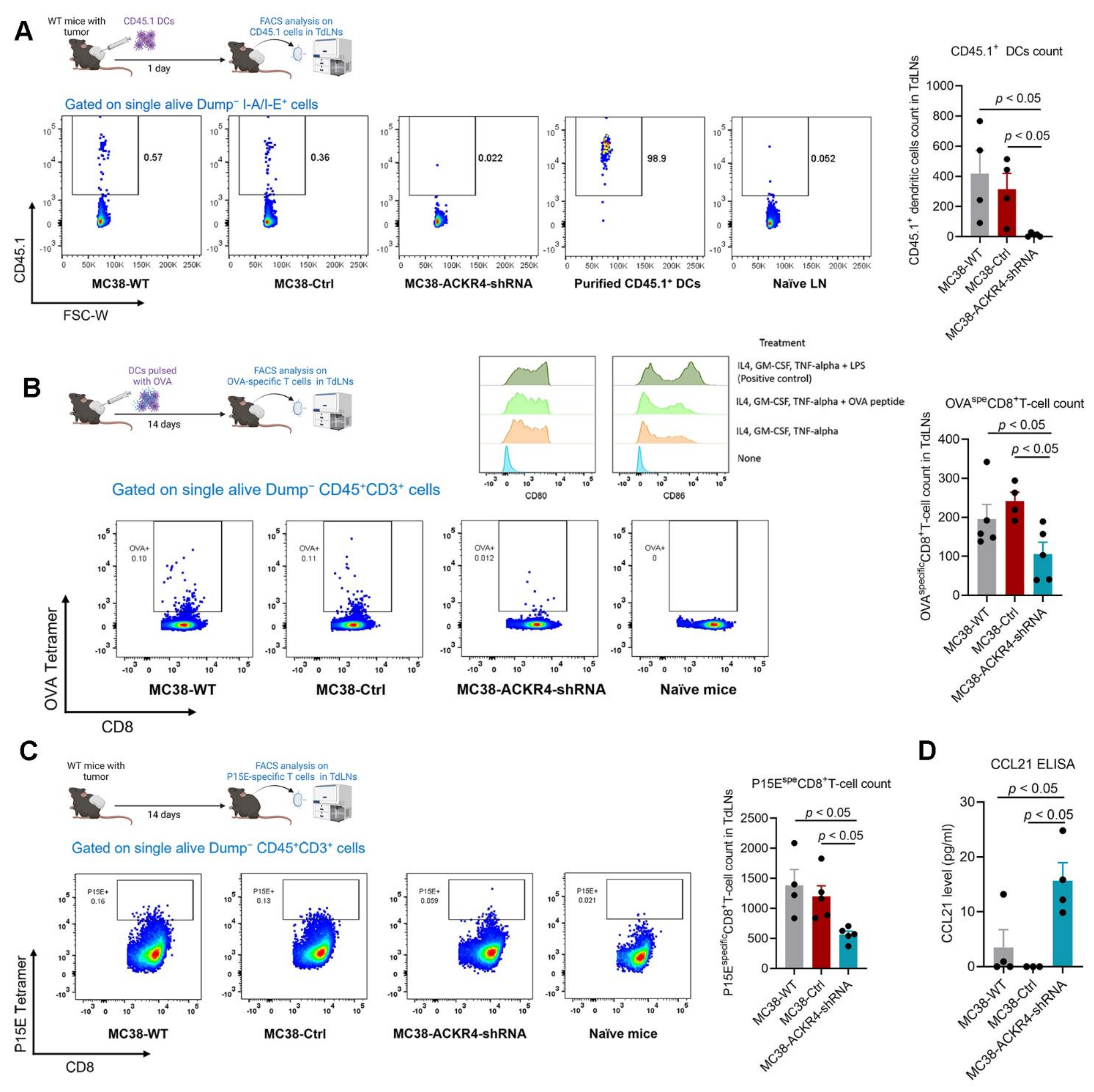
3.4. Loss of ACKR4 Impairs DC Migration to Tumor-Draining Lymph Nodes and Tumor-Specific T-Cell Expansion
3.5. Loss of ACKR4 Weakens Tumor Response to Immune Checkpoint Blockade
3.6. MicroRNA miR-552 Downregulates ACKR4 in CRC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Torre, L.A.; Soerjomataram, I.; Hayes, R.B.; Bray, F.; Weber, T.K.; Jemal, A. Global patterns and trends in colorectal cancer incidence in young adults. Gut 2019, 68, 2179–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, A.; Moehler, M.; Trojan, J.; Goekkurt, E.; Vogel, A. Immuno-oncology in GI tumours: Clinical evidence and emerging trials of PD-1/PD-L1 antagonists. Crit. Rev. Oncol./Hematol. 2018, 130, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Lumish, M.A.; Cercek, A. Immunotherapy for the treatment of colorectal cancer. J. Surg. Oncol. 2021, 123, 760–774. [Google Scholar] [CrossRef]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. eLife 2019, 8, e49020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.W.; Peng, X.C.; Liu, X.Q.; Wang, D.; Li, N.; Cheng, J.T.; Lyv, Y.N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Mayoux, M.; Roller, A.; Pulko, V.; Sammicheli, S.; Chen, S.; Sum, E.; Jost, C.; Fransen, M.F.; Buser, R.B.; Kowanetz, M.; et al. Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaav7431. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T-cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T-cell Immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef]
- Bastow, C.R.; Bunting, M.D.; Kara, E.E.; McKenzie, D.R.; Caon, A.; Devi, S.; Tolley, L.; Mueller, S.N.; Frazer, I.H.; Harvey, N.; et al. Scavenging of soluble and immobilized CCL21 by ACKR4 regulates peripheral dendritic cell emigration. Proc. Natl. Acad. Sci. USA 2021, 118, e2025763118. [Google Scholar] [CrossRef]
- Bryce, S.A.; Wilson, R.A.; Tiplady, E.M.; Asquith, D.L.; Bromley, S.K.; Luster, A.D.; Graham, G.J.; Nibbs, R.J. ACKR4 on Stromal Cells Scavenges CCL19 To Enable CCR7-Dependent Trafficking of APCs from Inflamed Skin to Lymph Nodes. J. Immunol. 2016, 196, 3341–3353. [Google Scholar] [CrossRef] [Green Version]
- Hjorto, G.M.; Larsen, O.; Steen, A.; Daugvilaite, V.; Berg, C.; Fares, S.; Hansen, M.; Ali, S.; Rosenkilde, M.M. Differential CCR7 Targeting in Dendritic Cells by Three Naturally Occurring CC-Chemokines. Front. Immunol. 2016, 7, 568. [Google Scholar] [CrossRef] [Green Version]
- Nibbs, R.J.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Matti, C.; Salnikov, A.; Artinger, M.; D’Agostino, G.; Kindinger, I.; Uguccioni, M.; Thelen, M.; Legler, D.F. ACKR4 Recruits GRK3 Prior to beta-Arrestins but Can Scavenge Chemokines in the Absence of beta-Arrestins. Front. Immunol. 2020, 11, 720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Yuan, C.; Wangmo, D.; Subramanian, S. Tumor-Secreted Extracellular Vesicles Regulate T-Cell Costimulation and Can Be Manipulated To Induce Tumor-Specific T-Cell Responses. Gastroenterology 2021, 161, 560–574.e11. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.; Huang, C.; Wang, X.; Petyuk, V.A.; Savage, S.R.; Wen, B.; Dou, Y.; Zhang, Y.; Shi, Z.; Arshad, O.A.; et al. Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 2019, 177, 1035–1049.e1019. [Google Scholar] [CrossRef] [Green Version]
- Sarver, A.L.; French, A.J.; Borralho, P.M.; Thayanithy, V.; Oberg, A.L.; Silverstein, K.A.; Morlan, B.W.; Riska, S.M.; Boardman, L.A.; Cunningham, J.M.; et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer 2009, 9, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Subramanian, S. Intrinsic Resistance of Solid Tumors to Immune Checkpoint Blockade Therapy. Cancer Res. 2017, 77, 817–822. [Google Scholar] [CrossRef] [Green Version]
- Perez-Ruiz, E.; Melero, I.; Kopecka, J.; Sarmento-Ribeiro, A.B.; Garcia-Aranda, M.; De Las Rivas, J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: Targets, biomarkers, and remedies. Drug Resist. Updates: Rev. Comment. Antimicrob. Anticancer Chemother. 2020, 53, 100718. [Google Scholar] [CrossRef]
- Hou, T.; Liang, D.; Xu, L.; Huang, X.; Huang, Y.; Zhang, Y. Atypical chemokine receptors predict lymph node metastasis and prognosis in patients with cervical squamous cell cancer. Gynecol. Oncol. 2013, 130, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.Y.; Ou, Z.L.; Wu, F.Y.; Shen, Z.Z.; Shao, Z.M. Involvement of a novel chemokine decoy receptor CCX-CKR in breast cancer growth, metastasis and patient survival. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 2962–2970. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.; Sun, C.; Wang, X. Loss of atypical chemokine receptor 4 facilitates C-C motif chemokine ligand 21-mediated tumor growth and invasion in nasopharyngeal carcinoma. Exp. Ther. Med. 2019, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Y.; Yang, L.X.; Wang, Z.C.; Wang, L.Y.; Zhou, J.; Wang, X.Y.; Shi, G.M.; Ding, Z.B.; Ke, A.W.; Dai, Z.; et al. CC chemokine receptor-like 1 functions as a tumour suppressor by impairing CCR7-related chemotaxis in hepatocellular carcinoma. J. Pathol. 2015, 235, 546–558. [Google Scholar] [CrossRef]
- Lewandowska, P.; Wierzbicki, J.; Zawadzki, M.; Agrawal, A.; Krzystek-Korpacka, M. Biphasic Expression of Atypical Chemokine Receptor (ACKR) 2 and ACKR4 in Colorectal Neoplasms in Association with Histopathological Findings. Biomolecules 2020, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.E.; Osman, M.; Kara, E.E.; Abbott, C.; Foeng, J.; McKenzie, D.R.; Fenix, K.A.; Harata-Lee, Y.; Foyle, K.L.; Boyle, S.T.; et al. ACKR4 restrains antitumor immunity by regulating CCL21. J. Exp. Med. 2020, 217, e20190634. [Google Scholar] [CrossRef] [Green Version]
- Lucas, B.; White, A.J.; Ulvmar, M.H.; Nibbs, R.J.; Sitnik, K.M.; Agace, W.W.; Jenkinson, W.E.; Anderson, G.; Rot, A. CCRL1/ACKR4 is expressed in key thymic microenvironments but is dispensable for T lymphopoiesis at steady state in adult mice. Eur. J. Immunol. 2015, 45, 574–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckert, N.; Werth, K.; Willenzon, S.; Tan, L.; Forster, R. B cell hyperactivation in an ACKR4-deficient mouse strain is not caused by lack of ACKR4 expression. J. Leukoc. Biol. 2020, 107, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Werth, K.; Hub, E.; Gutjahr, J.C.; Bosjnak, B.; Zheng, X.; Bubke, A.; Russo, S.; Rot, A.; Forster, R. Expression of ACKR4 demarcates the “peri-marginal sinus,” a specialized vascular compartment of the splenic red pulp. Cell Rep. 2021, 36, 109346. [Google Scholar] [CrossRef]
- Zhao, X.; Kassaye, B.; Wangmo, D.; Lou, E.; Subramanian, S. Chemotherapy but Not the Tumor Draining Lymph Nodes Determine the Immunotherapy Response in Secondary Tumors. iScience 2020, 23, 101056. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wangmo, D.; Premsrirut, P.K.; Yuan, C.; Morris, W.S.; Zhao, X.; Subramanian, S. ACKR4 in Tumor Cells Regulates Dendritic Cell Migration to Tumor-Draining Lymph Nodes and T-Cell Priming. Cancers 2021, 13, 5021. https://doi.org/10.3390/cancers13195021
Wangmo D, Premsrirut PK, Yuan C, Morris WS, Zhao X, Subramanian S. ACKR4 in Tumor Cells Regulates Dendritic Cell Migration to Tumor-Draining Lymph Nodes and T-Cell Priming. Cancers. 2021; 13(19):5021. https://doi.org/10.3390/cancers13195021
Chicago/Turabian StyleWangmo, Dechen, Prem K. Premsrirut, Ce Yuan, William S. Morris, Xianda Zhao, and Subbaya Subramanian. 2021. "ACKR4 in Tumor Cells Regulates Dendritic Cell Migration to Tumor-Draining Lymph Nodes and T-Cell Priming" Cancers 13, no. 19: 5021. https://doi.org/10.3390/cancers13195021