Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications



1
Department of Crystallography, Geological Faculty, M.V. Lomonosov Moscow State University, Leninskie Gory 1, 119991 Moscow, Russia
2
Chemical Faculty, M.V. Lomonosov Moscow State University, Leninskie Gory 1, 119991 Moscow, Russia
3
Skolkovo Institute of Science and Technology, 3 Nobel Street, 143025 Moscow, Russia
*
Author to whom correspondence should be addressed.
Minerals 2020, 10(6), 524; https://doi.org/10.3390/min10060524
Submission received: 28 April 2020
/
Revised: 2 June 2020
/
Accepted: 5 June 2020
/
Published: 7 June 2020
(This article belongs to the Special Issue Battery Minerals)
Abstract
:For successful development of novel rechargeable batteries, considerable efforts should be devoted to identifying suitable cathode materials that will ensure a proper level of energy output, structural stability, and affordable cost. Among various compounds explored as electrode materials, structural analogues of minerals–natural stable inorganic solids–occupy a prominent place. The largest number of varieties of phosphate minerals occurs in rare metal granite pegmatites, and many of which contain transition metals as essential components. Transition metal phosphates are promising candidates for exploration as cathode materials due to a perfect combination of easily scalable synthesis, moderate-to-high voltage operation, thermal/chemical stability, and environmental safety. However, impurities usually presented in natural objects, and often inappropriate sample morphologies, do not permit the use of minerals as battery electrode materials. Nevertheless, the minerals of different classes, especially phosphates, are considered as prototypes for developing novel materials for battery applications. The crystal chemical peculiarities of the phosphate representatives that are most relevant in this aspect and the electrochemical characteristics of their synthetic analogues are discussed here.
1. Introduction
“They created a rechargeable world”: the 2019 Nobel Prize in Chemistry was awarded to John B. Goodenough, M. Stanley Whittingham, and Akira Yoshino for their contribution to the development of lithium-ion batteries, which revolutionized our daily life and laid the foundation of a wireless fossil fuel-free society [1].
Ever-growing demands for energy and depleting of fossil fuels require utilizing renewable energy resources (solar, wind, geothermal). For accumulating the energy from these intermittent sources, we need highly efficient energy conversion and storage systems. Rechargeable batteries are the most efficient energy storage technology for a wide range of devices from portable electronics to electric vehicles and grids [2]. Meeting the future needs of our society requires a significant improvement of existing Li-ion technology and development of alternative Na- and K-ion batteries, and the main research task is to find suitable materials for electrodes. The discovered ability of synthetic triphylite LiFePO4 to reversibly de/intercalate lithium ions [3] has brought great attention to transition metal phosphates as electrode materials for Li-ion batteries.
Phosphates are the most widespread minerals in the Earth’s crust after silicates and oxides. Their remarkable structural diversity allows for a large range of technological applications such as magnetic and nonlinear optical materials, catalysts, adsorbents, molecular sieves, phosphors, and biomaterials, to name but a few [4]. The advance of battery technologies promotes more sustainable and environmentally friendly energy production, storage, and consumption. Transition metal phosphates are attractive here due to their enhanced thermodynamic and kinetic stability compared to oxides, higher operating potentials owing to the inductive effect of the phosphate group for the same cation redox couples, and the environmental friendliness of their robust crystal structures.
However, the mineral phases may not be suitable for research due to the unstable impurities typically present in natural samples. At the same time, their crystal structures, often with such promising features as low dimensionality, lack of a center of symmetry, ion diffusion channels may serve as excellent reference points for the development of new materials. The use of information on chemical compositions, structural features, and conditions of mineral formation and alteration in the Earth’s crust is essential for understanding and adequate interpretation of the properties of their synthetic analogues. Thus, crystal chemical study of transition metal phosphates with alkali atoms makes it possible to determine the miscibility limits of solid solutions and to find ways to control cation ordering, defect concentration, and ion oxidation, which are important for creating electrochemically active materials. Moreover, phosphate-based materials provide ease of synthesis along with reliable electrochemical characteristics.
In this review, we will demonstrate the definite attractiveness of minerals, mainly phosphates, as a source of inspiration for researchers working in the field of battery materials.
2. Background Information
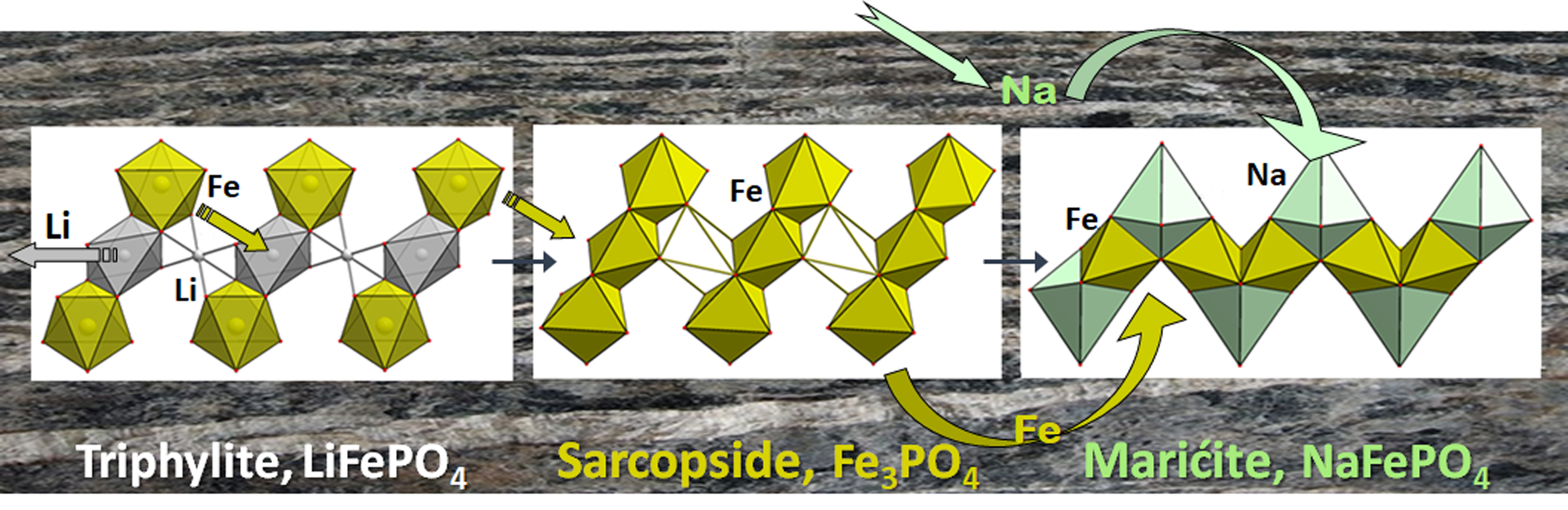
Modern technologies require a tremendous amount of energy, and the problem of energy storage is now key for the world energy supply. A secondary (or rechargeable) battery is an electrochemical energy storage device, which converts chemical energy into electrical energy and vice versa in a repeated way. Among the present batteries, Li-ion technology is the best due to high delivered energy density, long cycle life, rate capability, and energy efficiency. The Li-ion battery (LIB) comprises one or more sequential or parallel single electrochemical cells consisting of two electrodes (anode and cathode) and a non-aqueous electrolyte, which supports the transport of charge carriers between the separated electrodes. The process of energy storage is carried out through the reversible de/intercalation of alkali metal ions into the electrode materials. During charge, the electrons are pumped “uphill”, thermodynamically speaking, from a positive (cathode) to a negative (anode) electrode through an external circuit; ions also flow toward the anode but through the electrolyte to complete the circuit and keep the reaction running (Figure 1). The opposite process occurs during discharge: the favorable free energy of the redox pair allows the reaction to proceed “downhill”, thereby providing energy to the device that receives power from the battery.
Battery performance is evaluated by power and energy density parameters. Power refers to the amount of energy that can be instantaneously released by a battery, while the energy density of a battery is defined as its specific capacity (the amount of charge passed between the cathode and the anode per unit mass) multiplied by the potential difference between these electrodes. These parameters depend on the choice of active electrode materials (particularly the cathode material), which must meet severe chemical and electrochemical requirements. They must demonstrate a high specific capacity, fast reversible de/intercalation of alkali ions, and sufficient electrical conductivity. In addition, they must undergo reversible structural changes with minimal volume changes upon cycling. Of particular note is the requirement for a high specific capacity defined by the formula:
where n is the number of electrons (or alkali metal ions) involved in the reaction, M is the molar weight of the compound, and 26.8 is the Faraday constant, normalized to Ah units. The smaller the molar mass of the compound and the greater the number of alkali metal ions involved in the reversible process, the higher the specific capacity obtained. This limits the choice of electrochemically active metal by 3d and 4d transition metals (Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Nb, Mo), which are capable of demonstrating multiple oxidation states that can be easily converted from one to the other. The working potential of the electrode material is determined by the electrochemically active transition metal and its coordination environment [5].
CT = 26.8 × n/M (Ah·g−1),
First-generation cathode materials for LIBs based on layered LiMO2 oxides (M = Mn, Co, Ni) with a NaCl-derivative structure and a spinel LiMn2O4 are still widely used in different portable devices [6]. Despite the high specific capacity (up to 180–200 mAh·g−1) delivered by the mixed oxides LiNixMnxCo1−2xO2 with a layered structure, application of these cathodes in large-scale devices is limited due to their thermal instability and due to their high cost [7].
The development and commercial success of LiFePO4-based cathode materials has drawn attention to compounds with various anion groups, (XOm)n− [3]. Despite the smaller theoretical gravimetric capacity owing to the presence of heavy oxoanion groups in the composition, these materials provide a number of advantages, which make them a good alternative to oxide materials for use in large-scale applications. Thus, their robust framework supports long-term structural stability and contributes to the superior cycle life and high safety. Due to the inductive effect of the (XOm)n− group, these materials exhibit higher operating potentials compared to that in oxides, resulting in enhanced specific energy density [5]. Compounds with complex anions adopt a large number of atomic arrangements, providing a rich variety of crystal structures. In the last two decades, compounds with different tetrahedral oxoanion groups, such as silicates, phosphates, sulfates, pyrophosphates, and their fluorinated compounds, have been widely investigated [8,9].
For most of the LIB cathode materials used, the potential for further enhancement of their performance characteristics has already been exhausted, so the search for new cathode materials with high specific energy is an important task from both fundamental and practical points of view. For large power supplies, when the price per kilowatt-hour becomes an important parameter, the low relative abundance of the constituent elements should be taken into account [10]. It imposes restriction on the use of rare metals, lithium, as well as cobalt, which functions as a redox center within the battery. For this reason, the development of alternative metal-ion technologies based on Na-, K-, Mn-, and Fe-containing compounds may considerably reduce the cost and environmental impact.
Na and K are widely distributed in nature; in comparison to the occurrence of Li (20 ppm) in the Earth’s crust, the abundance of Na (23,000 ppm) and K (17,000 ppm) seems to be limitless, and they occupy, respectively, seventh and eighth places in the elemental content of the crust [2]. Considering the quite similar chemistry of Li, Na, and K alkali metals, the alternative metal-ion batteries can take advantage of the working principles and manufacturing technologies of mature LIB. Sodium (Na+) and potassium (K+) ions are heavier and have larger ionic radii than lithium ions (Li+) and, therefore, Na-ion batteries (NIBs) and K-ion batteries (KIBs) are not expected to compete with LIBs for volumetric and gravimetric energy density, but it is anticipated that their abundant resources will be put to use to ensure price competitiveness and to complement LIBs in large-scale applications [2,11]. It should also be mentioned that Na and K ions may exhibit higher diffusion rates in crystal frameworks compared to Li-ions; thus, leading to the higher specific power of these types of batteries. While the KIB finds itself at an early stage of development, the NIB technology is already approaching a satisfactory level for commercialization considering the energy density and cycle life: today NIBs are considered as a low-cost solution to replace toxic lead-acid batteries.
KIBs have recently aroused great interest because of the certain advantages they can provide. Among these three alkali metals, K+ has the smallest Stoke’s radius of solvated ions, resulting in the highest ion mobility and ion conductivity in liquid electrolytes. More importantly, in non-aqueous electrolytes, K demonstrates a lower standard redox potential than Na and even Li, and therefore potentially KIBs can offer higher operating voltage, providing higher energy density [2].
For the successful development of new technologies, considerable efforts should be devoted to identifying suitable cathode materials that will ensure a proper level of energy output, structural stability, and affordable cost. In the search for electrochemically active transition metals, iron and manganese prevail over other metals. Iron is one of the rock-forming elements; it is extremely widely distributed in the Earth’s crust (56,300 ppm) and is the cheapest non-toxic transition metal. Manganese is another rock-forming element that is widespread: an enormous amount of manganese nodules have been discovered recently on the seabed. These metals form various compounds which demonstrate electrochemical activity with appropriate operating potentials corresponding to Fe2+/Fe3+ and Mn2+/Mn3+ redox couples; moreover, Mn-based compounds can offer an ideal combination of high-voltage performance, low cost, and environmental safety.
Among various compounds with oxoanion groups explored as electrode materials, structural analogues of minerals occupy a prominent place. This suggests that mineral-like compounds, primarily phosphates and silicates, might be prototypes of novel materials with attractive electrochemical properties. Phosphates are evident leaders among complex anions, having a perfect combination of easily scalable synthesis, moderate-to-high voltage operation, and thermal/chemical and operational stability.
3. Phosphate Minerals in Pegmatites
Granite pegmatites present a source of raw materials for glass and ceramics, silicon for microprocessors, and industrial metals, for example, Li, Cs, Be, Nb, Ta, Sn, REEs, and U. This type of rock is unique in having a variety of economically significant minerals and colored gemstones in such a concentrated form [12]. Although the largest amount of phosphorus is concentrated in apatite deposits, the greatest number of varieties of phosphate minerals occurs in rare metal granite pegmatites. According to Moore [13], about 190 phosphates are associated with granite pegmatites and at least 40 of them are primary phases formed directly from pegmatite fluid. Analysis of the chemical formulae of more than 200 phosphate minerals showed that the most common cations in phosphates are Fe2+ and Fe3+ (95 compounds), Al3+ (60), Ca2+ (56), Mn2+, and Mn3+ (45) [14]. A novel classification of granitic pegmatites, including phosphate minerals, was presented in [15].
The high-temperature primary minerals that crystallize in the core of pegmatite (triplite-zwieselite, amblygonite-montebrasite, triphylite-lithiophilite series, etc.) have the more complicated three-dimensional frameworks formed by octahedra or/and five vertex polyhedra [16]. As shown in [17], 75% of the high-temperature phosphates possess densely packed or “denser-than-densely-packed” structures. Their frameworks incorporate “clusters” of octahedra sharing edges and faces, while PO4 tetrahedra, in these structures, have edge contacts with metal-centered polyhedra. Giant crystals, over one meter in dimension (graftonite, apatite, arrojadite, and triphylite) are known for the primary phosphates [18].
Greatly “polymerized” associations of octahedra sharing edges and vertices are the basis of crystal structures of early metasomatic phosphates that crystallize at the boundary of the pegmatite core and an intermediate zone. They form along the initial metasomatic replacements as a result of the alteration of the earlier phosphates under the action of the later fluid and usually they are presented by smaller crystals [19]. A distinctive typomorphic feature of these phosphate minerals is the presence of mixed valence transition metals, resulting in the dark brown-black or green-black colors due to a M2+ ↔ M3+ charge transfer. Their 3D frameworks built by amphoteric oxocomplexes contain hole defects (arrojadite, lipscombite, wyllieite), or their constructions present one-dimensional (1D) tapes and two-dimensional (2D) layers (alluaudite, kryzhanovskite) of edge-sharing octahedra.
Depending on the conditions of crystal genesis, amphoteric complexes can form the cationic part of the crystal structure or act as anions together with acid tetrahedral groups. In other words, in combination with, for instance, acid (PO4)3− oxocomplexes, amphoteric metal complexes can form mixed anionic networks similar to organic polyanions [20]. The amphotericity of oxocomplexes of transition metals and some others is well known in chemistry. This property turned out to be the defining geochemistry of many natural processes. In the alkaline conditions of mineral genesis (agpaitic pegmatites, hydrothermalites, etc.), the alkaline metals quantitatively dominate in the system of crystallization. Under conditions of increased alkalinity, the cationic fragment of the crystal structure is usually built by alkaline elements, while amphoteric oxocomplexes together with acid tetrahedra form its anionic part in the presence of a significant amount of more electropositive cations. These anionic constructions may be formed by octahedra and/or five vertex polyhedra around an amphoteric metal, sharing vertices with acid PO4 tetrahedra, or by chemically different tetrahedral oxocomplexes. The phosphate minerals containing significant amount of alkali cations may have zeolite-like microporous architecture [21,22,23].
4. Mineral Prototypes for Battery Design
4.1. Phosphates of the Triphylite Li(Fe, Mn)PO4–Lithiophilite Li(Mn, Fe)PO4 Series and Their Derivatives
4.1.1. The Triphylite Li (Fe, Mn) PO4–Lithiophilite Li (Mn, Fe) PO4 Series: Occurrence, Crystal Chemistry, and Electrochemical Behavior
Phosphate of the triphylite Li (Fe, Mn) PO4–lithiophilite Li (Mn, Fe) PO4 series [olivine-type minerals with an ordered distribution of Li and Fe (Mn) among M1 and M2 sites surrounded by oxygen octahedra] are the most abundant among the accessory minerals in granite pegmatites. These phosphates may form crystals with sizes up to several meters. Field studies show that the minerals in the series are primary phases associated with alkali feldspar and quartz in the middle and inner parts of zoned granite pegmatites [13,24]. Subsequently attacked by aqueous rest liquid during consolidation of the pegmatite, members of the triphylite-lithiophilite solid solution series are subject to stepwise alteration. First, the oxidation of Fe2+ to Fe3+ together with reductions in the lithium content and the formation of the ferrisicklerite Li1-x(Fe3+xFe2+1−x)PO4–sicklerite Li1-x(Mn3+xMn2+1−x)PO4 minerals, and second, Mn2+ to Mn3+ oxidation followed by complete extraction of the lithium and the formation of phosphates of the isomorphous heterosite-purpurite series (Fe3+,Mn3+) PO4 [25]. The small Li+ cations are quite leachable and the primary crystals easily suffer hydrothermal attack without structure collapse: the crystal structure of heterosite (purpurite) is based on the same hexagonal close packing of oxygen atoms as triphylite (lithiophilite) but with ordered vacancies in the cation positions formerly occupied by lithium [17] (Figure 2).
The first single crystal triphylite end member, LiFePO4 (crystal characteristics of synthetic analogues of minerals, studied as cathode materials, are given in Table 1 in the conclusion section), was hydrothermally synthesized and structurally characterized in 1977 [26]. Twenty years later, Goodenough’s group discovered the electrochemical activity of LiFePO4 and stated this phosphate as a candidate for the cathode of rechargeable lithium batteries [3]. LiFePO4 satisfies many of the criteria for an electrode material. It reversibly de/intercalates Li, exhibiting a flat plateau around 3.45 V (vs. Li/Li+) because of a two-phase redox reaction mechanism with the formation of fully delithiated FePO4. This material has good gravimetric capacity (170 mAh·g−1) and is stable against overcharge or discharge and compatible with commercially available electrolyte systems. It is also environmentally friendly and cheap. Nevertheless, there are several disadvantages associated with the moderate operating potential and, consequently, relatively low energy density (volumetric especially), large volume change during cycling (~7%), poor electronic conductivity and a two-phase mechanism of Li+ ion de/intercalation.
The electrochemical behavior of the natural triphylite LiFe1−yMnyPO4 has been investigated [27] at elevated temperature (80 °C) before and after high-temperature carbon-coating treatment. It was shown that the discharge capacity, kinetics, and cycling stability were all improved after the heating process. The observed capacity values of 80 mAh·g−1 (> 54% of the theoretical value) were much smaller than for the synthetic samples, suggesting that impurities present in natural triphylites as well as inappropriate sample morphology might create problems; for that reason, all battery cathode materials are synthetic [7].
Today, lithium-iron phosphate, LiFePO4, stands as a commercialized cathode material in LIBs for hybrid vehicles and grid storage systems. The development of new approaches for the synthesis of materials such as the transition to nanostructured LiFePO4 material, creating a conductive carbon coating on the surface of the active material particles and surface doping, significantly improved the mass transfer rate. These state-of-the-art methods led to the high-rate LiFePO4 material, which demonstrates a specific capacity close to its theoretical value [28,29,30]. Computational methods (density functional theory and molecular dynamics) also contributed to exploration of the new synthesis approaches. It was found that Li-ion diffusion in LiFePO4 is strongly anisotropic and occurs through a curved path along the {010} direction, and later on, Nishimura et al. provided experimental evidence for this curved one-dimensional (1D) path for lithium motion [31,32,33]. These findings prompted the development of LiFePO4 material with specific morphology, which was prepared by hydrothermal and solvothermal approaches [34,35].
Further development of the functional properties of LiFePO4 material are directed towards increasing specific energy and power characteristics: today, materials based on Li(Fe,Mn)PO4 are actively investigated. It is found that the partial substitution of iron by manganese not only leads to an increase of the working potential of the material and consequently its energy density but also changes the mechanism of Li extraction/insertion [36].
4.1.2. Lithiophilite—Purpurite—Niahite: Comparative Crystal Chemistry and Structure Transformation
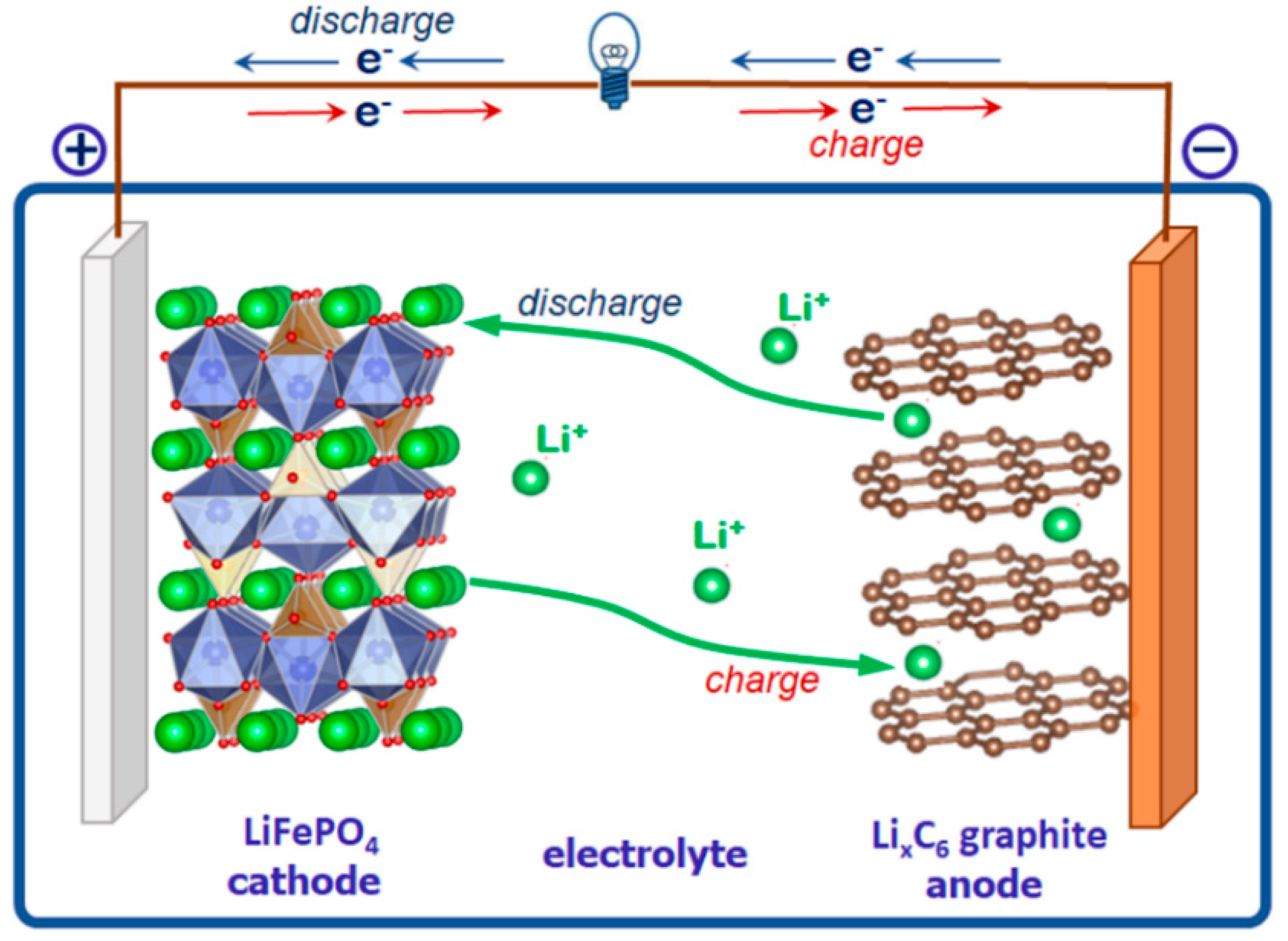
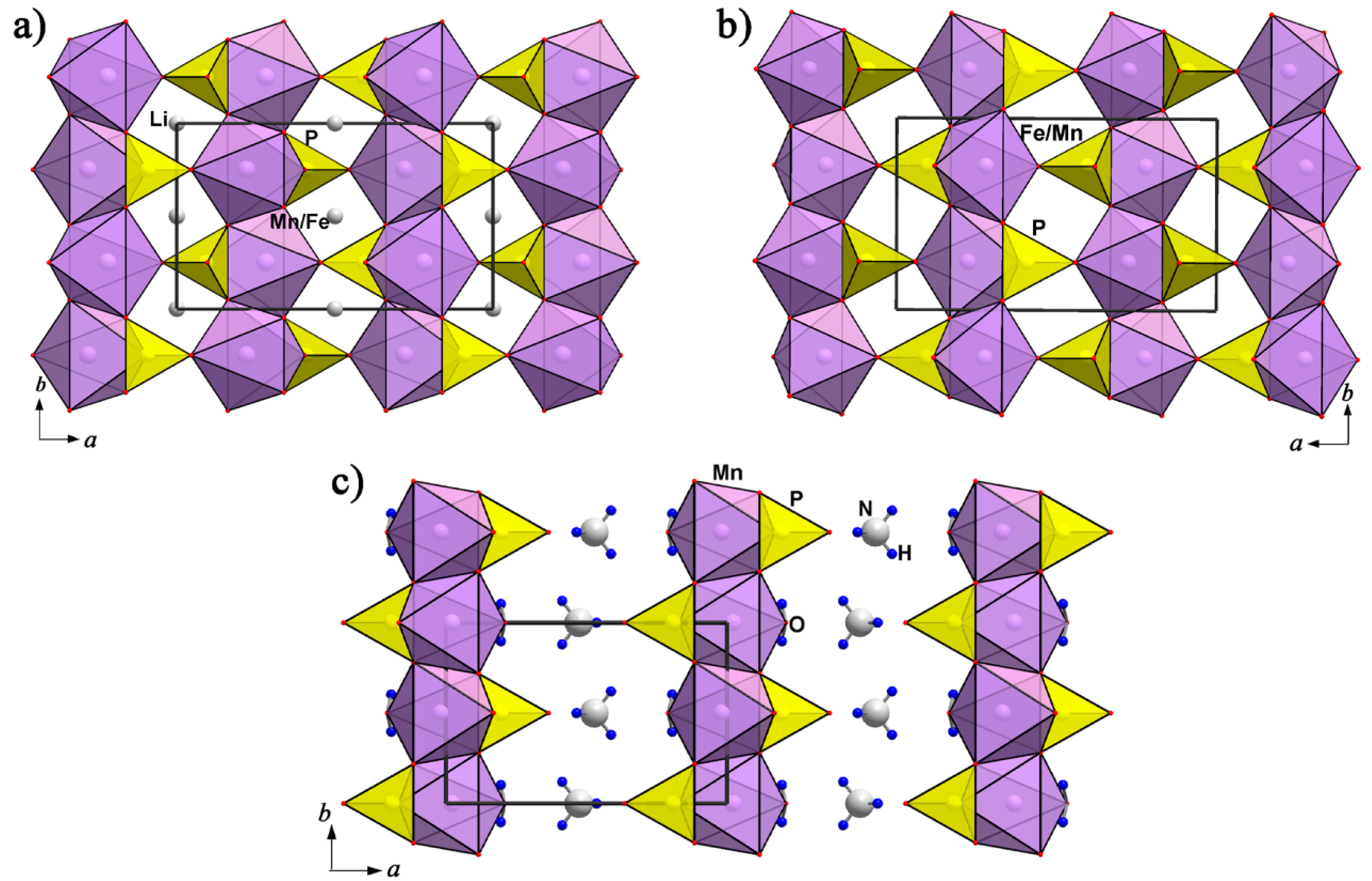
A topological analysis reveals similar fragments forming crystal structure types of the triphylite-lithiophilite and niahite, (NH4)MnPO4(H2O), mineral groups [37,38]. Niahite has a biogenic nature; it was found in a limestone cave with bat guano in Malaysia [39]. The mineral crystal structure was not solved because of the very soft and fragile nature of niahite. The crystal structures of related synthetic varieties with formulae NH4[Fe(PO4)]HF and (NH4)CoPO4.H2O have been studied. Lately, a full formula analogue of mineral NH4MnPO4(H2O) obtained under low-temperature hydrothermal conditions, so-called “chemie douce” [40], was structurally investigated [41].
In the crystal structure of niahite, Mn2+ has octahedral coordination where five vertices are occupied by phosphate O atoms and one by the O atom of the H2O molecule. The Mn-centered octahedra share common O vertices to form two-dimensional (2D) quadratic layers parallel to the bc plane of the unit cell (Figure 2). The PO4 tetrahedra share one edge and one vertex with MnO6 octahedra. They are placed at capping positions alternating above and below the square meshes of the MnO6 layer. One of four vertices of each PO4 tetrahedron is not coordinated to Mn but is directed towards the interlayer space where NH4 groups are placed (Figure 3). As this terminal O atom is bonded only to P5+ in the structure and therefore has a formal bond valence of 1.25, which is too small, it becomes an acceptor of four possible hydrogen bonds from the ammonium ions. The mixed Mn, P sheets are linked along the {100} direction by three N_H…..O hydrogen bonds in a three-dimensional construction. Additional hydrogen bonding occurs along the layer planes from the aqua ligands towards the phosphate oxygen atoms. These O_H…..O hydrogen bonds are bifurcated.
The plate-like habitus of the crystals corresponds to the layer structure; the a axis is normal to the crystal-plate. The mixed Mn, P layers in the niahite NH4MnPO4(H2O) structure can be interpreted as fragments of the lithiophilite, Li(Mn,Fe)PO4, or purpurite, (Mn,Fe)PO4, structure. All minerals are orthorhombic, their space groups differ within the inversion center, and the unit cell volumes are close. There are two Mn, P layers parallel to the bc plane of the same topology in the unit cell of purpurite (and lithiophilite) (Figure 2). These layers connect in the {100} direction through common vertices of Mn- and P- centered polyhedra. Every second layer of MnO6 octahedra with adjacent PO4 tetrahedra may be extracted from the structure of purpurite to provide a model of the [Mn(PO4)] layer in niahite, stabilized by terminal aqua ligands. The NH4+ cations between the sheets supply the interlayer connection along the {100} direction to form the crystal structure of niahite, NH4MnPO4(H2O) (Figure 3). In nature, this mechanism may occur during a bioorganic process under reducing conditions with formation of the mineral niahite, (NH4)MnPO4(H2O), after purpurite, MnPO4, and lithiophilite, Li(Mn,Fe)PO4. As in the case of the triphylite–heterosite conversion in hydrothermal conditions, this process can run without a rearrangement of the mother structure: the newly forming compound adopts its basic fragments.
A supposition regarding possible lithiophilite → niahite transformation [37,38] was indirectly supported by experimental works on the preparation of synthetic analogues of minerals of the triphylite–lithiophilite series using the “niahite” matrix NH4MPO4⋅H2O (M = Fe, Mn, Co, or Ni) as precursor [42,43,44]. The LiFePO4/C (coated with carbon) material, synthesized using the “niahite” precursor, demonstrated high rate capability with capacity values close to the theoretical one, while the LiMnPO4/C sample showed much lower capacity values. The preservation of precursor morphology was also observed upon replacement of NH4+ groups by Li+ cations through solid-state reaction at elevated temperature: the obtained LiMPO4 particles completely repeat the shape of the intermediate NH4MPO4·H2O crystals (M = Fe, Mn) [44].
4.1.3. Simferite, Li(Mg,Fe3+,Mn3+)2(PO4)2 and Tavorite, LiFe(PO4)(OH,F) Based Materials and Their Electrochemical Potential
The secondary pegmatite mineral simferite, Li(Mg,Fe3+,Mn3+)2(PO4)2, is structurally very close to triphylite or lithiophilite, although it contains Mg and atoms of transition metals in 3+ oxidation state. The crystal structure refinement of simferite established that Li+ statistically occupies one half of M1 positions at the center of symmetry, while the octahedral M2 position is populated by Mg, Fe3+, and Mn3+ in a ratio of 0.5:0.3:0.2 [45]. Simferite happened to be another mineral, which motivated researchers to synthesize and study its laboratory analogous.
To understand the phase transformation behavior of triphylite electrode materials upon Li-deintercalation, the effect of Mg substitution in LiFe0.6Mn0.4PO4 has been investigated [46]. It was demonstrated that, in contrast to the equilibrium two-phase nature of LiFePO4, the simferite-like compound LixMg0.5Fe0.3Mn0.2PO4 undergoes a single-phase reaction mechanism, providing faster rate kinetics. Moreover, a continuous increase in the voltage of the Mn2+/Mn3+ redox couple on the addition of Mg to LiMn0.4Fe0.6PO4 was reported.
A rare mineral of lithium pegmatites, tavorite, LiFePO4(OH,F), is a product of hydrothermal transformation of triphylite and usually occurs with barbosalite, ferrisicklerite, and heterosite [13]. The tavorite crystal structure (Figure 4) is composed of centrosymmetric Fe3+- centered octahedra sharing OH vertices to form columns running along the b axis. Along the a and c directions, these columns are joined through PO4 tetrahedra in a framework with Li+ cations in its cavities. The LiO5 polyhedra share edges to form pairs linked along the c axis by phosphate tetrahedra.
Black crystals of the octahedral shape, obtained under hydrothermal conditions and identified by X-ray diffraction as tavorite, have been studied in order to understand a supposal Fe2+ → Fe3+ charge transfer responsible for the crystals’ coloring [47]. A precise crystal structure analysis of a spherical sample, including investigation of the deformation electron density, revealed an additional position in the octahedral surrounding of O atoms (Figure 4a). This position with small occupancy was identified as statistically populated by Fe2+ ions [48]. An Fe–Fe distance of 2.556 Å between the centers of three octahedra sharing faces (so-called “h clusters”), which form when additional octahedra are populated by Fe3+ ions, led to the charge transfer between neighboring Fe atoms. The crystal chemical formula of the Fe3+-bearing tavorite (for simplicity of presentation of the material, we use mineral names when identifying synthetic analogues that are employed in battery technology) was established as LiFe3+Fe2+x[PO4](OH)1−2xO2x]; this formula describes a correlation between amounts of Fe2+ and H+ cations. It is important to mention that the crystal structure allows a maximal number of Fe2+ in the additional position at the center of symmetry (0, 0.5, 0.5) of triclinic space group P up to an x value equal to 0.5. If this limit is achieved, the Fe2+ ions would become ordered in their site, the hydroxyl groups would disappear, and the Fe3+: Fe2+ ratio would become equal to 2:1.
Synthetic LiFePO4(OH) was tested as the positive electrode, and electrochemical activity with a reversible capacity of around 100 mAh·g−1, corresponding to the insertion of additional Li, was observed at an average voltage of 2.3 V vs Li/Li+ [49]. LiVPO4F is one of the most studied fluoride-phosphates with the tavorite structure. The incorporation of a fluorine ion in the coordination sphere of the transition metal “enhances” the inductive effect of (PO4)-groups, increasing the potential of the V3+/V4+redox transition up to 4.2 V vs. Li/Li+. Furthermore, LiVPO4F demonstrates the feasibility of multi-electronic activity based on the multivalent nature of vanadium with the reversible insertion of additional Li (V3+/V2+ redox transition) at 1.8 V [50].
4.1.4. Na-Bearing Triphylite-Lithiophilite Structural Analogous and Their Derivatives as Promising Cathode Materials
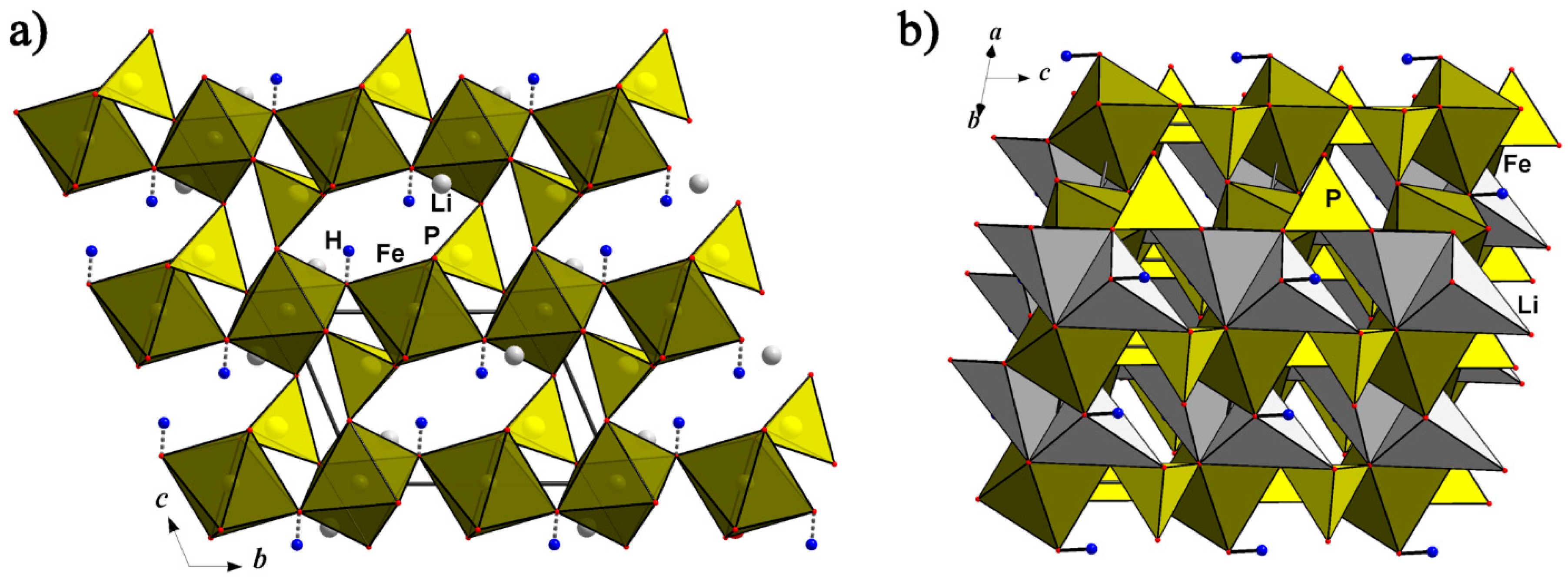
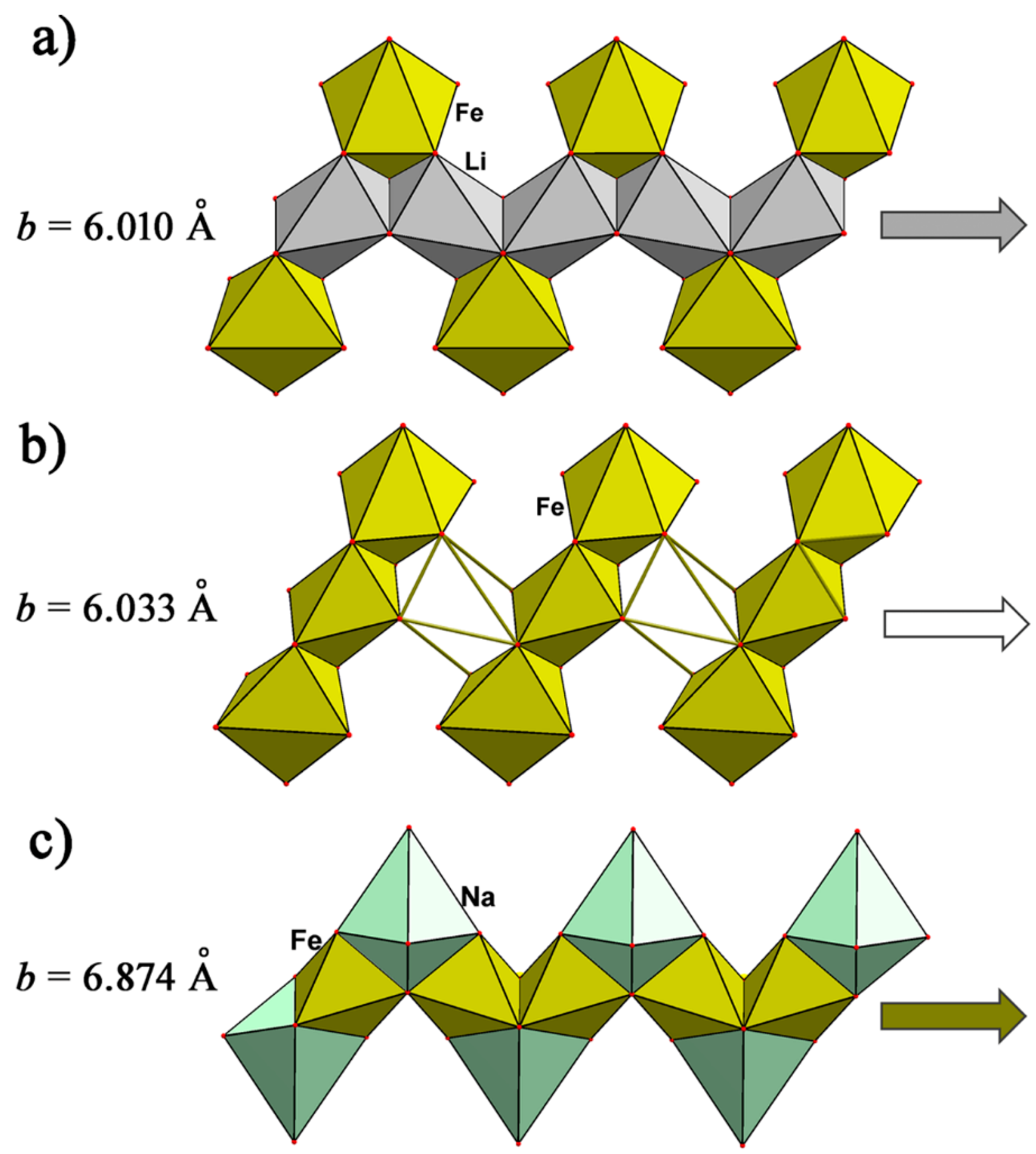
The olivine (Mg,Fe)2(SiO4) structure turned out to be very stable; many compounds of different compositions, such as silicates, phosphates, germanates, borates, and so on, crystallize in this structure type. It is based on hexagonal close packing of O atoms with cations partly occupying octahedral and tetrahedral voids. Several pegmatite phosphates, namely the above-discussed minerals of the triphylite Li(Fe,Mn)PO4–lithiophilite Li(Mn,Fe)PO4, ferrisicklerite Li1-x(Fe3+xFe2+1-x)PO4 –sicklerite Li1-x(Mn3+xMn2+1-x)PO4, and heterosite (Fe3+,Mn3+)PO4–purpurite (Mn3+, Fe3+)PO4 series, besides the Na analogues natrophilite, Na(Mn,Fe)PO4, and karenwebberite, Na(Fe,Mn)PO4, have olivine-type structures with alkali cations occupying positions in the core of “olivine tape” and atoms of transition metal in the “teeth” (Figure 5a). The movement of alkali ions along these tapes defines intercalation chemistry of the corresponding phosphates.
There are two natural sodium phosphates that are isotypic with lithiophilite-triphylite series’ members, namely karenwebberite, Na(Fe,Mn)PO4, and natrophilite, Na(Mn,Fe)PO4. Karenwebberite is an extremely rare mineral; it was found in 2011 as a late-magmatic-stage exsolution product within graftonite nodules in association with Na-bearing ferrisicklerite and a heterosite-like phase [51]. Karenwebberite is not a thermodynamically stable phase, which means it cannot be obtained using conventional chemical methods. The obvious reason for the thermodynamic instability of both the mineral and its synthetic analogues is that Na+ ions are too large to occupy positions in the centers of the octahedra forming the olivine tape core. In the structures of the minerals of the triphylite-lithiophilite series, these sites are populated by smaller Li+ ions. The same paradox, which was already analyzed in 1972, is connected with the crystal structure of natrophilite, NaMnPO4. At that moment it was the only representative of an ordered olivine type structure with the M1 sites populated by cations (Na+) that are significantly larger than the cations in the M2 sites (Mn2+), with a radius ratio of rM1:rM2 = 1.24 [52]. Similar to karenwebberite, the natrophilite deposits are single. They are known in granitic pegmatites, where the mineral is associated with lithiophilite in accordance with the conclusion of Moore [52]: “Since Mn2+ is more resistant than Fe2+ to oxidation in crystals and in solution, potential natrophilite occurrences should be found only where the lithiophilite member prevails; such localities are few and the products are at least partially oxidized”.
Triphylite-type NaFePO4 can be prepared by an ion-exchange method from LiFePO4 [53]: Li-ions are chemically or electrochemically extracted from triphylite, LiFePO4, forming heterosite, FePO4. Further chemical (and electrochemical) Na intercalation into FePO4 heterosite results in the formation of triphylite-type NaFePO4. This phase is stable below 480 °C; above this temperature, it transforms into marićite, NaFePO4 [54]. In a Na-cell, it reversibly de/intercalates nearly one mole of Na ions with two voltage plateaus on the discharge curve (at 2.87 and 2.97 V), which originate from the appearance of an intermediate sodium-ordered phase [55].
A karenwebberite dimorph, the thermodynamically stable orthorhombic mineral marićite, Na(Fe,Mn)PO4 [56], may be considered, from the structural point of view, as a triphylite derivative with Fe2+ ions occupying positions at the symmetry centers (M1) and Na+ ions placed in the positions with m symmetry (M2), just opposite to the distribution of these atoms in the crystal structure of karenwebberite (Figure 5). This kind of structure transformation leads to strong distortion of hexagonal close packing of O atoms inherent to the triphylite structure. The reasons for this distortion are the large Na+ cations, which transform the olivine tape when incorporated in its teeth through sharing two neighboring faces of NaO6 with FeO6 octahedra (Figure 5c). The reconstruction leads to the growth of the unit cell b parameter parallel to the olivine tape, compared to that of the triphylite unit cell, and lowering of the a parameter perpendicular to the tape [57]. As shown experimentally in [58], the polymorphic transformation from marićite-type to olivine-type crystal structure occurs in the region of 325 °C (P = 100 bars). According to [51], following the Fe2+ oxidation mechanism, karenwebberite transforms into a dark brown Na analog of ferrisicklerite, Na1−x(Fe3+,Mn2+)(PO4).
Synthetic marićite has been considered as electrochemically inactive because sodium sites are isolated in the structure, and a large barrier to sodium migration is expected [54]. The structural transformation associated with the opposite distribution of alkali and transition metal atoms to that of karenwebberite locks the channels along the {010} direction preventing movements of Na-ions. The alternative channels parallel to the {011} direction with small open windows (Figure 6) are also characterized by high activation energy for Na-ion diffusion (>1.5 eV, in contrast to values ranging between 0.2 and 0.6 eV in conventional battery materials) [59]. It was found that upon first charge in a Na-cell, marićite, NaFePO4, with nanoparticle morphology undergoes structural transformation, resulting in nucleation and growth of the amorphous FePO4, and this transformed phase delivers a capacity of 142 mAh·g−1 (92% of the theoretical value) and shows remarkable cyclability with a negligible capacity fade [59].
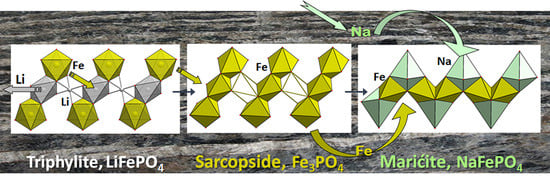
One more mineral structurally related to triphylite, sarcopside, Fe3[PO4]2, has never been observed as a primary crystal phase [60]. According to Moore, 1972 [60], sarcopside is an exsolved phase associated either with graftonite or triphylite. The sarcopside crystal structure is based on hexagonal close packing of oxygen atoms in which three-eighths of the octahedral and one-eighth of the tetrahedral voids are filled. It may be interpreted as the triphylite’s derivative. The structural transformation from triphylite to sarcopside is reconstructed as Li leaching from the M1 positions in the octahedra of the “olivine tape” and subsequent ordered occupation of one half of them by Fe atoms (Figure 5b). The ordered distribution of Fe inside the core of the tape dictates a decrease in symmetry from orthorhombic (in triphylite) to monoclinic (Figure 5b). The crystal chemical ordering relationship between triphylite and sarcopside may be described after Moore, 1972, as:
triphylite, Li4Fe4(PO4)4 (sp. group Pnma)→sarcopside (□2Fe2)Fe4(PO4)4 (sp. group P1121/a), where □ indicate ordered vacancies; Z = 1.
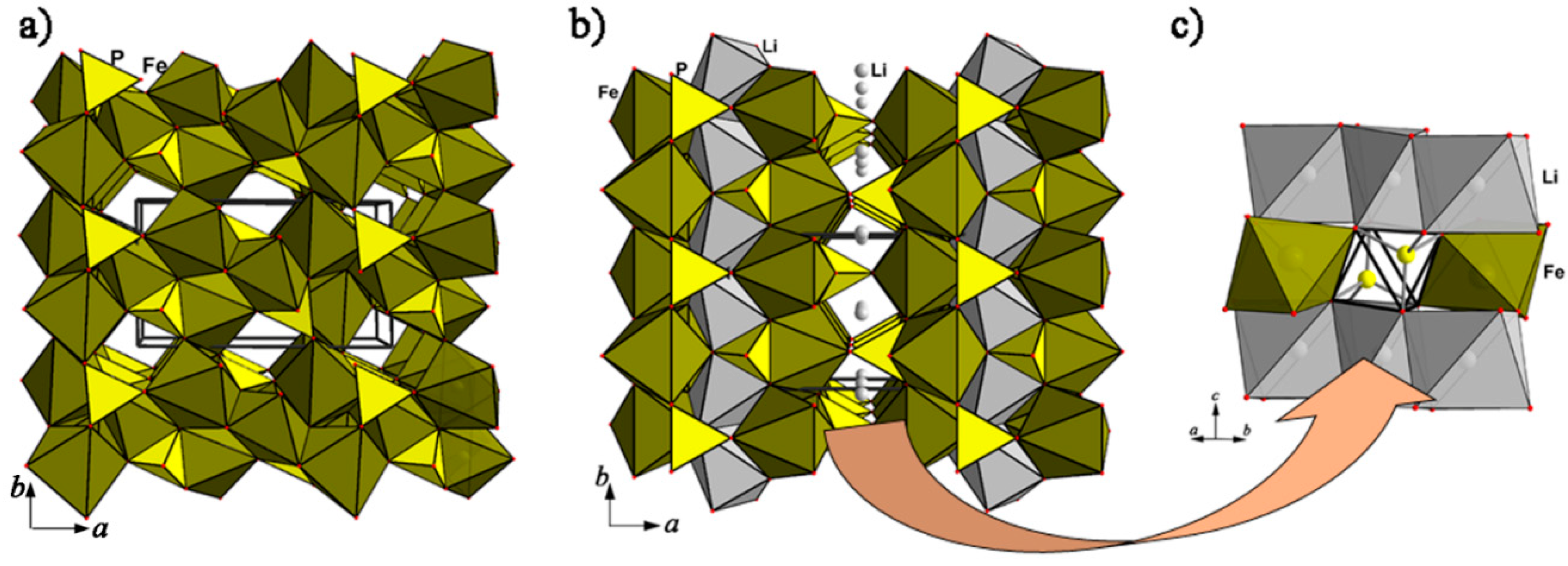
The similarity of triphylite, sarcopside, and heterosite crystal structures is an obvious starting point for studies of the electrochemical characteristics of their synthetic analogues. Thus, to explain the positive effect of vanadium doping on Li-ion transport, the formation of sarcopside defects in LiVyFe1−3/2yPO4 material was suggested. Whittingham’s group confirmed that upon doping, vanadium indeed occupies Fe sites, but some Fe exists at Li sites within sarcopside clusters, leaving a large percentage of the tunnels free for lithium diffusion [61]. Sarcopside defects were also reported in a transmission electron microscopy study of single crystals of LiFePO4 with some excess iron (no more than 1%) compared to the stoichiometric composition [62].
As one can see on comparing the triphylite and sarcopside crystal structures presented in the same projection (Figure 7), the closure of Li migration paths along the b axis during heterovalent substitution by Fe can be at least partly compensated by the remaining channels parallel to the {001} direction (Figure 7a,b). These channels are formed by Li-centered octahedra alternating along the c axis with small interstitial cavities of an octahedral shape, which share two opposite faces with phosphate tetrahedra (Figure 7c). However, participation of these interstitial cavities in Li-ion diffusion is unlikely, even as intermediate positions, because of the high activation energy revealed by computational studies [31,32].
Peculiarities of the hydrothermal synthesis of the lithium iron phosphate cathode material were studied in situ, by Calvet calorimetry and appearance of the intermediate with sarcopside structure was indicated. It was found that depending on the initial precursor concentration, the formation of this “antisite iron phosphate” and the reaction with Li+ to produce the final LiFePO4 phase can take place as two consecutive stages or as merging of these two stages into one. The observed changes in the reaction mechanism result in profound difference in the unit cell parameters of the triphylite phase, particle morphology, and electrochemical properties of the obtained LiFePO4 samples [35].
According to [63], direct thermal oxidation of sarcopside, Fe3(PO4)2, above 450 °С, makes it possible to obtain heterosite, FePO4, according to the reaction Fe3(PO4)2 + ¾ O2 → 2 FePO4 + ½ Fe2O3, which leads to the extraction of iron from the sarcopside structure. The authors note that the oxidized product demonstrates electrochemical performances consistent with those of heterosite, FePO4.
4.2. Other Phosphate-Based Series
4.2.1. Mineral Kosnarite and Structurally Related Phosphates
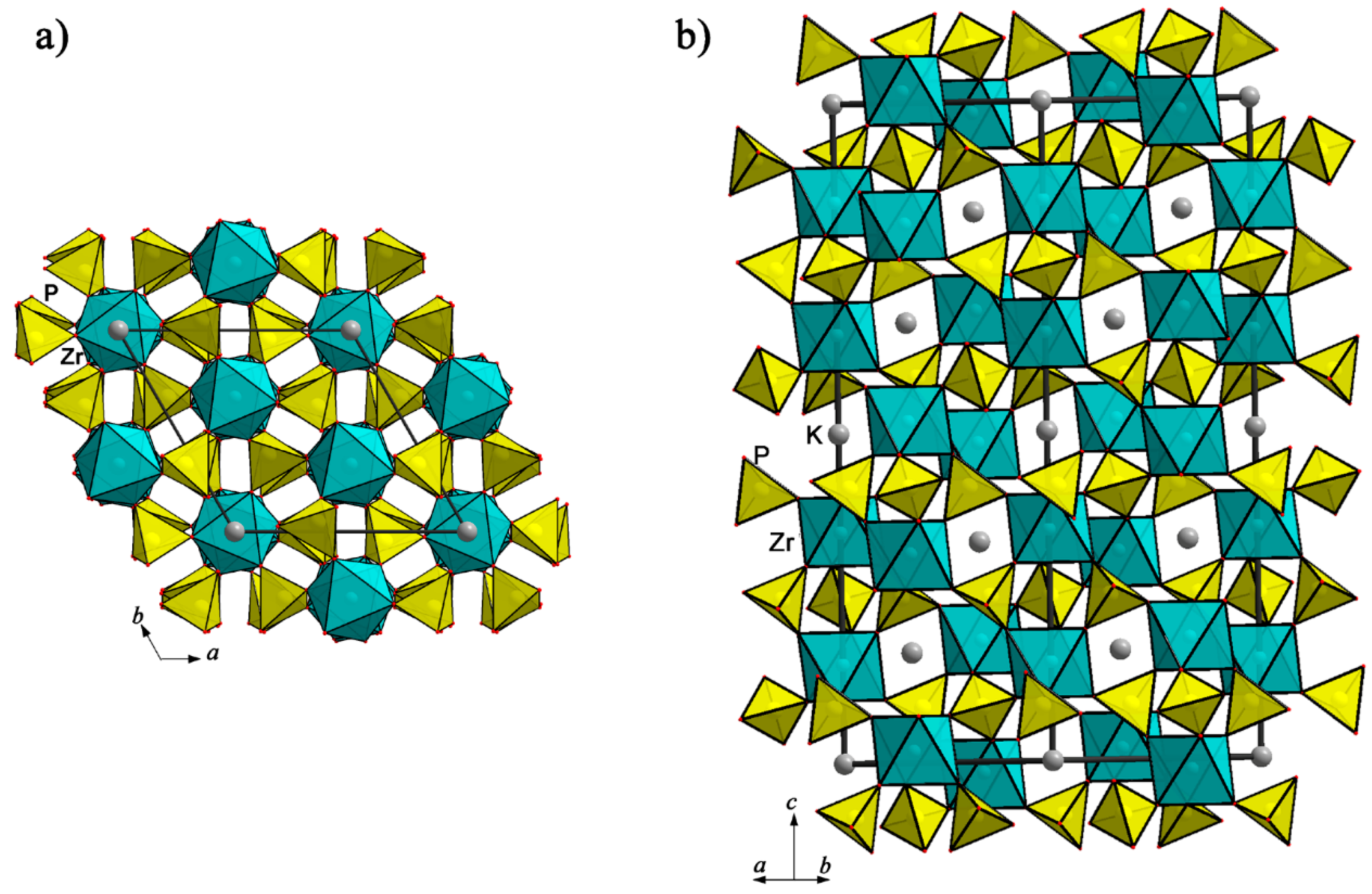
A late alteration mineral of granitic pegmatites, kosnarite, KZr2(PO4)3, was discovered in the early 1990s as part of a late-stage, secondary phosphate mineral assemblage [64]. The crystal structure of its synthetic analogue was determined in 1969 [65]. The kosnarite crystal structure presents a 3D framework built from ZrO6 octahedra and PO4 tetrahedra linked through oxygen-bridge contacts (Figure 8). Each octahedron shares all six oxygen vertices with tetrahedra, and conversely, each tetrahedron shares four vertices with the surrounding octahedra. The mixed-type anionic framework [Zr2P3O12]− is dense in the whole unit cell except at , where holes exist between the zirconium polyhedra. The K+ cations are located in these holes at the centers of trigonal antiprisms surrounded by six O.
A large group of synthetic phases with crystal structures close to that of kosnarite belongs to the so-called NASICON family (Na Super Ionic CONductor). Depending on the valence state of the atoms, forming a mixed anionic framework in association with PO4 tetrahedra, and on their size, the number of alkali cations in the kosnarite-type structure changes from zero to five per formula unit. Li-containing phosphate compositions, Li3M2(PO4)3 (M = Fe, V), adopt two distinct crystallographic forms: rhombohedral (R) referred to as NASICON, and monoclinic (P21/n), referred to as anti-NASICON, which differ in the way the M2(PO4)3 lantern units are connected [8]. The monoclinic phase is the stable form, prepared via solid-state reaction, whereas the rhombohedral Li3M2(PO4)3 is obtained through ion exchange from the NASICON sodium analogues, Na3M2(PO4)3 (M = Fe, V) [66,67]. Thus, two rhombohedral crystal structures of homeotypes of kosnarite, Na3M2(PO4)3 [68] and Li3M2(PO4)3, have the same 3D framework [M2P3O12]. The main difference is connected with the quantity, distribution, and coordination numbers of Na or Li atoms in the framework’s cavities. According to [69], there are two symmetrically independent sites for Na atoms in the Na3M2(PO4)3 kosnarite-type structure crystallizing in the same Rc space group, both partly populated by Na atoms with six and eight oxygen atoms in their first coordination sphere. Differently, Li atoms in tetrahedral coordination occupy one general type position in the framework cavities. In recent years, polyanionic frameworks within the NASICON structural family were heavily investigated due to their specific 3D framework structure, stable long-term cycling ability, and high alkali ion diffusion rate [70].
Rhombohedral Li3V2(PO4)3 with the V3+/V4+ redox couple has been identified as a high voltage cathode material with a theoretical capacity of 131 mAh·g−1, corresponding to the extraction of two Li+ ions. It demonstrates discharge curves with the flat plateau at around 3.75 V (vs. Li/Li+), while the monoclinic phase exhibits a more complicated behavior with multiple transitions in the discharge curves [8]. The reversible Li-insertion into rhombohedral phase at an average potential of 1.75 V (vs. Li/Li+) was observed, indicating that it can be applied as an anode material as well. The material delivered capacity values close to theoretical values for both types of activities (121 mAh·g−1 for the cathode and 117 mAh·g−1 for the anode), with excellent rate capability and long cycling performance [71]. In contrast to the rhombohedral phase, monoclinic Li3V2(PO4)3 exhibits reversible extraction of three Li ions, with activation of the V3+/V4+ and V4+/V5+ redox couples, which corresponds to high specific capacity (197 mAh·g−1). Nevertheless, properties of this material will not be discussed, because although the structure of monoclinic Li3V2(PO4)3 is closely related, it still differs from the structure of kosnarite.
Na3V2(PO4)3 was shown to be active toward Na+ extraction and insertion of additional sodium. Approximately 2 mol of Na is extracted from Na3V2(PO4)3 with a flat operating voltage of 3.3 V (vs. Na/Na+). Insertion of one additional mole of Na+ into Na3V2(PO4)3 based on the V3+/V2+ redox at around 1.5 V may be applied for the negative electrode performance. Today, this material has been identified as interesting NIB cathode material, possessing satisfactory energy density and high power for an extended cycle life. A large number of special approaches (carbon coating, control of particle shape) have been proposed and applied to improve its electrochemical performances [70]. Complete extraction of 2 Na per formula unit, providing a theoretical capacity of 117 mAh·g−1, is based on the V3+/V4+ redox couple, and deeper deintercalation with activation of the V4+/V5+ redox couple at higher voltages may also contribute to increasing the energy density of Na3V2(PO4)3 materials. To enhance specific energy storage characteristics, various substitutions on the V-site have been investigated, and a new Na4MnV(PO4)3 cathode material has been suggested. In Na4MnV(PO4)3, two redox couples of Mn2+/Mn3+ and V3+/V4+ were accessed with two voltage plateaus located at 3.6 and 3.3 V (vs. Na/Na+) and a maximum capacity value of 101 mAh·g−1. This material demonstrated long cycling stability (over 1000 cycles) and excellent rate performance, attributed to the uniform morphology of the nanosize particles and to high-speed Na+ diffusion in the NASICON framework structure [72]. It was found that an additional Na+ can be extracted from the Na4MnV(PO4)3 structure, reaching a higher capacity of 156 mAh·g−1 with a new redox activity located at around 3.86 V (vs. Na/Na+) [73]. It is associated with “unlocking” of the Na1 site in the rhombohedral phase. However, this additional activity is accompanied by relatively fast capacity degradation upon cycling and should be further investigated [74].
4.2.2. Alluaudite- and Wyllieite-Type Compounds
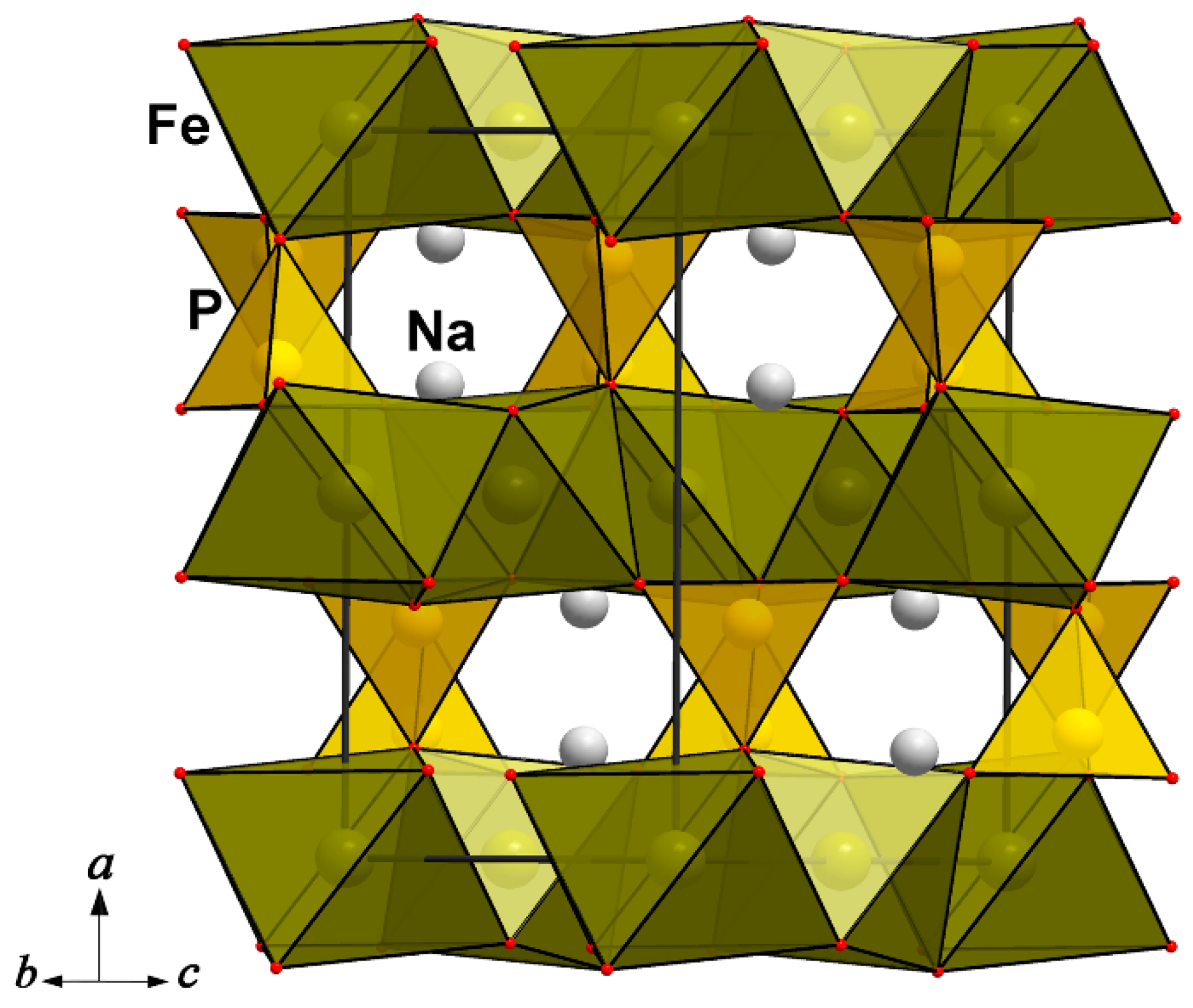
At the moment, the alluaudite mineral group counts 30 phosphate and arsenate members, mainly of alkaline and transition metals [75]. Phosphate representatives of the group are associated with pegmatites, while arsenates are found in volcanic deposits and zones of hypergenesis. The first solution of the crystal structure of alluaudite, NaCaMnFe2(PO4)3, was performed by P.B. Moore [76]. According to Moore’s paragenetic tree, alluaudites are derivatives of triphylite, lithiophilite, ferrisicklerite, and heterosite by Na-Li metasomatic exchange; most natural samples involve mixed Fe valences and are derivatives of the composition Na2Mn2+(Fe2+Fe3+)(PO4)3. This conclusion is supported by a well-known paragenesis of triphylite, LiFePO4, and alluaudite in pegmatites, and the mineral ferroalluaudite, Na2Fe3+Fe2+2(PO4)3, can be considered the end product of this transformation. Many experimental proofs of the triphylite → alluaudite transformation [77,78] lie in Moore’s paradigm of the continuous alteration of triphylite into alluaudite following the scheme «Li3Fe2+3(PO4)3 + 2Na → Na2Fe3+Fe2+2(PO4)3 + 3Li». This process is accompanied by a triple increase in the unit-cell volume of alluaudite (in comparison with triphylite) and a lowering of symmetry from orthorhombic (in triphylite) to monoclinic (in alluaudite). During the conversion process, layers of FeO6 octahedra sharing vertices in the triphylite structure became transformed into columns of edge-sharing octahedra in the crystal structure of alluaudite. Despite the difference in the assemblies of the Fe-centered octahedra (2D in triphylite and 1D in alluaudite), their 3D heteropolyhedral frameworks contain open channels elongated in one direction (Figure 9).
The first alluaudite analog, Na2(Fe3+0.5Fe2+0.5)2Fe2+(PO4)3, was obtained in the laboratory under hydrothermal conditions in 1977 [79], and analysis of its crystal structure revealed the new cation site on the twofold axis, populated by Na atoms. Synthetic materials with the alluaudite-type compounds are being actively studied today. The alluaudite-type structure provides not only fast transport of alkali ions (both Li+ and Na+) but also allows for substantial variations in the chemical composition (from Na2M2(SO4)3 to Na2M3(PO4)3, M = Fe, Mn) through substitutions in the anionic and cationic positions. The most famous representative, Na2Fe2(SO4)3, delivers a reversible capacity of 100 mAh·g−1 (85% of the theoretical one) with the working potential located at 3.8 V (versus Na/Na+), offering excellent rate kinetics and cycling stability [80]. However, its application is limited by the chemical instability (air sensitivity) and difficult preparation. By contrast, phosphate alluaudite-type materials can be easily synthesized using solid-state reaction or hydrothermal methods and have high chemical stability but demonstrate poorer electrochemical performance. Na2Mn2V(PO4)3 exhibits electrochemical activity at an average voltage of 3.5 V (vs. Na/Na+), associated with the combination of Mn2+/Mn3+ and V3+/V4+ redox couples; the discharge capacity of 97 mAh·g−1 corresponds to 89% of the theoretical one and fades quickly at different current rates [81]. The electrochemical properties of the Li-containing “alluaudites” Li0.5Na0.5MnFe2(PO4)3 and Li0.75Na0.25MnFe2(PO4)3 as positive electrodes in Li-cells were also studied: at an average potential of 2.7 V, they demonstrated reversible insertion of 0.7 and 1.3 Li+, respectively; these values are smaller than 1.7 Li+ intercalated into the non-lithiated NaMnFe2(PO4)3 [82].
The mineral wyllieite with the ideal formula Na2Fe2+2Al(PO4)3 was firstly described in [83] as a primary crystallized phase in pegmatites. Later, this type of wyllieite received the name ferrowyllieite [84] and the real wyllieite occurs as a Mn2+ variety, namely Na2Mn2+Fe2+Al(PO4)3. Its crystal structure has been interpreted as very close to that of alluaudite, Na2Mn2+(Fe2+Fe3+)[PO4]3. The main crystallographic difference between alluaudite and wyllieite having similar parameters of monoclinic unit cells is related to the type of Bravais lattices: the space group P21/n (P21/c) typical for wyllieite is a subgroup of C2/c (I2/a) of alluaudite. The space group reduction results from ordering over equivalent positions in the alluaudite structure type. The chains of edge sharing octahedra are traditionally considered as a basic fragment of the wyllieite and alluaudite structures. The way of cation distribution in the octahedral sites within these chains causes the formation of the first or the second structure type. The central M(1) octahedron in the triplet of edge sharing polyhedra in both structures is usually occupied by M2+ cations. The terminal M(2) position, which is jointly occupied by M2+ and M3+ cations in alluaudite, splits into M(2a) and M(2b) in wyllieite, with M(2b) filled mainly by M3+ cations(Figure 10a). In most crystal structures of the wyllieite mineral group, which counts seven members, this terminal position contains Al3+ and Fe3+ cations, with only one exception, that of fupingqiuite, (Na,Mn2+,□)2Mn2+2Fe3+(PO4)3, where it is fully populated by Fe3+ cations [85].
A synthetic member in the wyllieite group with an ideal formula of NaMn2+2.5Mn3+(PO4)3 was obtained under hydrothermal conditions. It is a new variety containing exclusively Mn atoms, besides Na+ as alkaline cations. Triplets of edge sharing Mn(2)-Mn(1)-Mn(3) octahedra form chains parallel to the {210} direction. These chains are linked by Mn(4) polyhedra to form an octahedral framework. The PO4 tetrahedra strengthen the framework by sharing all vertices with Mn octahedra. Na atoms occupy interstices of the structure (Na(2)) and channels parallel to the a axis (Na(1)) (Figure 10). The accommodation of Mn(4) atoms in the structure leads to the transformation of the octahedral layer motif to that of a framework in synthetic Mn-wyllieite [86]. Unlike the minerals of the wyllieite group, the NaMn2+2.5Mn3+(PO4)3 contains Mn3+ cations in the position usually occupied by Al (or Fe3+). As can be seen from Figure 10, the wyllieite crystal structure allows the migration of Na+ ions through the open channels. The theoretical capacity value of NaMn2+2.5Mn3+(PO4)3 is calculated to be 107 mAh·g−1 if processes related to the deintercalation of Na+ as well as the insertion of additional sodium into the structure are considered. The relatively low capacity might be compensated by a high potential associated with the Mn2+/ Mn3+ redox couple, thus making this material quite interesting for further exploration.
There are two synthetic arsenate representatives of the wyllieite family, to our knowledge, Ag1.09Mn3.46(AsO4)3 [87] and Na1.25Co2+2.187Al1.125(AsO4)3 [88], obtained by solid state reaction. The crystal structure of the second compound with an ideal formula of NaCo2+2.5Al3+(AsO4)3 is based on a 3D anionic framework built by (Al/Co)O6 octahedra and AsO4 tetrahedra, with Na+ cations in channels. According to Marzouki et al. [88], the wyllieite-type arsenate crystal structure encloses an additional new Na+ site as compared to other members of the family. The modeling of pathways for the Na+ migration through the anionic framework demonstrated that only the sodium called Na1 can diffuse through an infinite 1D pathway along the {100} direction. Considering the high activation energy and rather low theoretical capacity of this synthetic arsenate (87 mAh·g−1), and more importantly, the environmental impact, further investigation of these compounds as electrode materials for NIBs is not feasible.
4.2.3. Minerals with Two Sorts of Complex Anions: Sidorenkite, Na3Mn(PO4)(CO3) and Bonshtedtite, Na3Fe(PO4)(CO3)
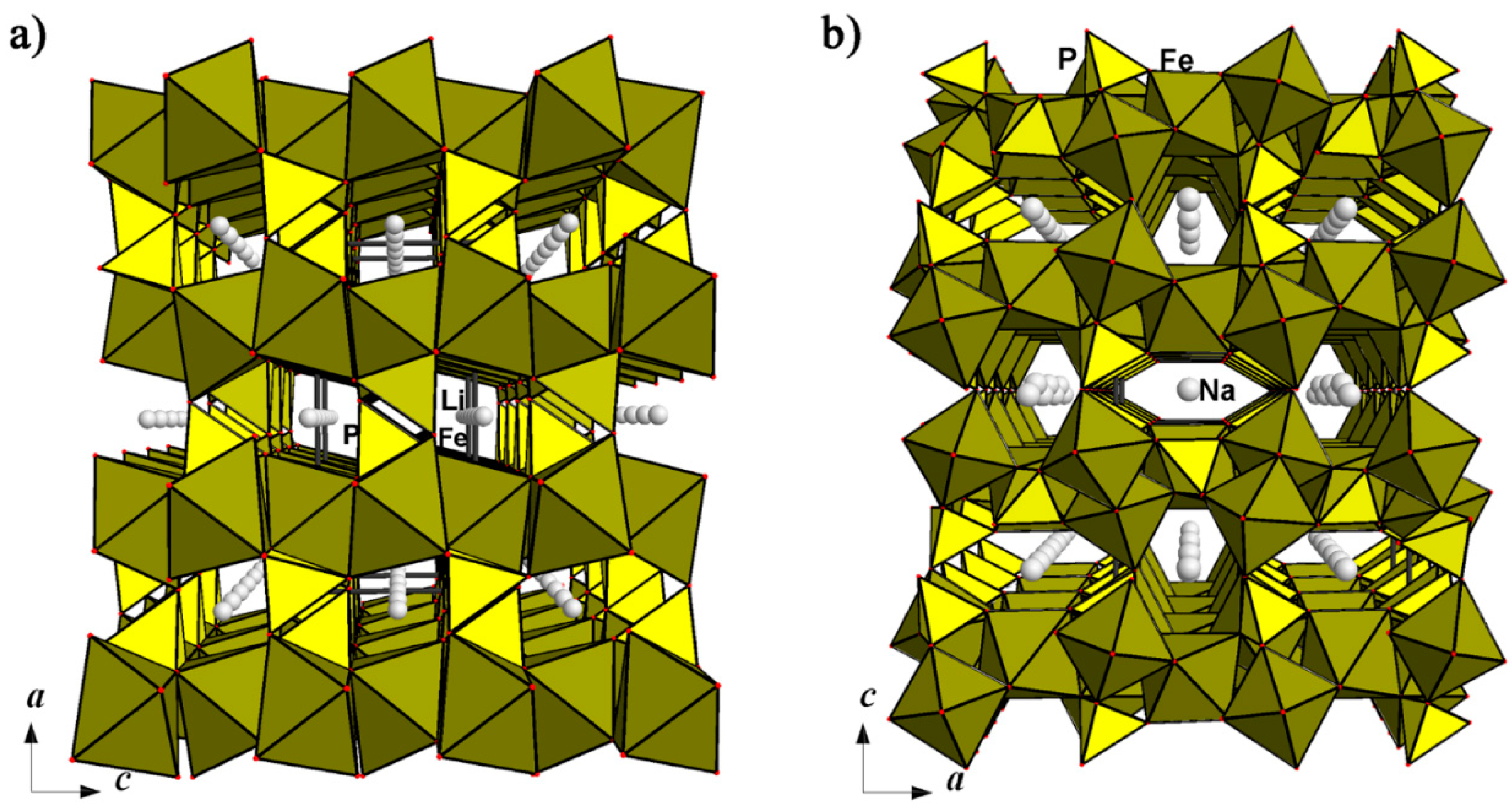
Sidorenkite and bonshtedtite are members of the bradleyite mineral group, which unites phosphate-carbonates with the general formula Na3M(PO4)(CO3). Sidorenkite, Na3Mn(PO4)(CO3), and its iron analogue bonshtedtite, Na3Fe(PO4)(CO3), are low-temperature hydrothermal minerals found in the highly alkaline hydrothermalites of the Khibiny massif, Kola Peninsula [89,90]. The minerals’ crystal structures were solved and refined by [91] and [92]. The MO6 octahedra and PO4 tetrahedra share oxygen vertices to form layers parallel to the yz plane of monoclinic (pseudo orthorhombic) structures. On the outside, along the a axis, these layers are inlaid with CO3 groups sharing an edge with M-centered octahedra (Figure 11).
Investigation of carbonate-phosphates as candidates for NIB electrode materials was initiated by ab initio computations, which predicted good intercalation characteristics of sidorenkite with the theoretical capacity of 191 mAh·g−1, owing to its capability to deliver the two-electron transfer via Mn2+/Mn3+ and Mn3+/Mn4+ redox reactions [93]. A series of carbonate-phosphates, Na3M(PO4)(CO3) (M = Mg, Mn, Fe, Co, Ni, Cu, Sr), were synthesized hydrothermally [94]. It was found that two Na-ions are extracted from Na3Mn(PO4)(CO3), resulting in a high initial charge capacity (∼200 mAh·g−1); however, the sample showed a relatively large irreversible capacity (∼75 mAh·g−1) at the initial discharge. At a slow rate, it delivered a stable capacity of over 125 mAh·g−1 at an average potential of 3.2 V (vs. Na/Na+) through a solid-solution-type structural evolution.
Na3Mn(PO4)(CO3) electrode materials with improved reversible capacity were reported [95,96]. Thus, Hassanzadeh et al. obtained a Na3Mn(PO4)(CO3)/rGO (reduced Graphene Oxide) nanocomposite with a reversible capacity of over 156 mAh·g–1; however, the electrode exhibited high charge/discharge polarization with fast capacity degradation.
A special hydrothermal technique was applied to obtain bonshtedtite, Na3Fe(PO4)(CO3), analogue with nanoplatelet morphology. The new material showed an initial capacity of 120 mAh·g−1 and demonstrated a stable capacity value of 96 mAh·g−1 after 50 cycles [97].
4.2.4. Phosphate Cathode Materials, Structurally Derived from Mineral Natisite, Na2TiSiO5
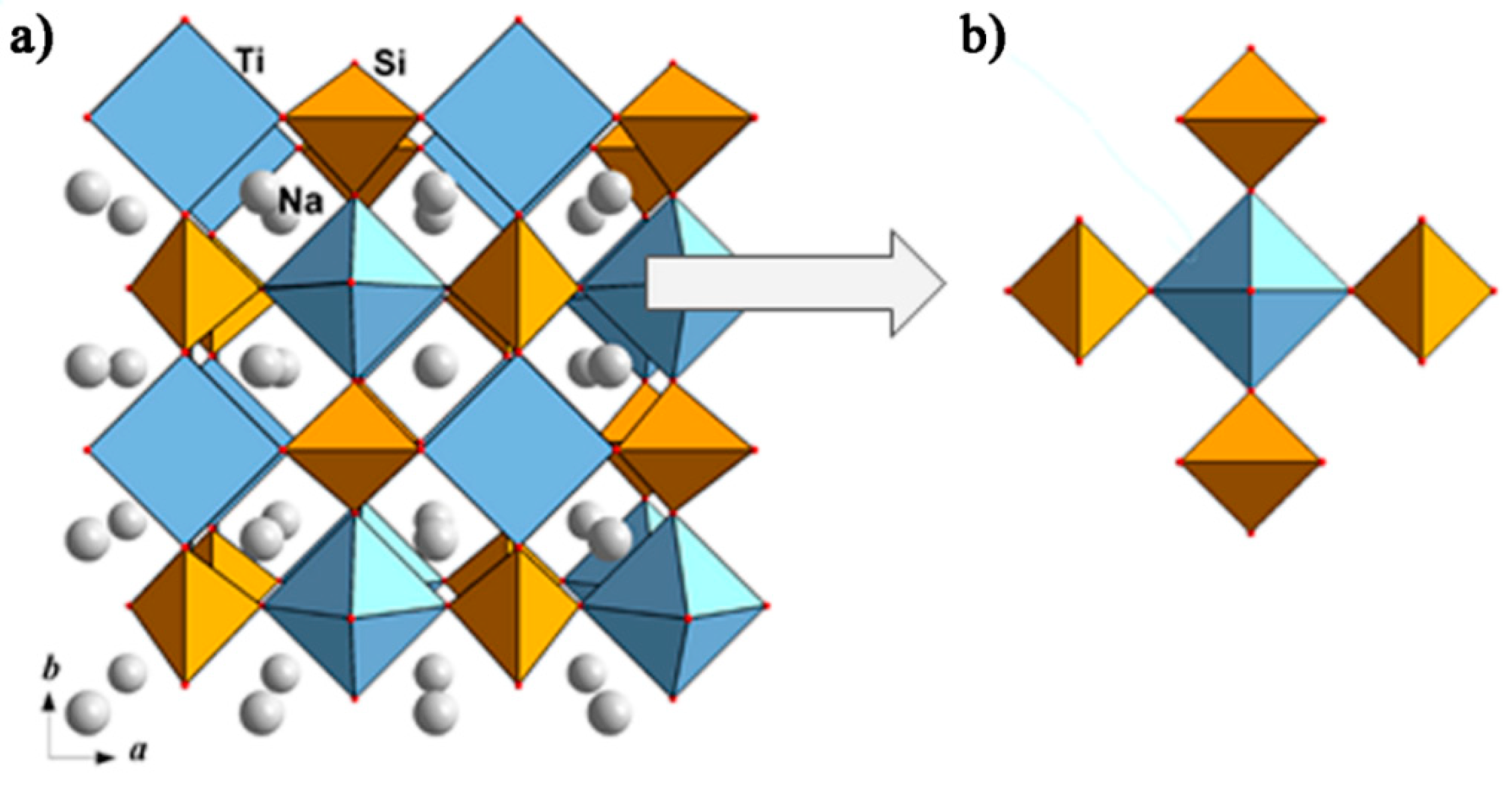
Natisite, Na2TiSiO5, is a typical mineral of ultra-agpaitic pegmatites and hydrothermalites; it can be treated as an indicator of highly alkaline conditions. It was found and described in [98]. The crystal structure of natisite contains layers of TiO5 hemi-octahedra (the sixth Ti-O distance is about 3.393 Å) and SiO4 tetrahedra with Na atoms between the layers [99]. The layer is a four-connected net (a square with alternating corner-sharing Ti pyramids and Si tetrahedra) (Figure 12a). Each Ti hemi-octahedron shares its four equatorial O atoms with four different SiO4 groups; thus, forming an essential building block (Figure 12b).
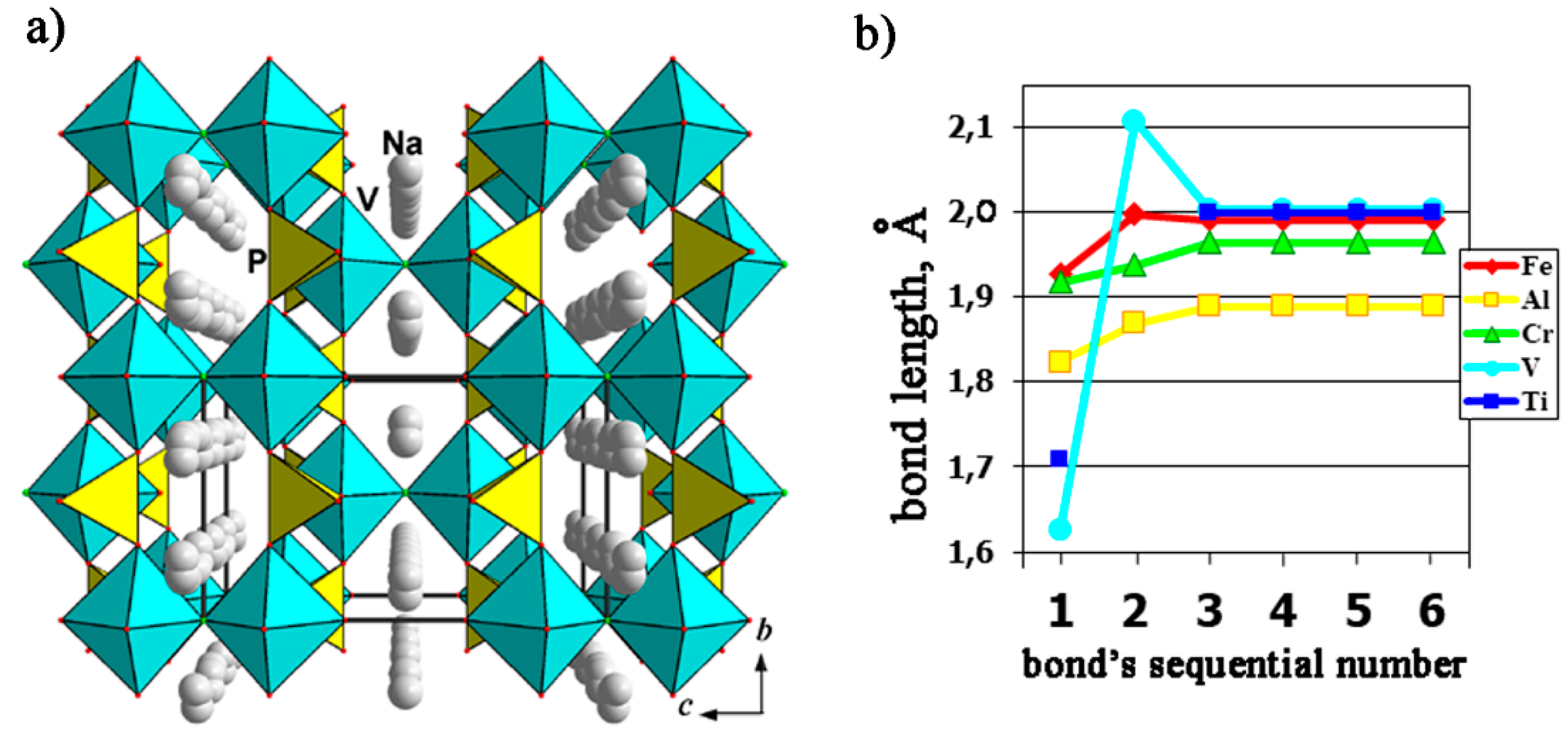
A number of phosphate crystal structures can be considered as derivatives of the mineral natisite, Na2TiSiO5, for example, Na3[Fe2(OH)2F(PO4)2] [100], Na3[Cr2F3(PO4)2] [101], Na3[Fe2F3(PO4)2] [102], Na3[Al2F3(PO4)2] [103], and Na3[V2O2F(PO4)2] [104]. They differ from natisite mainly by the degree of association of the mixed anionic substructure (layer or paraframework) and show symmetry relations of the “group–subgroup” type. The incorporation of F atoms in the interlayer space (with the simultaneous substitution of Si in tetrahedra for P, and Ti in octahedra for V) is accompanied by a transformation of 2D nets to 3D framework (layers connect along c trough common F-vertices of neighboring octahedra) in Na1.5{VOF0.5[PO4]}. Thus, the crystal structure of the V-containing member of the series, Na3[V2O2F(PO4)2], is formed by layers of alternating (VO5F) octahedra and (PO4) tetrahedra sharing O vertices. These layers are loosely interconnected along the c axis over the F-ligands, while the vanadyl O atoms remain terminal, and Na+ cations are disordered in the framework channels (Figure 13a). Strongly distorted (VO5F) octahedra with one short distance, V–O of 1.625 Å, characterizing the vanadyl group, correspond to rather regular MO4F2 (Fe, Al, and Cr) octahedra in other phosphate analogs. Figure 13b demonstrates the distortion of the octahedra in the structures under discussion.
Sodium-rich natisite-related Fe- and V-bearing fluoride phosphates, Na3V2O2(PO4)2F and Na3V2(PO4)2F3, are isostructural and form the Na3V2O2x(PO4)2F3−2x solid solution (0 ≤ x ≤ 1). They have drawn much attention as promising cathode materials for sodium-ion batteries: fluorine introduction effectively increases the voltage due to enhancement of the ionicity of the anionic framework; the reversible extraction of two Na+ corresponds to a theoretical capacity of 128 mAh·g−1. Although these compounds are active in both Li- and Na-cells, the electrochemical properties in the latter are the most attractive. Thus, the Na3V2O2x(PO4)2F3−2x materials were shown to demonstrate outstanding performance in the Na-cell, providing high energy densities (>500 W h kg−1) which are competitive with that delivered by LiFePO4 in LIBs [105,106]. According to [106], the reversible sodium de/intercalation in Na3V2O1.6(PO4)2F1.4 taking place at 3.65 and 4.02 V (vs. Na+/Na) is characterized by small volume changes (less than 3%) and low activation barriers for Na diffusion. It results in remarkable cycling stability and excellent rate capability (with capacity of more than 100 mAh·g−1 delivered at a 3 min charge/discharge rate). Comprehensive investigation of Na3V2O2x(PO4)2F3−2x revealed that the initial composition and thus the vanadium oxidation state have a major impact on electrochemical performance, with the de/intercalation mechanism evolving from two-phase behavior with two voltage plateaus for х = 0 to solid-solution type reactions with an “S-shape” for the two voltage domains for х = 1 [107].
4.2.5. Phosphate Analogues of Katiarsite, KTiOAsO4 and Yurgensonite, K2SnTiO2(AsO4)2, as Successive Electrode Materials for K-Ion Batteries
Two arsenate minerals, katiarsite, KTiOAsO4, and yurgensonite, K2SnTiO2(AsO4)2, were discovered recently in the volcanic fumaroles’ environment [108]. According to [109], yurgensonite, K2SnTiO2(AsO4)2, is the Sn/Ti-ordered analogue of katiarsite.
As it happens, their many synthetic structural analogues, collectively known as KTP (short for KTiPO5), were obtained, and studied more than 35 years ago. They are described by the common formula AM[XO4]Z, where A = K, Tl, Rb, Cs, Ag, and NH4; M = Ti, Fe, Al, Cr, Ge, Ga, Sb, Sn, Ta, Zr, and V; X = P, As, Si, and Ge; and Z = O and F. These compounds, being of interest as nonlinear-optical, ferroelectric, and superionic materials, recently began to be studied as possible candidates for batteries’ cathodes. A mixed-type anionic framework is built by chains of M-centered octahedra sharing cis- and trans-vertices and XO4 tetrahedra, linking these chains in a 3D construction through vertex-bridging contacts. The voids of the framework are filled by large single-charge cations (Figure 14).
Known as a good ionic conductor, this type of compounds has attracted attention as a possible material for metal-ion batteries. For the first time, unique electrochemical properties of KFeSO4F cathode material with KTP structure were discovered by Tarascon’s group [110]. Later, the V-bearing phosphates, KVPO4F and KVOPO4, were found to demonstrate attractive electrochemical performance. Thus, the K0.15VPO4F electrode (“VPO4F”) obtained from KVPO4F through electrochemical oxidation exhibited a reversible uptake of 0.7 Li per formula unit at an average voltage of ∼4 V vs. Li/Li+ and excellent rate capability, maintaining more than 50% of the theoretical capacity (> 80 mAh·g−1) at 90 second charge-discharge rate [111]. Moreover, the KTP-related “VPO4F” framework sustains reversible extraction/insertion of four alkali ions (Li+, Na+, K+, and even Rb+) with fast ionic mobility and high diffusion coefficients [112]. Solid-state transport properties of this framework were found to be very different for these alkali cations: the lowest values of the apparent diffusion coefficient were observed for lithium ions, while the diffusivity of K+ was shown to be the highest in the series. These findings confirmed the attractiveness of employing this material in sodium-ion and especially potassium-ion batteries [111].
The electrochemical performance of KVPO4F and KVOPO4 in K-cells was investigated [113,114]. These materials were reported to deliver reversible capacities of 92 and 84 mAh·g−1, respectively, with very good rate performance attributed to the open framework of the KTP type crystal structure. The average working voltages attributed to the V3+/V4+ and V4+/ V5+ redox couples were found to be very close: at 4.02 V for KVPO4F and 3.95 V for KVOPO4 [113].
The reversible K extraction/insertion from KVPO4F was confirmed by ex situ X-ray diffraction characterization and ab initio calculations, unveiling the formation of intermediate compounds at x = 0.75, 0.625, and 0.5 [114]. The authors also explained the effect of the F/O ratio on the electrochemical properties of KVPO4+xF1−x, suggesting that the oxygenation of KVPO4F resulted in a more disordered structure around the K and V sites and in a smooth voltage profile. In addition, they claimed that oxygen substitution for fluorine in KVPO4F decreases the operating voltage because of a reduced inductive effect in the compound with lower F-content.
Recently, a titanium-based electrode material, KTiPO4F, exhibiting a superior electrode potential of 3.6 V in a potassium-ion cell has been reported. KTiPO4F demonstrates a complex charge-discharge behavior characterized by three distinct reversible processes at 2.7, 3.1, and 3.6 V (vs. K/K+), and all three processes display plateau-like voltage profiles indicating a two-phase transformation mechanism. Carbon-coated KTiPO4F delivered a capacity of 94 mAh·g−1 (uptake of 0.8 K) at a rate of C/20 with high cycling stability. The authors suggested that a combination of chemical (inductive effect) and structural (charge/vacancy ordering, electrostatics) peculiarities of the electrode material results in extraordinarily high potential for the Ti3+/Ti4+ redox activity, thus making it an attractive cathode material for K-ion batteries; before this finding, the Ti3+/Ti4+ redox couple was considered for anode materials [115].
4.2.6. Arrojadite–Type Compounds
Mineral arrojadite, first described in the late eighteenth century [116] as an unknown mineral “near triphylite” from the Nickel Plate pegmatite in South Dakota, was not structurally characterized until the 1980s. After a first publication devoted to the structural study of arrojadite [117], the crystal structures of this mineral of diverse composition from different pegmatites have been investigated. According to [118], arrojadite is normally found as unaltered and unleached species.
The basis of the crystal structure of arrojadite, KNa4Ca(Fe,Mn)14Al(PO4)12(OH,F)2, presents a dense 3D framework built by Fe-, Mn-, and Al-centered polyhedra sharing edges. PO4 tetrahedra strengthen the framework by sharing vertices and edges with octahedra and five-vertex polyhedra populated by transition metal atoms or Al. Different cations occupy positions in the framework voids or channels, where they are often statistically distributed, having static or dynamic disorder [119].
To the best of our knowledge, there is only one report regarding the synthetic analog of arrojadite obtained. Brown-black prismatic crystals up to 1 mm in size were synthesized under hydrothermal conditions at T = 450 °C and P = 1000 atm [120]. As noted in [118], the role of statistics in the distribution of atoms is unusually important in the structure of arrojadite, as confirmed by the structure of its synthetic analogue, with fourteen disordered cation and two disordered anion positions. A main difference between the mineral and its synthetic counterpart lies in the chemical composition. Thus, in synthetic arrojadite, K2Na5Fe2+14 Fe3+(PO4)12(OH)2, the Fe atoms in a oxidation state of 3+ correspond to the Al atoms that usually occur in the composition of mineral phases. The largest negatively charged channels are open along the b axis through six-membered windows; they include Na+ and K+ cations. There are four such channels in the unit cell, parallel to each other (Figure 15). The theoretical capacity of K2Na5Fe2+14Fe3+(PO4)12(OH)2 is only 84 mAh·g−1, since half of the transition metals are not involved in the redox process, in addition to the weight penalty of the large oxoanion part.
The electrochemical activity of natural arrojadite Na3K1.22AlMn6Fe8.35(PO4)12(OH)2 was investigated for the LixAlMn6Fe8.35(PO4)12(OH)2 composition prepared by substitution of Na+ and K+ ions for Li+ [121]. In a Li-cell, the obtained sample delivered quite low capacity, probably due to the improper morphology of the electrode material. The author reported high thermal and cycling stability of the arrojadite material assigned to its framework crystal structure. Because of their small theoretical capacity, arrojadite-based materials can hardly be used as commercial cathodes; however, the features of their crystal structure might inspire hope for new, stable objects [121].
5. Conclusions
The given examples of the successful usage of mineralogical and crystal chemical information concerning phosphates (Table 1) to create modern battery materials can be supplemented by lipscombite, (Fe2+,Fe3+)3(PO4)2(OH,O)2, its ordered polymorph barbosalite, Fe2+Fe3+2(PO4)2(OH)2, minerals of the triplite-zwieselite, (Mn,Fe)2(PO4)F, and triploidite-wolfeite, (Mn,Fe)2(PO4)OH series, lithiophosphate, Li3PO4, and some others. In particular, we have highlighted above two candidates to test as possible cathode materials for Me-ion batteries: the wyllieite-like NaMn2+2.5Mn3+(PO4)3 and the arrojadite K2Na5Fe2+14 Fe3+(PO4)12(OH)2, along with their derivatives. Many recently discovered phosphate minerals may also stand as prototypes to create new battery electrode materials.
Although most current cathode materials for metal-ion batteries are phosphates, representatives of other mineral classes with complex anions, for example, silicates, sulfates, borates, and vanadates, might be considered in this respect; the above-discussed mineral natisite, Na2TiSiO5, is a good example. One more illustration may be given in connection with the rare minerals grandidierite, MgAl3(SiO4)(BO3)O2, and ominelite, FeAl3O2(SiO4)(BO3). Synthetic fluoride phosphates with the common formula A2M(PO4)F, where A = Li, Na and M = Ni, Fe, Co, Mn, Mg, extensively studied as promising cathodes for LIBs and NIBs (see, for instance one of the latest papers: [122]) are structural analogues of the mentioned natural silicate-borates [123].
Coming back to the problem of the search of novel materials for a battery cathode design, let us emphasize that data about mineral genesis contain important information on the conditions under which a particular structure was developed. The chemical compositions of mineral species indicate the types of atoms participating in their stable crystal structures, and the results of X-ray and neutron diffraction studies reveal the preferred positions for their population by definite ions. Subsequent topological analysis of crystal structures will facilitate identification of possible migration routes of ions and help to predict the electrochemical properties of compounds. Crystallographic and mineral databases provide useful information on phases with rigid framework structures, their chemical stability, and possible ion diffusion pathways within the framework. This suggests that mineral-like compounds, primarily phosphates and silicates, might be prototypes of novel electrode materials for metal-ion batteries. The widespread prevalence of silicates and phosphates in nature makes it possible, on the one hand, to obtain their synthetic analogues and, on the other hand, to guarantee the low cost and environmental safety of these materials.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Composition and crystal characteristics of synthetic analogues of minerals, studied as cathode materials.
Table 1.
Composition and crystal characteristics of synthetic analogues of minerals, studied as cathode materials.
| Formula (Mineral Name) | Unit-Cell Parameters a, b, c, Å and α, β, γ,o | Space Group, V Å3, g/cm3, Z | Ref. |
|---|---|---|---|
| 1. The olivine (Fe,Mg)2(SiO4) Structure Type and Its Derivatives | |||
| Fe End Members | |||
| LiFe(PO4) (triphylite) | a = 10.332(4), b = 6.010(5), c = 4.787(4) | Pnma, 297.25 3.53, 4 | [124] |
| NaFe(PO4) (karenwebberite) | a = 10.4063(6), b = 6.2187(3), c = 4.9469(3) | Pnma, 320.13 3.61, 4 | [53] |
| NaFe(PO4) (marićite) | a = 9.001(8), b = 6.874(3), c = 5.052(4) | Pnma, 312.58 3.69, 4 | [125] |
| Fe3(PO4)2 (sarcopside) | a = 10.450(11), b = 6.033(6), c = 4.782(5) γ = 90.96(8) | P1121/a*, 301.44 3.94, 2 | [126] |
| Fe(PO4) (heterosite) | a = 9.839(1), b = 5.809(3), c = 4,776(1) | Pnma, 272.97 3.67, 4 | [127] |
| NH4Fe(PO4)HF (Fe variety of niahite) | a = 8.958(2), b = 5.649(1), c = 4.811(1) | Pnm21, 243.45 2.58, 2 | [37] |
| Mn End Members | |||
| LiMn(PO4) (lithiophilite) | a = 10.46(3), b = 6. 10(2), c = 4.744(10) | Pnma, 302.7 3.44, 4 | [128] |
| NaMn(PO4) (natrophilite) | a = 10.523(5), b = 6.312(3), c = 4.987(2) | Pnma, 330.48 3.47, 4 | [52] |
| NaMn(PO4) (Mn analogue of marićite) | a = 9.0882(1), b = 6.9041(1), c = 5.1134(1) | Pnma, 320.84 3.58, 4 | [129] |
| Mn(PO4) (purpurite) | a = 9.6237(7), b = 5.9019(3), c = 4.7711(3) | Pnma, 270.99 3.67, 4 | [130] |
| NH4Mn(PO4)(H2O) (niahite centrosymmetric variety) | a = 17.5822(3), b = 5.7310(1), c = 4.9090(1) | Pnma, 494.65 2.50, 4 | [41] |
| 2. The Tavorite, LiFe(PO4)(OH,F) Structure Type | |||
| LiFe(PO4)(OH) (tavorite) | a = 5.3528(6), b=7.2896(7), c = 5.1187(5) α= 109.359(4), β= 97.733(5), γ=106.359(5) | P, 174.98 3.22, 2 | [49] |
| LiV(PO4)F (tavorite) | a = 5.3094(2), b=7.4994(2), c =5.1688(2) α= 112.93 (2), β= 81.66(2), γ=113.12(1) | P, 174.3 3.27, 2 | [9] |
| 3. The Kosnarite, KZr2(PO4)3 (NASICON) Structure Type (Rombohedral Analogues) | |||
| Na3Fe2(PO4)3 | a = 8.7270(2), c = 21.8078(5) | Rc, 1438.38 3.22, 6 | [69] |
| Na3V2(PO4)3 | a = 8.7288(2), c = 21.8042(7) | Rc, 1438.73 3.16, 6 | [68] |
| Li3Fe2(PO4)3 | a = 8.3162(4), c = 22.459(1) | R, 1345.15 3.09, 6 | [69] |
| Na4MnV(PO4)3 | a = 8.96354(2), c = 21.448319(7) | Rc, 1494.82 3.25, 6 | [73] |
| 4. The Alluaudite, Na2Mn2+(Fe3+, Fe2+)2(PO4)3 Structure Type | |||
| Na2Fe2+(Fe2+Fe3+) (PO4)3 (ferroalluaudite) | a = 10.923(1), b = 6.500(1), c = 12.538(2) γ = 98.3(1) | I112/a **, 890.19 3.71, 4 | [79] |
| (Li,Na)MnFe2(PO4)3 | a = 11.9892(2), b =12.4927(2), c = 6.3859(1) β = 114.639(1) | C2/c, 869.34 3.56, 4 | [82] |
| Na2Fe2(SO4)3 | a = 12.65847(7), b =12.77062(7) c =6.51210(3) β = 115.5391(4) | C2/c, 949.86 3.11, 4 | [80] |
| 5. The Wyllieite, Na2Fe2+2Al(PO4)3 Structure Type | |||
| NaMn2+2.5Mn3+(PO4)3 | a = 6.5291(6), b = 12.653(1), c = 10.952(1) β = 97.18(1) | P21/c, 897.68 3.74, 4 | [86] |
| Ag1.09Mn3.46(AsO4)3 | a = 6.7470(7), b =12.9820(9), c = 11.2970(8) β = 98.85(3) | P21/c, 977.72 4.92 4 | [87] |
| Na1.25Co2+2.187Al1.125(AsO4)3 | a = 6.532(2), b =12.492(2), c = 11.060(2) β = 99.44(2) | P21/c, 890.3 4 | [88] |
| 6. The bradleyite, Na3Mg(CO3)(PO4) Structure Type | |||
| Na3Mn(CO3)(PO4), (sidorenkite) | a = 8.9859(1), b=6.73577(5), c = 5.15998(4) β = 90.123(1) | P21/m, 312.31 2.97, 2 | [94] |
| Na3Fe(CO3)(PO4), (bonshtedtite) | a = 8.9542(1), b=6.61129(5), c = 5.16023(5) β = 89.600(1) | P21/m, 305.47 3.04, 2 | [97] |
| 7. The Natisite, Na2TiSiO5 Homeotypes | |||
| Na3Fe2(OH)2F(PO4)2 | a =9.050(2), c = 10.679(2) | P42/mnm, 874.64 3.22, 8 | [100] |
| Na3Fe2F3(PO4)2 | a =6.399(1), c= 10.679(3) | I4/mmm, 429.11 3.39, 2 | [102] |
| Na3V2O2F(PO4)2 | a =6.3856(2), c = 10.6119(9) | I4/mmm, 431.05 3.17, 2 | [104] |
| 8. The Katiarsite, KTiO(AsO4) (KTP) Structure Type | |||
| KFeF(PO4) | a = 12.885(6), b = 6.370(2), c = 10.656(4) | Pna21, 874.62 3.17, 8 | [131] |
| KVF(PO4) | a = 12.8200(3), b = 6.3952(1), c = 10.6115(2) | Pna21, 869.99 3.11, 8 | [110] |
| KVO(PO4) | a = 12.816(5), b = 6.388(2), c = 10.556(5) | Pna21, 864.21 3.09, 8 | [113] |
| KTiF(PO4) | a = 13.0020(2), b = 6.43420(8), c = 10.7636(2) | Pna21, 900.45 3.01, 8 | [115] |
| 9. The Arrojadite, KNa4Ca(Fe,Mn)14Al(PO4)12(OH,F)2 Structure Type | |||
| KNa5Fe2+14Fe3+(PO4)12(OH)2 (arrojadite) | a = 16.53(2), b = 10.084(9), c = 24.64(3) β = 105.75(7) | C12/c1, 3953 3.69, 4 | [120] |
* Crystal characteristics are given in non-standard setting of the space group P21/c for comprehensible comparison of sarcopside and triphylite crystal structures (see Figure 7). ** Crystal characteristics are given in non-standard setting of the space group C2/c for comprehensible comparison of ferroalluaudite and triphylite crystal structures (see Figure 9).
Author Contributions
Conceptualization, O.Y.; validation, N.K., E.A.; writing, original draft preparation, O.Y., N.K.; writing, review and editing, O.Y., N.K., E.A. All authors contributed equally. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Foundation for Basic Research (RFBR grant No. 18-29-12076 mk).
Conflicts of Interest
The authors declare no conflict of interest.
References
- The Nobel Prize. Available online: https://www.nobelprize.org/prizes/chemistry/2019/press-release/ (accessed on 22 April 2020).
- Kubota, K.; Dahbi, M.; Hosaka, T.; Kumakura, S.; Komaba, S. Towards K-Ion and Na-Ion Batteries as “Beyond Li-Ion”. Chem. Rec. 2018, 18, 459–479. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy K., S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Phosphates: Geochemical, Geobiological and Materials Importance. In Mineralogical Society of America and Geochemical Society Reviews in Mineralogy and Geochemistry; Kohn, M.J.; Rakovan, J.; Hughes, J.M. (Eds.) Walter de Gruyter GmbH & Co KG: Berlin, Germany, 2002; p. 742. [Google Scholar]
- Goodenough, J.B.; Kim, Y. Challenges for Li Rechargeable Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Mizushima, K.; Jones, P.C.C.; Wiseman, P.J.; Goodenough, J.B. LixCoO2 (0<x<−1): A new cathode material for batteries of high energy density. Mater. Res. Bull. 1980, 15, 783–789. [Google Scholar]
- Whittingham, M.S. Ultimate Limits to Intercalation Reactions for Lithium Batteries. Chem. Rev. 2014, 114, 11414–11443. [Google Scholar] [CrossRef]
- Masquelier, C.; Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef] [PubMed]
- Antipov, E.V.; Khasanova, N.R.; Fedotov, S.S. Perspectives on Li and transition metal fluoride phosphates as cathode materials for a new generation of Li-ion batteries. IUCr J. 2015, 2, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.C.; Bianchini, M.; Seo, D.-H.; Rodriguez-Garcia, J.; Ceder, G. Recent Progress and Perspective in Electrode Materials for K-Ion Batteries. Adv. Energy Mater. 2018, 8, 1702384. [Google Scholar] [CrossRef] [Green Version]
- London, D.; Kontak, D.J. Granitic Pegmatites: Scientific wonders and economic bonanzas. Elements 2012, 8, 257–261. [Google Scholar] [CrossRef]
- Moore, P.B. Pegmatite phosphates: Descriptive mineralogy and crystal chemistry. Mineral. Rec. 1973, 4, 103–130. [Google Scholar]
- Fisher, D.G. Geochemistry of Phosphorus Containing Minerals. In Environmental Phosphorus Handbook; Griffith, E., Beeton, A., Spenser, J., Eds.; Wiley: New York, NY, USA, 1973. [Google Scholar]
- Černý, P.; Ercit, S. The classification of granitic pegmatites revisited. Can. Mineral. 2005, 43, 2005–2026. [Google Scholar] [CrossRef] [Green Version]
- Keller, P.; Fransolet, A.-M.; Fontan, F. Triphylite–lithiophilite and triplite–zwieselite in granitic pegmatites: Their textures and genetic relationships. N. Jahrb. Mineral. Abh. 1994, 168, 127–145. [Google Scholar]
- Moore, P.B. Crystallochemical Aspects of the Phosphate Minerals. In Phosphate Minerals; Nriagu, J.O., Moore, P.B., Eds.; Springer: Berlin/Heidelberg, Germany, 1984; pp. 155–170. [Google Scholar]
- Moore, P.B. The primary pegmatite phosphate minerals. The mineralogy of pegmatites, Вrown, G.Е., Jr. Аmer. Miner. 1982, 67, 180–189. [Google Scholar]
- Mücke, A. The paragenesis of the phosphate minerals of the Hagendorf pegmatite—A general view. Chem. Erde. 1981, 40, 217–237. [Google Scholar]
- Sandomirskii, P.A.; Belov, N.V. Crystal Chemistry of Mixed Anionic Radicals; Nauka: Moscow, Russia, 1984. (In Russian) [Google Scholar]
- Yakubovich, O.V.; Urusov, V.S. The genetic crystal chemistry of pegmatite phosphates. Mosc. Univ. Geol. Bull. 1996, 51, 18–40. [Google Scholar]
- Yakubovich, O.V. Phosphates with amphoteric oxo-complexes: From structural features to genetic conclusions. Z. Krist. 2008, 223, 126–131. [Google Scholar] [CrossRef]
- Yakubovich, O.V. Phosphates with Amphoteric Oxocomplexes: Crystal Chemical Features and Expected Physical Properties In Minerals as Advanced Materials I; Krivovichev, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; p. 255. [Google Scholar]
- Shigley, J.E.; Brown, G.E. Lithiophilite formation in granitic pegmatites: A reconnaissance experimental study of phosphate crystallization from hydrous aluminosilicate melts. Am. Mineral. 1986, 71, 356–366. [Google Scholar]
- Eventoff, W.; Martin, R.; Peacor, D.R. The crystal structure of heterosite. Am. Mineral. 1972, 57, 45–51. [Google Scholar]
- Iakubovich, O.V.; Simonov, M.A.; Belov, N.V. Crystal structure of synthetic triphyline LiFe[PO4]. Dokl. Akad. Nauk SSSR 1977, 235, 93–95. [Google Scholar]
- Ravet, N.; Chouinard, Y.; Magnan, J.F.; Besner, S.; Gauthier, M.; Armand, M. Electroactivity of natural and synthetic triphilyte. J. Power Sources 2001, 97–98, 503–507. [Google Scholar] [CrossRef]
- Huang, H.; Yin, S.C.; Nazar, L.F. Approachim theoretical capacity of LiFePO4 at room temperature at high rates. Electrochem. Solid State Lett. 2001, 4, A170–A172. [Google Scholar] [CrossRef]
- Dominko, R.; Gaberšček, M.; Drofenik, J.; Bele, M.; Pejovnik, S. A Novel Coating Technology for Preparation of Cathodes in Li-Ion Batteries. Electrochem. Solid State Lett. 2001, 4, A187–A190. [Google Scholar] [CrossRef]
- Chung, S.-J.; Bloking, J.T.; Chiang, Y.M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mat. 2002, 1, 123–128. [Google Scholar] [CrossRef]
- Morgan, D.; Van der Ven, A.; Ceder, G. Li Conductivity in LixMPO4 (M =Mn, Fe, Co, Ni) Olivine Materials. Electrochem. Solid-State Lett. 2004, 7, A30–A32. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.; Slater, P.R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Nishimura, S.; Kobayashi, G.; Ohoyama, K.; Kanno, K.; Yashima, M.; Yamada, A. Experimental visualization of lithium diffusion in LixFePO4. Nat. Mater. 2008, 7, 707–711. [Google Scholar] [CrossRef]
- Qin, X.; Wang, X.; Xiang, H.; Xie, J.; Li, J.; Zhou, Y. Mechanism for Hydrothermal Synthesis of LiFePO4 Platelets as Cathode Material for Lithium-Ion Batteries. J. Phys. Chem. C 2010, 114, 16806–16812. [Google Scholar] [CrossRef]
- Sharikov, F.Y.; Drozhzhin, O.A.; Sumanov, V.D.; Baranov, A.N.; Abakumov, A.M.; Antipov, E.V. Exploring the Peculiarities of LiFePO4 Hydrothermal Synthesis Using In Situ Calvet Calorimetry. Cryst. Growth Des. 2018, 18, 879–882. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Xiang, K.; Xing, W.; Borkiewicz, O.J.; Wiaderek, K.M.; Gione, P.; Chapman, K.W.; Chupas, P.J.; Chiang, Y.-M. Extended Solid Solutions and Coherent Transformations in Nanoscale Olivine Cathodes. Nano Lett. 2014, 14, 1484–1491. [Google Scholar]
- Yakubovich, O.V.; Karimova, O.V.; Melnikov, O.K.; Urusov, V.S. Crystal structure of (NH4)(FePO4)HF-niahite synthetic variant. Dokl. Akad. Nauk SSSR 1995, 342, 40–44. [Google Scholar]
- Yakubovich, O.V.; Karimova, O.V.; Dimitrova, O.V.; Mass, A.W. Layer structure of (NH4)CoPO4 H2O. Acta Cryst. C 1999, 55, 151–153. [Google Scholar] [CrossRef]
- Bridge, P.J.; Robinson, B.W. Niahite—a new mineral from Malaysia. Mineral. Mag. 1983, 47, 79–80. [Google Scholar] [CrossRef]
- Livage, J. Chimie douce: From shake-and-bake processing to wet chemistry. New J. Chem. 2001, 25, 1. [Google Scholar] [CrossRef]
- Kiriukhina, G.V.; Yakubovich, O.V.; Dimitrova, O.V. Crystal structure of a new polymorphic modification of niahite, NH4MnPO4 H2O. Cryst. Rep. 2015, 60, 198–203. [Google Scholar] [CrossRef]
- Wu, C.; Xie, J.; Cao, G.; Zhao, X.; Zhang, S. Ordered LiMPO4 (M = Fe, Mn) nanorods synthesized from NH4MPO4·H2O microplates by stress involved ion exchange for Li-ion batteries. Cryst. Eng. Comm. 2014, 16, 2239–2245. [Google Scholar] [CrossRef]
- Bramnik, N.; Ehrenberg, H. Precursor-based synthesis and electrochemical performance of LiMnPO4. J. Alloys Compd. 2008, 464, 259–264. [Google Scholar] [CrossRef]
- Alyoshin, V.A.; Pleshakov, E.A.; Ehrenberg, H.; Michailova, D. Platelike LiMPO4 (M=Fe, Mn, Co, Ni) for Possible Application in Rechargeable Li Ion Batteries: Beyond Nanosize. J. Phys. Chem. C 2014, 118, 17426–17435. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Bairakov, V.V.; Simonov, M.A. Crystal structure of simferite Li(Mg,Fe3+,Mn3+)2[PO4]2. Soviet Physics Doklady. 1989, 34, 669–671. [Google Scholar]
- Omenya, F.; Miller, J.K.; Fang, J.; Wen, B.; Zhang, R.; Wang, Q.; Chernova, N.A.; Whittingham, M.S. Single-Phase Lithiation and Delithiation of Simferite Compounds Li(Mg,Mn,Fe)PO4. Chem. Mater. 2014, 26, 6206–6212. [Google Scholar] [CrossRef]
- Moore, P.B. Crystal chemistry of the basic iron phosphates. Am. Mineral. 1970, 55, 135–169. [Google Scholar]
- Yakubovich, O.V.; Urusov, V.S. The structure and electron density of Fe2+-bearing tavorite in relation to the genetic crystal chemistry of secondary phosphates of lithium pegmatites. Geochem. Intern. 1997, 35, 630–638. [Google Scholar]
- Marx, N.; Croguennec, L.; Carlier, D.; Wattiaux, A.; Le Cras, F.; Suard, E.; Delmas, C. The structure of tavorite LiFePO4(OH) from diffraction and GGA + U studies and its preliminary electrochemical characterization. Dalton Trans. 2010, 39, 5108–5116. [Google Scholar] [CrossRef]
- Barker, J.; Gover, R.K.B.; Burns, P.; Bryan, A. LiVPO4F: A new active material for safe lithium-ion batteries. Electrochem. Solid-State Lett. 2005, 8, A285–A287. [Google Scholar] [CrossRef]
- Vignola, P.; Hatert, F.; Fransolet, A.-M.; Medenbach, O.; Diella, V.; Andò, S. Karenwebberite, Na(Fe2+,Mn2+)PO4, a new member of the triphylite group from the Malpensata pegmatite, Lecco Province, Italy. Am. Mineral. 2013, 98, 767–772. [Google Scholar] [CrossRef]
- Moore, P.B. Natrophilite, NaMn(PO4), has ordered cations. Am. Mineral. 1972, 57, 1333–1344. [Google Scholar]
- Moreau, P.; Guyomard, D.; Gaubicher, J.; Boucher, F. Structure and Stability of Sodium Intercalated Phases in Olivine FePO4. Chem. Mater. 2010, 22, 4126–4128. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef]
- Casas-Cabanas, M.; Roddatis, V.V.; Saurel, D.; Kubiak, P.; Carretero-Gonzalez, J.; Palomares, V.; Serras, P.; Rojo, T. Crystal chemistry of Na insertion/deinsertion in FePO4–NaFePO4. J. Mater. Chem. 2012, 22, 1742–17423. [Google Scholar] [CrossRef]
- Le Page, Y.; Donnay, G. The crystal structure of the new mineral marićite, NaFePO4. Can. Mineral. 1977, 15, 518–521. [Google Scholar]
- Yakubovich, O.V.; Urusov, V.S. Electron density distribution in lithiophosphatite Li3PO4: Crystallochemical features of orthophosphates with hexagonal close packing. Cryst. Rep. 1997, 42, 261–268. [Google Scholar]
- Corlett, M.I.; Armbruster, T. Structural relations between marićite and natrophilite in the system NaFePO4-NaMnPO4. GAC-MAC Join Annu. Meet. 1979, 4, 1979. [Google Scholar]
- Kim, J.; Seo, D.-H.; Kim, H.; Park, I.; Yoo, Y.-K.; Jung, S.-K.; Park, Y.-U.; Goddard, W.A.; Kang, K. Unexpected discovery of low-cost maricite NaFePO4 as a high-performance electrode for Na-ion batteries. Energy Environ. Sci. 2015, 8, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.B. Sarcopside: Its atomic arrangement. Am. Mineral. 1972, 57, 24–35. [Google Scholar]
- Omenya, F.; Chernova, N.A.; Zhang, R.; Fang, J.; Huang, Y.; Cohen, F.; Dobrzynski, N.; Senanayake, S.; Xu, W.; Whittingham, M.S. Why Substitution Enhances the Reactivity of LiFePO4. Chem. Mater. 2013, 25, 85–89. [Google Scholar] [CrossRef]
- Janssen, Y.; Santhanagopalan, D.; Qian, D.; Chi, M.; Wang, X.; Hoffmann, C.; Meng, Y.S.; Khalifah, P. Reciprocal Salt Flux Growth of LiFePO4 Single Crystals with Controlled Defect Concentrations. Chem. Mater. 2013, 25, 4574–4584. [Google Scholar] [CrossRef]
- Crouzet, C.; Recham, N.; Brunet, F.; Findling, N. A novel route for FePO4 olivine synthesis from sarcopside oxidation. Solid State Sci. 2016, 62, 29–33. [Google Scholar] [CrossRef]
- Bnownfield, M.-E.; Foord, E.E.; Sutley, S.J.; Botinelly, T. Kosnarite, KZr2(PO4)3, a new mineral from Mount Mica and Black Mountain, Oxford County, Maine. Am. Mineral. 1993, 78, 653–656. [Google Scholar]
- Šljukić, M.; Matković, B.; Prodić, B.; Anderson, D. The crystal structure of KZr2(PO4)3. Z. Krist. 1969, 130, 148–161. [Google Scholar] [CrossRef]
- D’Yvoire, F.; Pintard-Screpel, M.; Bretey, E.; De la Roche`re, M. Phase transitions and ionic conduction in 3D skeleton phosphates A3M2(PO4)3: A = Li, Na, Ag, K; M = Cr, Fe. Solid State Ionics 1983, 9–10, 851–857. [Google Scholar] [CrossRef]
- Gaubicher, J.; Wurm, C.; Goward, G.; Masquelier, C.; Nazar, L.F. Rhombohedral Form of Li3V2(PO4)3 as a Cathode in Li-Ion Batteries. Chem. Mater. 2000, 12, 3240–3242. [Google Scholar] [CrossRef]
- Zatovsky, I.V. NASICON-type Na3V2(PO4)3. Acta Cryst. 2010, E66, i12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masquelier, C.; Wurm, C.; Rodriguez-Carvajal, J.; Gaubicher, J.; Nazar, L.F. A Powder Neutron Diffraction Investigation of the Two Rhombohedral NASICON Analogues: γ-Na3Fe2(PO4)3 and Li3Fe2(PO4)3. Chem. Mater. 2000, 12, 522–532. [Google Scholar] [CrossRef]
- Chen, S.; Wu, C.; Shen, L.; Zhu, C.; Huang, Y.; Xi, K.; Maier, J.; Yu, Y. Challenges and Perspectives for NASICON-Type Electrode Materials for Advanced Sodium-Ion Batteries. Adv. Mater. 2017, 29, 170043. [Google Scholar] [CrossRef]
- Jian, Z.; Han, W.; Liang, Y.; Lan, Y.; Fang, Z.; Hu, Y.-S.; Yao, Y. Carbon-coated rhombohedral Li3V2(PO4)3 as both cathode and anode materials for lithium-ion batteries: Electrochemical performance and lithium storage mechanism. J. Mater. Chem. A 2014, 2, 20231–20236. [Google Scholar] [CrossRef]
- Zhou, W.; Xue, L.; Lü, X.; Gao, H.; Li, Y.; Xin, S.; Fu, G.; Cui, Z.; Zhu, Y.; Goodenough, J.B. NaxMV(PO4)3 (M = Mn, Fe, Ni) Structure and Properties for Sodium Extraction. Nano Lett. 2016, 16, 7836–7841. [Google Scholar] [CrossRef]
- Chen, F.; Kovrugin, V.M.; David, R.; Mentré, O.; Fauth, F.; Chotard, J.-N.; Masquelier, C. A NASICON-Type Positive Electrode for Na Batteries with High Energy Density: Na4MnV(PO4)3. Small Methods 2018, 3, 1800218. [Google Scholar] [CrossRef]
- Zakharkin, M.V.; Drozhzhin, O.A.; Tereshchenko, I.V.; Chernyshov, D.; Abakumov, A.M.; Antipov, E.V.; Stevenson, K.J. Enhancing Na+ Extraction Limit through High Voltage Activation of the NASICON-Type Na4MnV(PO4)3 Cathode. Appl. Energy Mater. 2018, 1, 5842–5846. [Google Scholar] [CrossRef]
- Mindat.org. Available online: https://www.mindat.org/min-142.html (accessed on 22 April 2020).
- Moore, P.B. Crystal chemistry of the alluaudite structure type: Contribution to the paragenesis of pegmatite phosphate giant crystals. Am. Mineral. 1971, 56, 1955–1975. [Google Scholar]
- Hatert, F. The crystal chemistry of lithium in the alluaudite structure: A study of the (Na1-xLix)1.5Mn1.5Fe1.5(PO4)3 solid solution (x=0 to 1). Mineral. Petrol. 2004, 81, 205–217. [Google Scholar] [CrossRef]
- Hatert, F.; Ottolini, L.; Schmid-Beurmann, P. Experimental investigation of the alluaudite+triphlyite assemblage, and development of the Na-in-triphylite geothermometer: Application to natural pegmatite phosphates. Contrib. Mineral. Petrol. 2011, 161, 531–546. [Google Scholar] [CrossRef]
- Iakubovich, O.V.; Simonov, M.A.; Egorov-Tismenko, I.K.; Belov, N.V. Crystal structure of Na2(Fe0.53+Fe0.52+)2Fe2+[PO4]3-synthetic variety of alluaudite. Dokl. Akad. Nauk SSSR. 1977, 236, 1123–1126. [Google Scholar]
- Barpanda, P.; Oyama, G.; Nishimura, S.I.; Chung, S.C.; Yamada, A. A 3.8-V earth- abundant sodium battery electrode. Nat. Commun. 2014, 5, 4358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yang, K.; Song, L.; Feng, H.-J.; Gao, J. An alluaudite-type sodium-ion battery cathode candidate Na2Mn2V(PO4)3: Crystal growth, preparation, structure and electrochemical properties. J. Alloys Compd. 2019, 783, 409–415. [Google Scholar] [CrossRef]
- Tard, K.; Carlier, D.; Croguennec, L.; Wattiaux, A.; Ben Amara, M.; Delmas, C. Structural study of the Li(0.5)Na(0.5)MnFe2(PO4)3 and Li(0.75)Na(0.25)MnFe2(PO4)3 alluaudite phases and their electrochemical properties as positive electrodes in lithium batteries. Inorg. Chem. 2010, 49, 10378–10389. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.B.; Ito, J. Wyllieite, Na2Fe2+2Al[PO4]3, a new species. Mineral. Rec. 1973, 4, 131–136. [Google Scholar]
- Moore, P.B.; Ito, J. Alluaudites, wyllieites, arrojadites: Crystal chemistry and nomenclature. Mineral. Mag. 1979, 6, 227–235. [Google Scholar] [CrossRef]
- Yang, H.; Downs, R.T.; Gu, X.; Xie, X.; Kobsch, A. Fupingqiuite, IMA 2016-087. CNMNC Newsletter No. 35, February 2017, 211. Mineral. Mag. 2017, 81, 209–213. [Google Scholar]
- Yakubovich, O.V.; Massa, W.; Gavrilenko, P.; Dimitrova, O. The crystal structure of new synthetic member in the wyllieite group: Na1.265Mn2+2.690Mn3+0.785(PO4)3. Eur. J. Mineral. 2005, 17, 741–747. [Google Scholar] [CrossRef]
- Frigui, W.; Zid, M.F.; Driss, A. Wyllieite-type Ag1.09Mn3.46(AsO4)3. Acta Cryst. 2012, E68, i40–i41. [Google Scholar] [CrossRef]
- Marzouki, R.; Ben Smida, Y.; Avdeev, M.; Alghamdi, M.M.; Zid, M.F. Synthesis, structure and Na+ migration pathways of new Wylleite-type Na1.25Co2.187Al1.125(AsO4)3. Mater. Res. Express 2019, 6, 126313. [Google Scholar] [CrossRef]
- Khomyakov, A.P.; Semenov, E.I.; Kazakova, M.E.; Shumyatskaya, N.G. Sidorenkite, Na3Mn(PO4)(CO3), a new mineral. Zapiski VMO 1979, 108, 56–59. (In Russian) [Google Scholar] [CrossRef]
- Khomyakov, A.P.; Aleksandrov, V.V.; Krasnova, N.I.; Ermilov, V.V.; Smolyaninova, V.V. Bonshtedtite, Na3Fe(PO4)(CO3), a new mineral. Zapiski VMO 1982, 111, 486–490. (In Russian) [Google Scholar] [CrossRef]
- Kurova, T.A.; Shumyatskaya, N.G.; Voronkov, A.A.; Pyatenko, Y.A. Crystal structure of sidorenkite Na3Mn(PO4)(CO3). Dokl. Akad. Nauk. SSSR 1980, 251, 605–607. [Google Scholar]
- Krivovichev, S.V.; Chernyatieva, A.P.; Britvin, S.N.; Yakovenchuk, V.N.; Krivovichev, V.G. Refinement of the crystal structure of bonshtedtite, Na3Fe(PO4)(CO3). Geol. Ore Depos. 2013, 55, 669–675. [Google Scholar] [CrossRef]
- Hautier, G.; Jain, A.; Chen, H.; Moore, C.; Ong, S.P.; Ceder, G. Novel mixed polyanions lithium-ion battery cathode materials predicted by high-throughput ab initio computations. J. Mater. Chem. 2011, 21, 17147–17153. [Google Scholar] [CrossRef]
- Chen, H.; Hao, Q.; Zivkovic, O.; Hautier, G.; Du, L.-S.; Tang, Y.; Hu, Y.-Y.; Ma, X.; Grey, C.P.; Ceder, G. Sidorenkite (Na3MnPO4CO3): A New Intercalation Cathode Material for Na-Ion Batteries. Chem. Mater. 2013, 25, 2777–2787. [Google Scholar] [CrossRef]
- Wang, C.; Sawicki, M.; Emani, S.; Liu, C.; Shaw, L.L. Na3MnCO3PO4–A High Capacity, Multi-Electron Transfer Redox Cathode Material for Sodium Ion Batteries. Electrochim. Acta 2015, 161, 322–328. [Google Scholar] [CrossRef]
- Hassanzadeh, N.; Sadrnezhaad, S.K.; Chen, G. In-situ hydrothermal synthesis of Na3MnCO3PO4/rGO hybrid as a cathode for Na-ion battery. Electrochim. Acta 2016, 208, 188–194. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, J.; Li, B.; Ma, J.; Tao, S.; Xia, D.; Chu, W.; Wu, Z. Detailed investigation of Na2.24FePO4CO3 as a cathode material for Na-ion batteries. Sci. Rep. 2014, 4, 4188. [Google Scholar] [CrossRef]
- Men’shikov, Y.P.; Pakhomovskii, Y.A.; Goiko, E.A.; Bussen, I.V.; Mer´kov, A.N. A natural tetragonal titanosilicate of sodium, natisite. Zapiski VMO 1975, 104, 314–317. (In Russian) [Google Scholar]
- Nyman, H.; O’Keeffe, M.; Bovin, J.O. Sodium titanium silicate, Na2TiSiO5. Acta Cryst. 1978, B34, 905–906. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Simonov, M.A.; Melnikov, O.K. The mixed Fe-P framework in the crystal structure of Na3Fe23+[PO4]2(OH)2F. Kristallografiya 1984, 29, 484–488. [Google Scholar]
- Slobodyanik, N.S.; Nagornyi, P.C.; Kornienko, Z.I.; Kapshuk, A.A.; Mitkevich, V.V. Preparation and crystal structure of Na3Cr2F3(PO4)2. Russ. J. Inorg. Chem. 1989, 34, 1413–1414. [Google Scholar]
- Yakubovich, O.V.; Melnikov, O.K.; Urusov, V.S. The framework of mixed nature in Na3{Fe2F3[PO4]2} crystal structure-a new homeotype in the series of natisite. Dokl. Akad. Nauk SSSR 1995, 342, 615–620. [Google Scholar]
- Yakubovich, O.V.; Melnikov, O.K. Anionic framework of a mixed type in Na3{Al2F3[PO4]2} crystalline structure. Kristallografiya 1996, 41, 663–668. [Google Scholar]
- Massa, W.; Yakubovich, O.V.; Dimitrova, O.V. Crystal structure of a new sodium vanadyl(IV) fluoride phosphate Na3{V2O2F[PO4]2}. Solid State Sci. 2002, 4, 495–501. [Google Scholar] [CrossRef]
- Serras, P.; Palomares, V.; Goni, A.; Gil de Muro, I.; Kubiak, P.; Lezama, L.; Rojo, T. High voltage cathode materials for Na-ion batteries of general formula Na3V2O2x(PO4)2F3−2x. J. Mater. Chem. 2012, 22, 22301–22308. [Google Scholar] [CrossRef]
- Park, Y.U.; Seo, D.H.; Kwon, H.S.; Kim, B.; Kim, J.; Kim, H.; Kim, I.; Yoo, H.-I.; Kang, K. A new high-energy cathode for a Na-ion battery with ultrahigh stability. J. Am. Chem. Soc. 2013, 135, 13870–13878. [Google Scholar] [CrossRef]
- Broux, T.; Bamine, T.; Fauth, F.; Simonelli, L.; Olszewski, W.; Marini, C.; Meneétrier, M.; Carlier, D.; Masquelier, C.; Croguennec, L. Strong impact of the oxygen content in Na3V2(PO4)2F3-yOy (0 ≤ y ≤0.5) on its structural and electrochemical properties. Chem. Mater. 2016, 28, 7683–7692. [Google Scholar] [CrossRef] [Green Version]
- Pekov, I.V.; Yapaskurt, V.O.; Britvin, S.N.; Zubkova, N.V.; Vigasina, M.F.; Sidorov, E.G. New arsenate minerals from the Arsenatnaya fumarole, Tolbachik volcano, Kamchatka, Russia. V. Katiarsite, KTiO(AsO4). Miner. Mag. 2016, 80, 639–646. [Google Scholar] [CrossRef]
- Pekov, I.V.; Zubkova, N.V.; Agakhanov, A.A.; Yapaskurt, V.O.; Belakovskiy, D.I.; Vigasina, M.F.; Britvin, S.N.; Turchkova, A.G.; Sidorov, E.G.; Pushcharovsky, D.Y. Yurgensonite, IMA 2019-059. CNMNC Newsletter No. 52. Mineral. Mag. 2019, 83. [Google Scholar] [CrossRef] [Green Version]
- Recham, N.; Gwenaelle, R.; Sougrati, M.T.; Chotard, J.-N.; Frayret, C.; Mariyappan, S.; Melot, B.C.; Jumas, J.-C.; Tarascon, J.-M. Preparation and Characterization of a Stable FeSO4F-Based Framework for Alkali Ion Insertion Electrodes. Chem. Mater. 2012, 24, 4363–4370. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Khasanova, N.R.; Samarin, A.S.; Drozhzhin, O.A.; Batuk, D.; Karakulina, O.M.; Hadermann, J.; Abakumov, A.M.; Antipov, E.V. AVPO4F (A = Li, K): A 4 V Cathode Material for High-Power Rechargeable Batteries. Chem. Mater. 2016, 28, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Nikitina, V.A.; Fedotov, S.S.; Vassiliev, S.Y.; Samarin, A.S.; Khasanova, N.R.; Antipov, E.V. Transport and Kinetic Aspects of Alkali Metal Ions Intercalation into AVPO4F Framework. J. Electrochem. Soc. 2017, 164, A6373–A6380. [Google Scholar] [CrossRef]
- Chihara, K.; Katogi, A.; Kubota, K.; Komaba, S. KVPO4F and KVOPO4 toward 4 volt-class potassium-ion batteries. Chem. Commun. 2017, 53, 5208–5211. [Google Scholar] [CrossRef]
- Kim, H.; Seo, D.-H.; Bianchini, M.; Clément, R.J.; Kim, H.; Kim, J.C.; Tian, Y.; Shi, T.; Yoon, W.-S.; Ceder, C. A new strategy for high-voltage cathodes for K-ion batteries: Stoichiometric KVPO4F. Adv. Energy Mater. 2018, 8, 1801591. [Google Scholar] [CrossRef] [Green Version]
- Fedotov, S.S.; Luchinin, N.D.; Aksyonov, D.A.; Morozov, A.V.; Ryazantsev, S.V.; Gaboardi, M.; Plaisier, J.R.; Stevenson, K.J.; Abakumov, A.M.; Antipov, E.V. Titanium-based potassium-ion battery positive electrode with extraordinarily high redox potential. Nat. Commun. 2020, 11, 1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headden, W.P. A phosphate near triphylite from the Black Hills. Amer. J. Sci. 1891, 41, 416–417. [Google Scholar]
- Krutik, V.M.; Puscharovskii, D.Y.; Pobedimskaya, E.A.; Belov, N.V. Crystal structure of arrojadite. Soy. Phys.-Crystallogr. 1979, 24, 425–429. [Google Scholar]
- Moore, P.B.; Araki, T.; Merlino, S.; Mellini, M.; Zanazzi, P.F. The arrojadite-dickinson series, KNa4Ca(Fe,Mn)2+14Al(OH)2(PO4)12: Crystal structure and crystal chemistry. Am. Mineral. 1981, 66, 1034–1049. [Google Scholar]
- Kallfaß, C.; Hoch, C.; Schier, H.; Simon, A.; Schubert, H. The transition metal-rich orthophosphate arrojadite with special structural features. Z. Naturforsch. B 2010, 65, 1427–1433. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Matvienko, E.N.; Simonov, M.A.; Mel’nikov, O.K. Crystal structure of synthetic Fe3+-arrojadite with ideal formula of K2Na5Fe2+14Fe3+[PO4]12(OH)2. Mosc. Univ. Geol. Bull. 1986, 41, 35–47. [Google Scholar]
- Hoch, C. Vom Mineral lernen. Nachr. Chem. 2012, 60, 1181–1184. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Aksyonov, D.A.; Samarin, A.S.; Karakulina, O.M.; Hadermann, J.; Stevenson, K.J.; Khasanova, N.R.; Abakumov, A.M.; Antipov, E.V. Tuning the Crystal Structure of A2CoPO4F (A = Li, Na) Fluoride-Phosphates: A New Layered Polymorph of LiNaCoPO4F. Eur. J. Inorg. Chem. 2019, 4365–4372. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Mass, W.; Kireev, V.V.; Urusov, V.S. Cation distribution in the crystal structure of Li2.32Fe0.563+[PO4]F-a synthetic structural analog of grandidierite. Dokl. Phys. 2006, 51, 474–480. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Belokoneva, E.L.; Tsirelson, V.G.; Urusov, V.S. Electron density distribution in synthetic triphylite LiFePO4. Mosc. Univ. Geol. Bull. 1990, 45, 93–99. [Google Scholar]
- Yakubovich, O.V.; Belokoneva, E.L.; Tsirelson, V.G.; Urusov, V.S. Electron density distribution and chemical bond in maricite NaFePO4. Mosc. Univ. Geol. Bull. 1992, 47, 46–56. [Google Scholar]
- Yakubovich, O.V.; Belokoneva, E.L.; Tsirelson, V.G.; Urusov, V.S. Distribution pattern of electronic density in Fe-sarcopside crystals Fe3(PO4)2. Mosc. Univ. Geol. Bull. 1991, 46, 77–84. [Google Scholar]
- Delacourt, C.; Rodríguez-Carvajal, J.; Schmitt, B.; Tarascon, J.-M.; Masquelier, C. Crystal chemistry of the olivine-type LixFePO4 system (0 ≤ x ≤ 1) between 25 and 370 °C. Solid State Sci. 2005, 7, 1506–1516. [Google Scholar] [CrossRef]
- Geller, S.; Durand, J.L. Refinement of the structure of LiMnPO4. Acta Cryst. 1960, 13, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Moring, J.; Kostiner, E. The crystal structure of NaMnPO4. J. Solid State Chem. 1986, 61, 379–383. [Google Scholar] [CrossRef]
- Mishima, Y.; Hojo, T.; Nishio, T.; Sadamura, H.; Oyama, N.; Moriyoshi, C.; Kuroiwa, Y. MEM charge density study of olivine LiMPO4 and MPO4 (M = Mn, Fe) as cathode materials for lithium-ion batteries. J. Phys. Chem. C 2013, 117, 2608–2615. [Google Scholar] [CrossRef]
- Matvienko, E.N.; Iakubovich, O.V.; Simonov, M.A.; Belov, N.V. Crystal structure of K, Fe3+-orthophosphate, KFe[PO4]F. Dokl. Akad. Nauk SSSR 1979, 246, 875–878. [Google Scholar]
Figure 1.
Schematics of Li-ion battery.

Figure 2.
Layers built by Mn/Fe-centered octahedra and PO4 tetrahedra parallel to yz plane in the crystal structures of lithiophilite, Li(Mn,Fe)PO4 (a) and purpurite, (Mn,Fe)PO4 (b).
Figure 2.
Layers built by Mn/Fe-centered octahedra and PO4 tetrahedra parallel to yz plane in the crystal structures of lithiophilite, Li(Mn,Fe)PO4 (a) and purpurite, (Mn,Fe)PO4 (b).

Figure 3.
Triphylite—(a), heterosite—(b), and niahite—(c) type crystal structures shown along the c axis of the orthorhombic unit cells.
Figure 3.
Triphylite—(a), heterosite—(b), and niahite—(c) type crystal structures shown along the c axis of the orthorhombic unit cells.

Figure 4.
The tavorite, LiFe(PO4)(OH), crystal structure in yz projection with Li+ ions shown as light gray spheres (a) and along {110} direction, with quasi chains of edge-sharing pairs of Li-centered five-vertex polyhedra parallel to the c axis (b).
Figure 4.
The tavorite, LiFe(PO4)(OH), crystal structure in yz projection with Li+ ions shown as light gray spheres (a) and along {110} direction, with quasi chains of edge-sharing pairs of Li-centered five-vertex polyhedra parallel to the c axis (b).

Figure 5.
“Olivine tapes” aligned in the {010} direction in the crystal structure types of triphylite, LiFePO4 (simferite with one half of LiO6 octahedra statistically vacant) (a), sarcopside, Fe1.5PO4 (b), and marićite, NaFePO4 (c).
Figure 5.
“Olivine tapes” aligned in the {010} direction in the crystal structure types of triphylite, LiFePO4 (simferite with one half of LiO6 octahedra statistically vacant) (a), sarcopside, Fe1.5PO4 (b), and marićite, NaFePO4 (c).

Figure 6.
The crystal structure of marićite projected along the {011} direction shows open channels with small windows.
Figure 6.
The crystal structure of marićite projected along the {011} direction shows open channels with small windows.

Figure 7.
The sarcopside (a) and triphylite (b) crystal structures in axonometric projection along the c axis. A fragment of the triphylite structure with small octahedral interstitium in constricted channels, parallel to the {001} direction (c).
Figure 7.
The sarcopside (a) and triphylite (b) crystal structures in axonometric projection along the c axis. A fragment of the triphylite structure with small octahedral interstitium in constricted channels, parallel to the {001} direction (c).

Figure 8.
The crystal structure of kosnarite in xy projection (a) and along {110} direction (b).

Figure 9.
Crystal structures of triphylite, LiFe2+PO4 (a) and ferroalluaudite, Na2Fe3+Fe2+2(PO4)3 (b) in an axonometric projection.
Figure 9.
Crystal structures of triphylite, LiFe2+PO4 (a) and ferroalluaudite, Na2Fe3+Fe2+2(PO4)3 (b) in an axonometric projection.

Figure 10.
Chains along the {201} built from Mn2+O6 (light pink) and Mn3+O6 (bright pink) octahedra (a) in the crystal structure of synthetic wyllieite, NaMn2+2.5Mn3+(PO4)3 (view from the {100} direction) (b).
Figure 10.
Chains along the {201} built from Mn2+O6 (light pink) and Mn3+O6 (bright pink) octahedra (a) in the crystal structure of synthetic wyllieite, NaMn2+2.5Mn3+(PO4)3 (view from the {100} direction) (b).

Figure 11.
Crystal structure of sidorenkite, Na3Mn(PO4)(CO3), in an axonometric projection.

Figure 12.
Crystal structure of the tetragonal natisite, Na2TiSiO5, in basic xy projection (a) and the block of the central TiO5 semi-octahedron sharing vertices with four SiO4 tetrahedra (b).
Figure 12.
Crystal structure of the tetragonal natisite, Na2TiSiO5, in basic xy projection (a) and the block of the central TiO5 semi-octahedron sharing vertices with four SiO4 tetrahedra (b).

Figure 13.
Crystal structure of Na3V2O2F(PO4) in axonometric projection; spheres: disordered Na atoms (a), and M-centered polyhedra distortion (b).
Figure 13.
Crystal structure of Na3V2O2F(PO4) in axonometric projection; spheres: disordered Na atoms (a), and M-centered polyhedra distortion (b).

Figure 14.
Polyhedral representation of the KTP (short for KTiPO5) structure type shown for the K(Ti,Cr)[PO4](O,F) solid solution.
Figure 14.
Polyhedral representation of the KTP (short for KTiPO5) structure type shown for the K(Ti,Cr)[PO4](O,F) solid solution.

Figure 15.
The arrojadite crystal structure of a synthetic Fe3+ variety exposed. Phosphate tetrahedra are shown in yellow, Fe3+-centered octahedra in light green, Fe2+-centered polyhedra in green, Na as light gray spheres, and K atoms as dark gray spheres.
Figure 15.
The arrojadite crystal structure of a synthetic Fe3+ variety exposed. Phosphate tetrahedra are shown in yellow, Fe3+-centered octahedra in light green, Fe2+-centered polyhedra in green, Na as light gray spheres, and K atoms as dark gray spheres.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yakubovich, O.; Khasanova, N.; Antipov, E. Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications. Minerals 2020, 10, 524. https://doi.org/10.3390/min10060524
AMA Style
Yakubovich O, Khasanova N, Antipov E. Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications. Minerals. 2020; 10(6):524. https://doi.org/10.3390/min10060524
Chicago/Turabian StyleYakubovich, Olga, Nellie Khasanova, and Evgeny Antipov. 2020. "Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications" Minerals 10, no. 6: 524. https://doi.org/10.3390/min10060524
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.