Exploring the Spectrum of Kidney Ciliopathies
by
Matteo Santoni
1,*, 
Francesco Piva
2,
Alessia Cimadamore
3,*,
Matteo Giulietti
2,
Nicola Battelli
1,
Rodolfo Montironi
3,
Laura Cosmai
4 and
Camillo Porta
5,6 1
Oncology Unit, Macerata Hospital, 62100 Macerata, Italy
2
Department of Specialistic Clinical and Odontostomatological Sciences, Polytechnic University of Marche, 60126 Ancona, Italy
3
Section of Pathological Anatomy, School of Medicine, United Hospitals, Polytechnic University of the Marche Region, 60126 Ancona, Italy
4
Division of Nephrology and Dialysis, ASST Fatebenefratelli-Sacco, Fatebenefratelli Hospital, 20121 Milan, Italy
5
Chair of Oncology, Department of Biomedical Sciences and Human Oncology, University of Bari ‘A. Moro’, 70121 Bari, Italy
6
Division of Medical Oncology, A.O.U. ConsorzialePoliclinico di Bari, 70124 Bari, Italy
*
Authors to whom correspondence should be addressed.
Diagnostics 2020, 10(12), 1099; https://doi.org/10.3390/diagnostics10121099
Submission received: 20 November 2020
/
Revised: 11 December 2020
/
Accepted: 14 December 2020
/
Published: 16 December 2020
(This article belongs to the Special Issue Novel Diagnostic and Predictive Strategies in Renal Cell Tumors)
Abstract
:Ciliopathies are a group of multi-organ diseases caused by the disruption of the primary cilium. This event leads to a variety of kidney disorders, including nephronophthisis, renal cystic dysplasia, and renal cell carcinoma (RCC). Primary cilium contributes to the regulation of the cell cycle and protein homeostasis, that is, the balance between protein synthesis and degradation by acting on the ubiquitin-proteasome system, autophagy, and mTOR signaling. Many proteins are involved in renal ciliopathies. In particular, fibrocystin (PKHD1) is involved in autosomal recessive polycystic kidney disease (ARPKD), while polycystin-1 (PKD1) and polycystin-2 (PKD2) are implicated in autosomal dominant polycystic kidney disease (ADPKD). Moreover, primary cilia are associated with essential signaling pathways, such as Hedgehog, Wnt, and Platelet-Derived Growth Factor (PDGF). In this review, we focused on the ciliopathies associated with kidney diseases, exploring genes and signaling pathways associated with primary cilium and the potential role of cilia as therapeutic targets in renal disorders.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Flagella and cilia in eukaryotic cells have been historically considered as organelles involved in cell motility [1]. The structure of the motile cilia, which are shorter and more numerous than flagella (>100 copies for each cell), is composed by a basal body and an axoneme with a 9 + 0 or 9 + 2 arrangement of microtubules. The function of cilia results in cell motility through water (typical of a single cell organism) or in moving water and its content across the cell surface (as for the motility of mucus within the human respiratory tract). Beyond motile cilia, Alexander Kowalevsky discovered in 1867 [1] the presence of a single nonmotile cilia, the primary cilium, which lacks the central pair of microtubules [2] and is present in almost all eukaryotic cells. Primary cilia can transduce extracellular signals acting as mechano- or photoreceptors, but they can also be involved in detecting, for example, osmolarity, chemicals, and temperature [3]. Moreover, primary cilia are associated with essential signaling pathways, such as Hedgehog, Wnt, and Platelet-Derived Growth Factor (PDGF) [4,5,6].
In the last decade, the advances in proteomics and genomics have led to the creation of a “ciliome” [7,8,9], a powerful collection of over 3000 genes encoding proteins involved in cilia assembly and functions [10]. For example, this list includes RFX-type transcription factor DAF-19, which regulates proteins implicated in intraflagellar transport [11,12], MKS1 (Meckel Syndrome Type 1), BBS3 (Bardet-biedl syndrome 3), and BBS5 (Bardet-biedl syndrome 5) [13,14,15,16]. Beyond these, a series of proteins that indirectly interact with ciliary components has been identified [17,18].
Ciliopathies are rare conditions that have emerged as a new challenge for worldwide researchers [19]. Alterations of ciliary motility can lead to a wide spectrum of specific diseases in humans, including, for example, sterility for defective sperm cilia and a condition known as “situs inversus”, in which internal organs are inverted due to defective embryonic cilia.
For the purposes of this review, we focused exclusively on the ciliopathies associated with kidney disorders, from renal cystic dysplasia to cancer, exploring proteins and signaling pathways associated with primary cilium and the potential role of cilia as therapeutic targets in renal diseases.
2. Genes and Signaling Pathways in the Primary Cilia
The cilium is formed by proteins and other molecules transported along the axoneme by the intraflagellar transport (IFT) particles, since protein translation does not occur in the cilia. Dynein 2/1b performs the intraflagellar transport from the base to the tip of cilium, whereas kinesin 2 carries the cargo in the opposite direction. The intraflagellar transport is important to ensure the normal functioning of the cilia. Indeed, the disruption of IFT88 (intraflagellar transport protein 88 homolog) results in polycystic kidney disease in mice [20]. Many other genes are involved in ciliopathies, in particular, fibrocystin (PKHD1) is implicated in autosomal recessive polycystic kidney disease (ARPKD), polycystin-1 (PKD1) and polycystin-2 (PKD2) are associated to autosomal dominant polycystic kidney disease (ADPKD), and nephrocystins are correlated with nephronophthisis. However, a list of genes involved in cilia-associated human cystic kidney disease can be found in the review by Dell et al. [21]. Among the genes not included in this list are cyclin-dependent-like kinase 5 (CDK5), a kinase regulated by p35 (cyclin-dependent kinase 5 activator 1) and p39 (cyclin-dependent kinase 5 activator 2). CDK5 hyper activation can promote renal cystogenesis, and its inhibition reduces ciliary length, restores cell differentiation, and attenuates disease progression [22].
The presence of cilia is regulated during cell cycle progression. Indeed, cilia are resorbed prior to entry into mitosis, allowing the centrioles’ detachment from the basal body. Therefore, it seems that primary cilia can prevent uncontrolled cell growth, as proved also by the role of IFT88 (intraflagellar transport protein 88 homolog) and NDE1 (nuclear distribution protein nudE homolog 1) proteins in controlling cell division. In particular, IFT88 is tightly associated with the centrosome, and influences the G1-S cell cycle progression [23]. On the other hand, centrosomal phosphoprotein NDE1 is a negative regulator of ciliary length that acts by modulating the IFT process. The NDE1 expression level is reduced when the cell enters G1 and is high in mitosis, thus ciliary length is regulated in a cell cycle-dependent manner. At the molecular level, CDK5 seems to regulate NDE1 degradation through FBW7 E3 ubiquitin ligase [24]. Another element that controls the cell cycle is NDEL1 (nuclear distribution protein nudE-like 1), a scaffold protein that regulates microtubule dynamics and microtubule-based transport. Its expression reduces cilia length in proliferating cells, which promotes cell cycle progression [25].
It is notable that, in patients with clear cell RCC with mutations in the VHL (von Hippel-Lindau disease tumor suppressor) gene, primary cilia have been lost, and the re-expression of VHL proteins restored cilia expression [26]. Although cell proliferation affects the presence of cilia, renal cancer cells do not contain primary cilia independently of levels of Ki67 cell proliferation marker [27]. In other words, the loss of cilia would be independent from the rate of cell proliferation.
Primary cilium also contributes to the regulation of protein homeostasis, that is, the balance between protein synthesis and degradation, by acting on the ubiquitin-proteasome system, autophagy, and mTOR signaling. In fact, several proteasome proteins are present in cilia, and contribute to their disassembly during the cell cycle. By autophagy, proteins and organelles are degraded in order to supply amino acids to sustain ciliogenesis, but autophagy can also limit ciliogenesis by eliminating components of ciliary transport; mTOR can promote ciliogenesis by limiting autophagy and increasing protein and lipid synthesis, glycolysis, and oxidative metabolism [28].
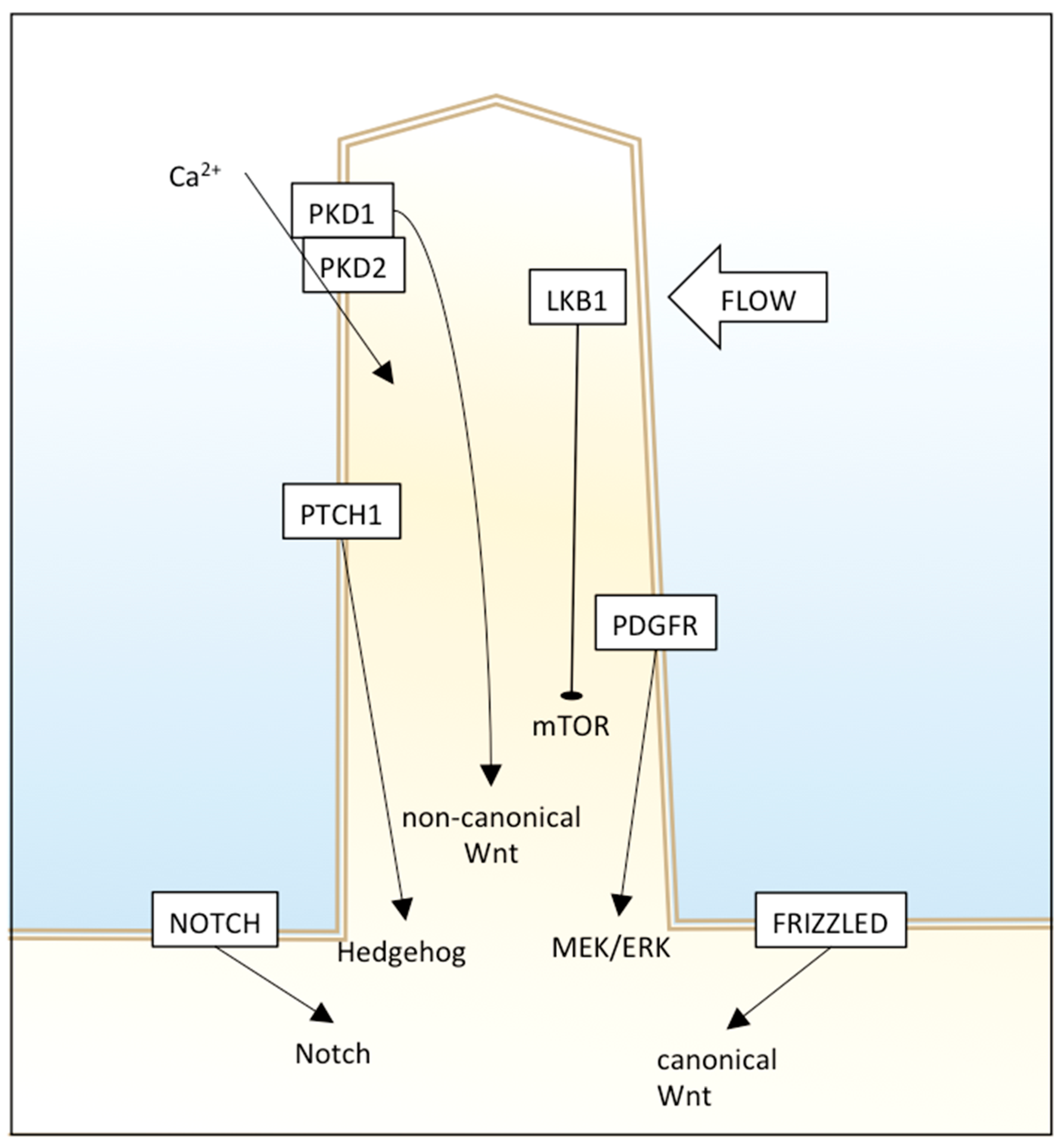
The primary cilium has different signaling receptors on the ciliary membrane, including those for Hedgehog (Hh), Notch, mTOR, platelet-derived growth factor receptor (PDGFR), and canonical and non-canonical Wnt pathways (Figure 1).
Hedgehog signaling is involved in vertebrate embryonic development and stem cell maintenance, and its dysregulation was found in many human tumors. Primary cilia have an essential role in Hh signaling; in fact, some genes are required both for Hh signaling and primary cilia formation. PTCH1 (protein patched homolog 1), the receptor for Hh ligands, resides in the cilium membrane and represses and averts SMO (smoothened homolog). In the unstimulated state, GLI transcription factors are suppressed by SUFU (suppressor of fused homolog). Upon binding of an Hh ligand to PTCH1, SMO is not repressed anymore, and enters the cilium instead of PTCH1. SMO represses SUFU, and this causes GLIs release and activation. The movement of these Hh proteins along cilium is facilitated by IFT proteins and motor proteins, therefore, loss of IFT proteins leads to Hh alteration [29]. However, the control of this pathway by cilia is complex and context-dependent, as nonfunctional or lack of cilia can result in inhibition or enhancement of Hh signaling. For example, in two Hh pathway-dependent mouse basal cell carcinoma models, cilia deletion was able to inhibit tumor growth induced by an activated form of SMO, but also promote tumor growth induced by activated GLI2 [30]. This suggests that primary cilia could promote or inhibit carcinogenesis through Hh depending on the nature of the oncogenic initiating event.
Wnt signaling is involved in cell migration, planar cell polarity, and organogenesis, but its alterations are related to different cancer types. Many published papers suggest that primary cilia have an important role in attenuation of a canonical and non-canonical (β-catenin independent) Wnt signaling pathway. Defects in cilia cause over-activation of Wnt, leading to cystic kidney disease in a mouse model [31] and Joubert syndrome in humans [32]. Moreover, Wnt ligands can bind to PKD1 and, through the PKD2 channel, induce Ca(2+) influx [33]. An interesting work showed that carcinogens ochratoxin A (OTA) and potassium bromate (KBrO3) induced loss of the primary cilium in a human proximal tubular epithelial cell line and the dysregulation of different pathways, including Wnt signaling and cytoskeletal remodeling [34]. Since beta-catenin is sequestered in the primary cilium, an undamaged primary cilium that retains the effector efficiently acts as a negative regulator of Wnt signaling, and deciliation could enhance Wnt, leading to the progression of malignant tumors.
The PDGF pathway is involved in cell growth, proliferation, migration, and embryonic development, and its dysregulation causes different diseases, including cancer.
PDGFRA is present in primary cilia membranes, and cells lacking normal cilia cannot activate MEK1/2 and ERK1/2 [35,36].
Notch signaling is involved in cell fate determination. Some Notch components reside in cilia, and loss of the latter alters Notch functioning, therefore, differentiation of basal cells to spinous cells. In particular, Notch3 localizes in the ciliary membrane and interacts with Presenilin-2, an enzyme localized to the ciliary basal body and responsible for Notch cleavage and activation. Notch signaling also regulates cilium length and trafficking of Hh mediators into primary cilium [35].
3. Renal Ciliopathies
3.1. Nephronophthisis
Nephronophthisis (NPHP) is inherited in an autosomal recessive mode and represents, although rare, the most frequent cause of end-stage renal disease (ESRD) in children and young adults [37], with a prevalence of 1/100,000 individuals. Although usually characterized by aspecific symptoms (i.e., urine concentration, polydipsia, and polyuria), in 10–20% of cases, NPHP presents additional features of ciliopathy syndrome, such as retinal defects, liver fibrosis, skeletal abnormalities, and brain developmental disorders [38].
Thus, diagnosis usually requires genetic tests, renal ultrasounds, and biopsies. At ultrasound, kidneys usually present normal size, with an increase in echogenicity sometimes associated with renal cysts. At biopsy, NPHP shows tubular basement lesions, which are thickened and multilayered, interstitial fibrosis, and inflammation [37].
Traditional classification of NPHP is based on the age of onset of ESRD: infantile, juvenile, or adolescent/adult. The most common form, juvenile NPHP, is also called classic NPHP [39].
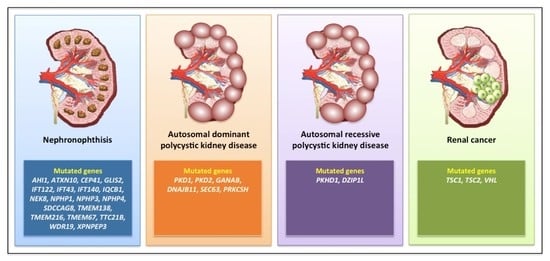
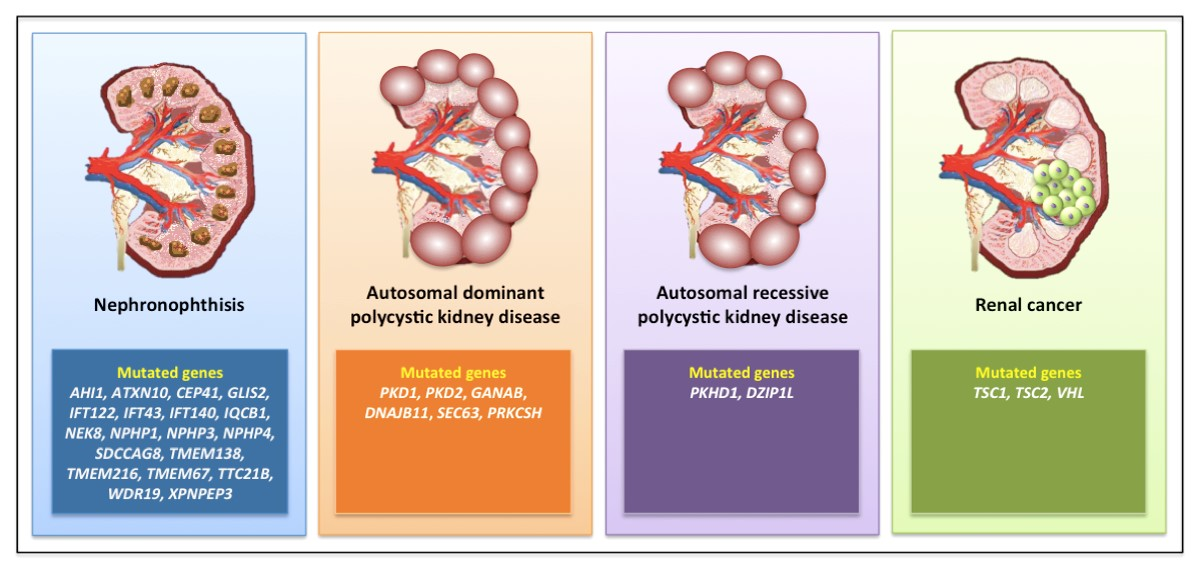
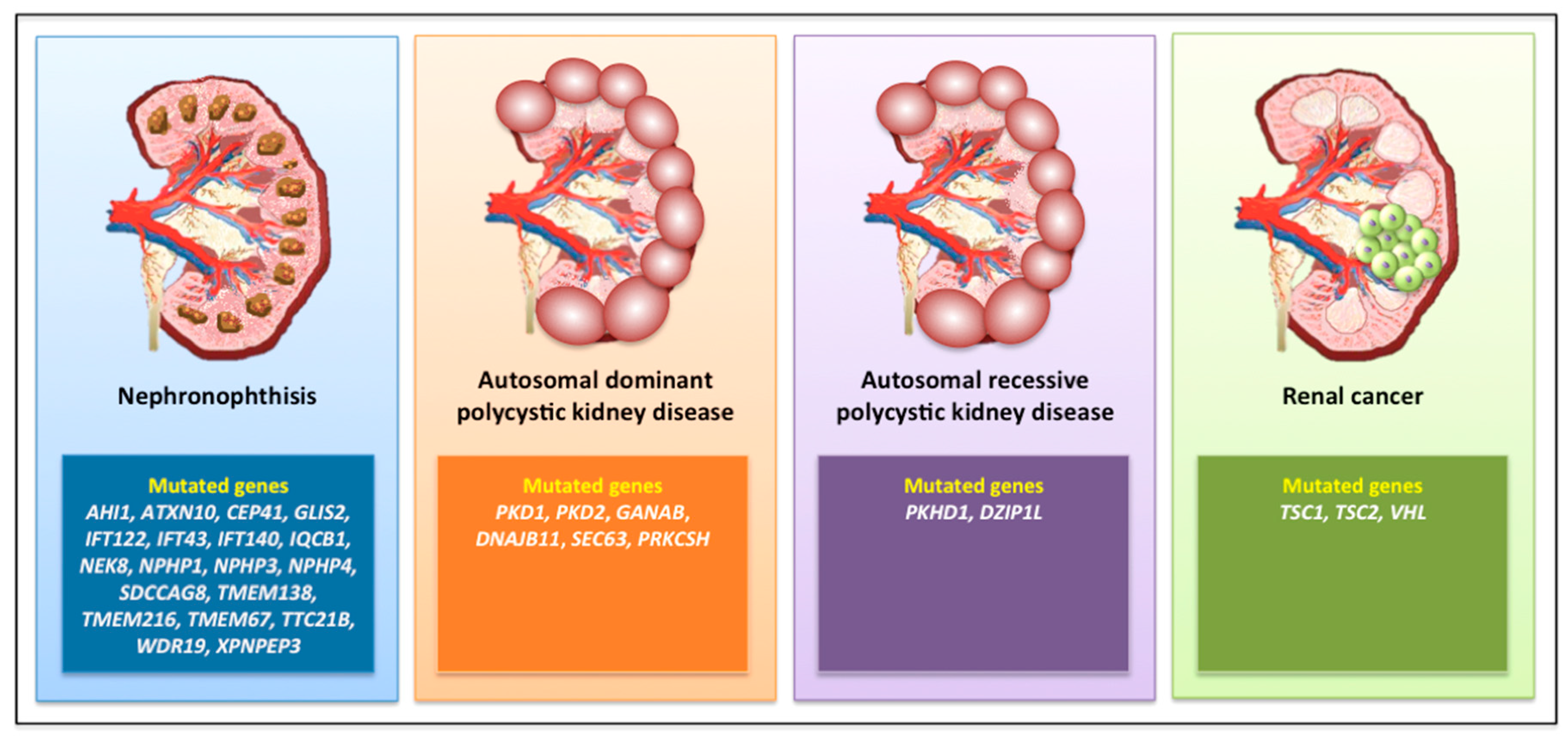
NPHP has been correlated with mutations in at least 25 different genes (Figure 2) involved in renal development and homeostasis [40].
The majority of these genes encode proteins interconnected in a nephrocystin protein complex that resides at the transition zone. Approximately 70% of NPHP are still genetically unclear, while mutations in the NPHP1 gene are responsible for 20% of all cases. This gene has been mapped to chromosome 2q13, firstly identified in 1997 [41], and codes for Nephrocystin-1, which is an adaptor protein mainly localized in primary cilia and the apical surface of renal epithelial cells, together with other adhesion and signaling transducer molecules. NPHP1 alterations are associated not only with NPHP, but also with neurological disorders, including Joubert syndrome, described by cerebellar vermis hypoplasia and brainstem abnormalities [42]; Senior–Løken syndrome, consisting of retinal degeneration to blindness; Bardet–Biedl syndrome, with intellectual disability and obesity [43]; and skeletal ciliopathies characterized by bone shortening (i.e., Jeune asphyxiating thoracic dystrophy [44] and Sensenbrenner syndrome [45]).
Meckel–Gruber syndrome is a highly lethal perinatal autosomal recessive congenital anomaly syndrome mapped to six different loci in different chromosomes; it has an extreme genetic heterogeneity, highly variable phenotype (due to multi-organ involvement), and displays allelism with other related ciliopathies, presenting significant challenges for diagnosis [46].
3.2. Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD), the most common inherited cause of renal failure, is generally an adult-onset condition with a prevalence of 1/1000, characterized by gradually growing fluid-filled renal cysts originating from all areas of the kidneys, associated with hepatobiliary changes, hypertension, and/or other extra-renal abnormalities (i.e., left ventricular hypertrophy and intracranial arterial aneurysms) [47,48].
The most common causes are mutations in PKD1 (80% of cases) or PKD2 (15% of cases), encoding two components of a heterodimeric complex (polycystin 1, PC1, and PC2) co-localized in the primary cilium, where they transduce the extracellular fluid flow shear stress into a Ca2+ signal [49,50]. Beyond PKD1 and PKD2 (which present with high levels of allelic heterogeneity [51]), a series of mutated genes has been described in ADPKD, such as neutral α-glucosidase AB (GANAB) [52], DNAJB11 [53], SEC63, and PRKCSH [54] (Figure 2).
Though fully penetrant, it has a considerable phenotypic variability, conditioned by factors such as gender, type of mutation, and concomitant acquired diseases, such as hypertension and chronic kidney disease [55].
Cysts arise from the epithelia of only 1 to 5% of all nephrons, and are localized mainly in the distal convoluted tubule and collecting duct. They are lined by a single layer of rapidly proliferating and less differentiated tubular cells. The inflammatory tissue surrounding these cysts is induced by the secretion of cytochines and chemokines from the epithelium of the cysts.
Clinical symptoms include hypertension, abdominal pain, haematuria, and urinary tract infections [48]. The radiologic diagnosis is based on ultrasonography, which should often be accompanied by CT or MRI in order to obtain more quantitative data. Indeed, in ADPKD, the kidneys are markedly enlarged with vascular remodeling and interstitial fibrosis, while benign adenomas are present in about 25% of patients [56]. Genetic diagnosis of ADPKD should be performed by next-generation sequencing (NGS), caused by the complexity of the PKD1 gene structure.
3.3. Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease (ARPKD) is much rarer (1/10,000) than ADPKD, and is generally a perinatal or childhood disease; indeed, 50% of the subjects develop ESRD from this disease at age 10 years or less. ARPKD is due to mutations in the Polycystic Kidney and Hepatic Disease 1 (PKHD1) gene, which encodes fibrocystin, and is localized to the primary cilium in the kidneys, liver, and pancreas, predominantly in the basal body of cilia present in renal tubular cells and biliary epithelial cells [57,58] (Figure 2). Fibrocystin is a key regulator of cell proliferation, apoptosis, and polarization [59]. Mutations in the gene encoding the ciliary transition zone protein DAZ-interacting protein 1-like protein (DZIP1L) have also been reported in ARPKD without evidence of PKHD1 mutations [60] (Figure 2).
The clinical symptoms of ARPKD include cysts localized in the collecting ducts, with large kidneys at ultrasound, and congenital hepatic fibrosis (CHF). The diagnosis of ARPKD is built on clinical imaging in serious neonatal or infantile cases, and should require NGS testing for a more efficient diagnostic approach.
3.4. Renal Cystic Dysplasia
Cystic renal dysplasia is a congenital condition characterized by the presence of cysts in the renal cortex, distended collecting ducts, and poorly developed medullary pyramids [37]. Fibrous tissue may also be present [61]. The diagnosis of congenital renal cystic dysplasia is usually performed by ultrasonography prenatally or during early childhood. Cystic renal dysplasia may be associated with several urologic abnormalities, including ureteropelvic and ureterovesicular junction obstruction, neurogenic bladder, ureterocele, and prune belly syndrome [37].
3.5. Rare Renal Phenotypes
3.6. Renal Cell Carcinoma
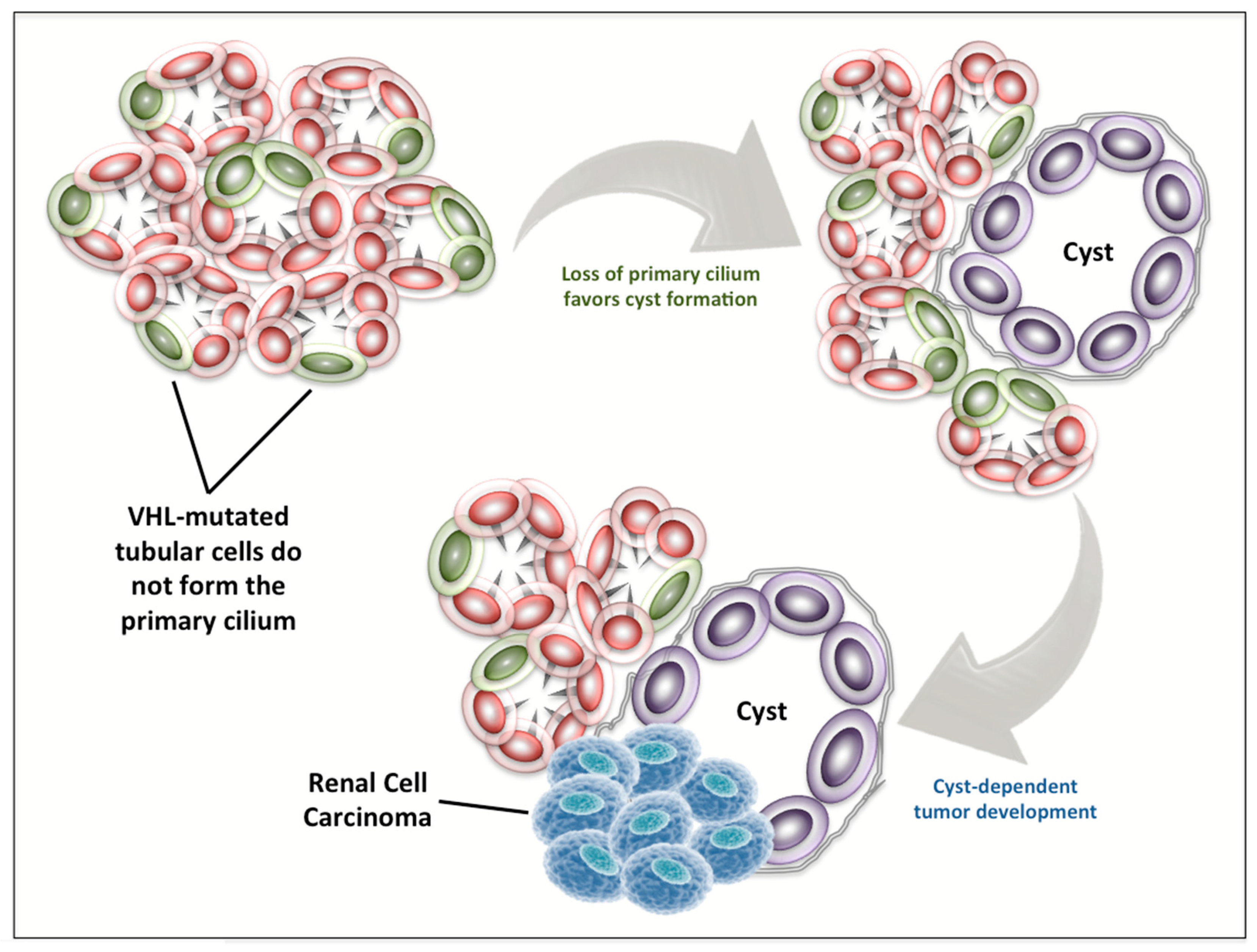
The connection between ciliogenesis, the formation of renal cysts, and the development of RCC is supported by a series of evidence published in the last 15 years [66,67,68,69,70,71,72,73]. Indeed, in 2004, Lolkema et al. [74] firstly showed that VHL protein can modulate tubulin turnover, thus influencing cellular migration, polarization, and cell–cell interactions. Two years later, it was reported that VHL can control ciliogenesis by orienting microtubule growth through the interaction with the Par3–Par6–atypical PKC complex [75]. In the same year, it became clear that VHL inactivation is associated with abrogation of the primary cilium in renal cysts of patients with VHL disease and in VHL-defective cell lines [76] (Figure 3), and this role of VHL for ciliogenesis is independent of hypoxia-induced factor (HIF)-α abundance, suggesting two distinct functions of VHL both necessary in RCC carcinogenesis [77].
In 2008, Lolkema et al. evidenced the essential activity of VHL for the initiation of ciliogenesis and in the maintenance of cilia in kidney cells [78]. They observed that reduced cellular levels of VHL led to a drop in ciliated cell frequency in mouse kidney cell lines. Moreover, only cells expressing VHL were able to respond to flow by rapidly increasing the intracellular Ca2+ concentrations, thus underlining the fundamental role of VHL in the mechanotransduction activity of primary cilia [78].
4. Conclusions
The advances in our knowledge about the genetic mechanisms underlying renal ciliopathies have improved the diagnosis and prognosis of these patients. The availability of NGS, together with bioinformatic analysis, will further expand our insights on ciliopathies, thus accelerating the route to the development of targeted approaches aimed at delaying or preventing the effects of this group of diseases. The fundamental role of ciliogenesis in RCC carcinogenesis opens a window to the possibility of developing strategies for the early phases of this disease. Nevertheless, the route to personalized approaches in patients with renal ciliopathies will probably require years of research and an even more straight collaboration between nephrologists, geneticists, and oncologists.
Author Contributions
Conceptualization, M.S. and C.P.; investigation, F.P., A.C., M.G.; resources, N.B., R.M.; writing—original draft preparation, M.S., L.C., C.P.; writing—review and editing, A.C., C.P.; supervision, M.S., C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Adams, M. The Primary Cilium: An Orphan Organelle Finds a Home. Nat. Educ. 2010, 3, 54. [Google Scholar]
- Adams, G.M.W.; Huang, B.; Piperno, G.; Luck, D.J.L. Central-pair microtubular complex of Chlamydomonas flagella: Polypeptide composition as revealed by analysis of mutants. J. Cell Biol. 1981, 91, 69–76. [Google Scholar] [CrossRef]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Krönig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Gherman, A.; Davis, E.E.; Katsanis, N. The ciliary proteome database: An integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006, 38, 961–962. [Google Scholar] [CrossRef]
- Inglis, P.N.; Boroevich, K.A.; Leroux, M.R. Piecing together a ciliome. Trends Genet. 2006, 22, 491–500. [Google Scholar] [CrossRef]
- Cervesato, A.; Raucci, R.; Buononato, D.; Marchese, E.; Capolongo, G.; Perna, A.; Capasso, G.; Zacchia, M. Application of proteomics and metabolomics to study inherited kidney disorders: From big data to precision medicine. Giornale Ital. Nefrol. 2020, 37, 1853–1861. [Google Scholar]
- Arnaiz, O.; Malinowska, A.; Klotz, C.; Sperling, L.; Dadlez, M.; Koll, F.; Cohen, J. Cildb: A knowledgebase for centrosomes and cilia. Database 2009, 2009. [Google Scholar] [CrossRef]
- Swoboda, P.; Adler, H.T.; Thomas, J.H. The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. Elegans. Mol. Cell 2000, 5, 411–421. [Google Scholar] [CrossRef]
- Piasecki, B.P.; Burghoorn, J.; Swoboda, P. Regulatory Factor X (RFX)-mediated transcriptional rewiring of ciliary genes in animals. Proc. Natl. Acad. Sci. USA 2010, 107, 12969–12974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badano, J.L.; Leitch, C.C.; Ansley, S.J.; May-Simera, H.; Lawson, S.; Lewis, R.A.; Beales, P.L.; Dietz, H.C.; Fisher, S.; Katsanis, N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 2006, 439, 326–330. [Google Scholar] [CrossRef]
- Chiang, A.P.; Nishimura, D.; Searby, C.; Elbedour, K.; Carmi, R.; Ferguson, A.L.; Secrist, J.; Braun, T.; Casavant, T.; Stone, E.M.; et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am. J. Hum. Genet. 2004, 75, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Kyttälä, M.; Tallila, J.; Salonen, R.; Kopra, O.; Kohlschmidt, N.; Paavola-Sakki, P.; Peltonen, L.; Kestilä, M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006, 38, 155–157. [Google Scholar] [CrossRef]
- Li, J.B.; Gerdes, J.M.; Haycraft, C.J.; Fan, Y.; Teslovich, T.M.; May-Simera, H.; Li, H.; Blacque, O.E.; Li, L.; Leitch, C.C.; et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004, 117, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Eley, L.; Gabrielides, C.; Adams, M.; Johnson, C.A.; Hildebrandt, F.; Sayer, J.A. Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 2008, 74, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.M.; Letteboer, S.J.F.; van Beersum, S.E.C.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A Is Mutated in Joubert Syndrome and Interacts with the Ciliopathy-Associated Basal Body Protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Dell, K.M. The role of cilia in the pathogenesis of cystic kidney disease. Curr. Opin. Pediatr. 2015, 27, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husson, H.; Moreno, S.; Smith, L.A.; Smith, M.M.; Russo, R.J.; Pitstick, R.; Sergeev, M.; Ledbetter, S.R.; Bukanov, N.O.; Lane, M.; et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis. Hum. Mol. Genet. 2016, 25, 2245–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, A.; Margall-Ducos, G.; Guidotti, J.E.; Brégerie, O.; Celati, C.; Bréchot, C.; Desdouets, C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J. Cell Sci. 2007, 120 Pt 4, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Maskey, D.; Marlin, M.C.; Kim, S.; Kim, S.; Ong, E.; Li, G.; Tsiokas, L. Cell cycle-dependent ubiquitylation and destruction of NDE 1 by CDK 5- FBW 7 regulates ciliary length. EMBO J. 2015, 34, 2424–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Goto, H.; Kasahara, K.; Kumamoto, K.; Yonemura, S.; Inoko, A.; Yamano, S.; Wanibuchi, H.; He, D.; Goshima, N.; et al. Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein-Aurora A pathway. J. Cell Biol. 2016, 212, 409–423. [Google Scholar] [CrossRef] [Green Version]
- Arjumand, W.; Sultana, S. Role of VHL gene mutation in human renal cell carcinoma. Tumor Biol. 2012, 33, 9–16. [Google Scholar] [CrossRef]
- Yuan, K.; Frolova, N.; Xie, Y.; Wang, D.; Cook, L.; Kwon, Y.J.; Steg, A.D.; Serra, R.; Frost, A.R. Primary cilia are decreased in breast cancer: Analysis of a collection of human breast cancer cell lines and tissues. J. Histochem. Cytochem. 2010, 58, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Wong, S.Y.; Seol, A.D.; So, P.L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Louie, C.M.; Silhavy, J.L.; Sintasath, L.; Decambre, M.; Nigam, S.K.; Willert, K.; Gleeson, J.G. Impaired Wnt-Β-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat. Med. 2009, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferland, R.J.; Walsh, C.A. Joubert Syndrome. In Encyclopedia of Neuroscience; Elsevier: Amsterdam, The Netherlands, 2009; pp. 249–256. ISBN 9780080450469. [Google Scholar]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca2+ signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.; Slattery, C.; Jennings, P.; Blaque, O.; Pfaller, W.; Gmuender, H.; van Delft, J.; Ryan, M.P.; McMorrow, T. Carcinogens induce loss of the primary cilium in human renal proximal tubular epithelial cells independently of effects on the cell cycle. Am. J. Physiol. Ren. Physiol. 2012, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary cilium-dependent signaling mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, M.; Obaidi, I.; McMorrow, T. Primary cilia and their role in cancer (Review). Oncol. Lett. 2019, 17, 3041–3047. [Google Scholar] [CrossRef]
- Arts, H.H.; Knoers, N.V.A.M. Current insights into renal ciliopathies: What can genetics teach us? Pediatr. Nephrol. 2013, 28, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Tao, Y.H. Nephronophthisis: A review of genotype–phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Simms, R.J.; Eley, L.; Sayer, J.A. Nephronophthisis. Eur. J. Hum. Genet. 2009, 17, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Novarino, G.; Akizu, N.; Gleeson, J.G. Modeling human disease in humans: The ciliopathies. Cell 2011, 147, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, F.; Otto, E.; Rensing, C.; Nothwang, H.G.; Vollmer, M.; Adolphs, J.; Hanusch, H.; Brandis, M. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet. 1997, 17, 149–153. [Google Scholar] [CrossRef]
- Caridi, G.; Dagnino, M.; Rossi, A.; Valente, E.M.; Bertini, E.; Fazzi, E.; Emma, F.; Murer, L.; Verrina, E.; Ghiggeri, G.M. Nephronophthisis type 1 deletion syndrome with neurological symptoms: Prevalence and significance of the association. Kidney Int. 2006, 70, 1342–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delvallée, C.; Nicaise, S.; Antin, M.; Leuvrey, A.S.; Nourisson, E.; Leitch, C.C.; Kellaris, G.; Stoetzel, C.; Geoffroy, V.; Scheidecker, S.; et al. A BBS1 SVA F retrotransposon insertion is a frequent cause of Bardet-Biedl syndrome. Clin. Genet. 2020. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Walczak-Sztulpa, J.; Eggenschwiler, J.; Osborn, D.; Brown, D.A.; Emma, F.; Klingenberg, C.; Hennekam, R.C.; Torre, G.; Garshasbi, M.; Tzschach, A.; et al. Cranioectodermal Dysplasia, Sensenbrenner Syndrome, Is a Ciliopathy Caused by Mutations in the IFT122 Gene. Am. J. Hum. Genet. 2010, 86, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel-Gruber syndrome: An update on diagnosis, clinical management, and research advances. Front. Pediatr. 2017, 5, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 1–24. [Google Scholar] [CrossRef]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Heyer, C.M.; Sundsbak, J.L.; Abebe, K.Z.; Chapman, A.B.; Torres, V.E.; Grantham, J.J.; Bae, K.T.; Schrier, R.W.; Perrone, R.D.; Braun, W.E.; et al. Predicted mutation strength of nontruncating PKD1 mutations AIDS genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 2872–2884. [Google Scholar] [CrossRef] [Green Version]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017, 127, 1772–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenth, J.P.H.; TeMorsche, R.H.M.; Smink, R.; Bonifacino, J.S.; Jansen, J.B.M.J. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003, 33, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [Green Version]
- Chebib, F.T.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am. J. Kidney Dis. 2016, 67, 792–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, C.J.; Yuan, D.; Masyuk, T.V.; Wang, X.; Punyashthiti, R.; Whelan, S.; Bacallao, R.; Torra, R.; LaRusso, N.F.; Torres, V.E.; et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003, 12, 2703–2710. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Mai, W.; Li, C.; Cho, S.Y.; Hao, C.; Moeckel, G.; Zhao, R.; Kim, I.; Wang, J.; Xiong, H.; et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2311–2316. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, W.E.; Avner, E.D. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res. 2006, 326, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef]
- Bergmann, C. Educational paper; ciliopathies. Eur. J. Pediatr. 2012, 171, 1285–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, C.; Kessler, K.; Giessl, A.; Dimmler, A.; Shalev, S.A.; Von Der Haar, S.; Zenker, M.; Zahnleiter, D.; Stöss, H.; Beinder, E.; et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011, 88, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavotinek, A.M.; Stone, E.M.; Mykytyn, K.; Heckenlively, J.R.; Green, J.S.; Heon, E.; Musarella, M.A.; Parfrey, P.S.; Sheffield, V.C.; Biesecker, L.G. Mutations in MKKS cause Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Beales, P.L.; Warner, A.M.; Hitman, G.A.; Thakker, R.; Flinter, F.A. Bardet-Biedl syndrome: A molecular and phenotypic study of 18 families. J. Med. Genet. 1997, 34, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Esmail, M.A.; Ansley, S.J.; Blacque, O.E.; Boroevich, K.; Ross, A.J.; Moore, S.J.; Badano, J.L.; May-Simera, H.; Compton, D.S.; et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat. Genet. 2004, 36, 989–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, F.; Santoni, M.; Matrana, M.R.; Satti, S.; Giulietti, M.; Occhipinti, G.; Massari, F.; Cheng, L.; Lopez-Beltran, A.; Scarpelli, M.; et al. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: Molecular diagnostics and possible targets for personalized therapies. Expert Rev. Mol. Diagn. 2015, 15, 1201–1210. [Google Scholar] [CrossRef]
- Montironi, R.; Santoni, M.; Scarpelli, M.; Piva, F.; Lopez-Beltran, A.; Cheng, L.; Briganti, A.; Montorsi, F. Re: Epithelial-to-mesenchymal transition in renal neoplasms. Eur. Urol. 2015, 68, 736–737. [Google Scholar] [CrossRef]
- Massari, F.; Ciccarese, C.; Santoni, M.; Brunelli, M.; Piva, F.; Modena, A.; Bimbatti, D.; Fantinel, E.; Santini, D.; Cheng, L.; et al. Metabolic alterations in renal cell carcinoma. Cancer Treat. Rev. 2015, 41, 767–776. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-De-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Occhipinti, G.; Santoni, M.; Massari, F.; Sotte, V.; Iacovelli, R.; Burattini, L.; Santini, D.; Montironi, R.; et al. Computational analysis of the mutations in BAP1, PBRM1 and SETD2 genes reveals the impaired molecular processes in Renal Cell Carcinoma. Oncotarget 2015, 6, 32161–32168. [Google Scholar] [CrossRef]
- Sabanovic, B.; Giulietti, M.; Piva, F. Role of primary cilium in pancreatic ductal adenocarcinoma (Review). Int. J. Oncol. 2020, 57, 1095–1102. [Google Scholar] [CrossRef]
- Yoder, B.K. Role of primary cilia in the pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lolkema, M.P.; Mehra, N.; Jorna, A.S.; Van Beest, M.; Giles, R.H.; Voest, E.E. The von Hippel-Lindau tumor suppressor protein influences microtubule dynamics at the cell periphery. Exp. Cell Res. 2004, 301, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Schermer, B.; Ghenoiu, C.; Bartram, M.; Müller, R.U.; Kotsis, F.; Höhne, M.; Kühn, W.; Rapka, M.; Nitschke, R.; Zentgraf, H.; et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J. Cell Biol. 2006, 175, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Harten, S.K.; Tran, M.G.; Maxwell, P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J. Am. Soc. Nephrol. 2006, 17, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.S.; Burk, R.D. Primary cilium formation requires von Hippel-Lindau gene function in renal-derived cells. Cancer Res. 2006, 66, 6903–6907. [Google Scholar] [CrossRef] [Green Version]
- Lolkema, M.P.; Mans, D.A.; Ulfman, L.H.; Volpi, S.; Voest, E.E.; Giles, R.H. Allele-specific regulation of primary cilia function by the von Hippel-Lindau tumor suppressor. Eur. J. Hum. Genet. 2008, 16, 73–78. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The primary cilium is a “central processing unit” of the cell that integrates different extracellular signals to regulate cell functioning. Different signaling pathways are shown.
Figure 1.
The primary cilium is a “central processing unit” of the cell that integrates different extracellular signals to regulate cell functioning. Different signaling pathways are shown.

Figure 2.
A selection of gene mutations associated with the development of renal ciliopathies.

Figure 3.
VHL loss in renal carcinogenesis. VHL-mutated tubular cells have been shown to not form the primary cilium. This event favors cyst formation and can lead to the development of renal cell carcinoma.
Figure 3.
VHL loss in renal carcinogenesis. VHL-mutated tubular cells have been shown to not form the primary cilium. This event favors cyst formation and can lead to the development of renal cell carcinoma.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Santoni, M.; Piva, F.; Cimadamore, A.; Giulietti, M.; Battelli, N.; Montironi, R.; Cosmai, L.; Porta, C. Exploring the Spectrum of Kidney Ciliopathies. Diagnostics 2020, 10, 1099. https://doi.org/10.3390/diagnostics10121099
AMA Style
Santoni M, Piva F, Cimadamore A, Giulietti M, Battelli N, Montironi R, Cosmai L, Porta C. Exploring the Spectrum of Kidney Ciliopathies. Diagnostics. 2020; 10(12):1099. https://doi.org/10.3390/diagnostics10121099
Chicago/Turabian StyleSantoni, Matteo, Francesco Piva, Alessia Cimadamore, Matteo Giulietti, Nicola Battelli, Rodolfo Montironi, Laura Cosmai, and Camillo Porta. 2020. "Exploring the Spectrum of Kidney Ciliopathies" Diagnostics 10, no. 12: 1099. https://doi.org/10.3390/diagnostics10121099
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.