
Using Data Mining and Network Analysis to Infer Arboviral Dynamics: The Case of Mosquito-Borne Flaviviruses Reported in Mexico

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Species Data
2.2. Data Analyses
2.3. Community Detection and Network Analysis
2.4. Predictive Models and Risk Maps
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roy, H.E.; Hesketh, H.; Purse, B.V.; Eilenberg, J.; Santini, A.; Scalera, R.; Stentiford, G.D.; Adriaens, T.; Bacela-Spychalska, K.; Bass, D.; et al. Alien pathogens on the horizon: Opportunities for predicting their threat to wildlife. Conserv. Lett. 2017, 10, 476–483. [Google Scholar] [CrossRef]
- Machado-Machado, E. Empirical mapping of suitability to dengue fever in Mexico using species distribution modeling. Appl. Geogr. 2012, 33, 82–93. [Google Scholar] [CrossRef]
- Samy, A.M.; Thomas, S.M.; Wahed, A.A.E.; Cohoon, K.P.; Peterson, A.T. Mapping the global geographic potential of Zika virus spread. Memorias do Instituto Oswaldo Cruz 2016, 111, 559–560. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, A.; Shackelton, L.; Holmes, E. Family level phylogenies reveal modes of macroevolution in RNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 238–243. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Health Organization [Internet]. Available online: http://www.who.int/en/ (accessed on 1 August 2017).
- Holzmann, I.; Agostini, I.; Areta, J.; Ferreyra, H.; Beldomenico, P.; di Bitetti, M. Impact of yellow fever outbreaks on two howler monkey species (Alouatta guariba clamitans and A. caraya) in Misiones, Argentina. Am. J. Primatol. 2010, 72, 475–480. [Google Scholar]
- Kilpatrick, A.M.; Peters, R.J.; Dupuis II, A.P.; Jones, M.J.; Daszak, P.; Marra, P.P.; Kramer, L.D. Predicted and observed mortality from vector-borne disease in wildlife: West Nile virus and small songbirds. Biol. Conserv. 2013, 165, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Ávila, J.E.; Rodríguez, M.H.; Santos-Luna, R.; Sánchez-Castañeda, V.; Román-Pérez, S.; Ríos-Salgado, V.H.; Salas-Sarmiento, J.A. Nation-wide, web-based, geographic information system for the integrated surveillance and control of dengue fever in Mexico. PLoS ONE 2013, 8, e70231. [Google Scholar] [CrossRef] [Green Version]
- Guerbois, M.; Fernandez-Salas, I.; Azar, S.R.; Danis-Lozano, R.; Alpuche-Aranda, C.M.; Leal, G.; Garcia-Malo, I.R.; Diaz-Gonzalez, E.E.; Casas-Martinez, M.; Rossi, S.L.; et al. Outbreak of Zika Virus Infection, Chiapas State, Mexico, 2015, and First Confirmed Transmission by Aedes aegypti Mosquitoes in the Americas. J. Infect. Dis. 2016, 214, 1349–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, A.M.; Fonseca, D.M.; Ebel, G.D.; Reddy, M.R.; Kramer, L.D. Spatial and temporal variation in vector competence of Culex pipiens and Cx. restuans mosquitoes for West Nile virus. Am. J. Trop. Med. Hyg. 2010, 83, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, S.; Tesh, R.; Vasilakis, N. The emergence of arthropod-borne viral diseases: A global prospective on dengue, chikungunya and zika fevers. Acta Trop. 2017, 166, 155–163. [Google Scholar] [CrossRef]
- Abundes-Gallegos, J.; Salas-Rojas, M.; Galvez-Romero, G.; Perea-Martínez, L.; Obregón-Morales, C.Y.; Morales-Malacara, J.B.; Chomel, B.B.; Stuckey, M.J.; Moreno-Sandoval, H.; García-Baltazar, A.; et al. Detection of Dengue virus in bat flies (Diptera: Streblidae) of common vampire bats, Desmodus rotundus, in Progreso, Hidalgo, Mexico. Vector-Borne Zoonotic Dis. 2018, 18, 70–73. [Google Scholar] [CrossRef]
- Aguilar-Setién, A.; Romero-Almaraz, M.L.; Sánchez-Hernández, C.; Figueroa, R.; Juárez-Palma, L.P.; Garcia-Flores, M.M.; Vázquez-Salinas, C.; Salas-Rojas, M.; Hidalgo-Martinez, A.C.; Pierlé, S.A.; et al. Dengue virus in Mexican bats. Epidemiol. Infect. 2008, 136, 1678–1683. [Google Scholar] [CrossRef] [PubMed]
- Loza-Rubio, E.; Rojas-Anaya, E.; Lopez-Ramirez, R.; Saiz, J. Prevalence of neutralizing antibodies against West Nile virus [WNV] in monkeys [Ateles geoffroyi and Alouatta pigra] and crocodiles [ Crocodylus acutus and C. acutus-C. moreletti hybrids] in Mexico. Epidemiol. Infect. 2016, 144, 2371–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cigarroa-Toledo, N.; Talavera-Aguilar, L.G.; Baak-Baak, C.M.; García-Rejón, J.E.; Hernandez-Betancourt, S.; Blitvich, B.J.; Machain-Williams, C. Serologic evidence of flavivirus infections in peridomestic rodents in Merida, Mexico. J. Wildl. Dis. 2015, 52, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Machain-Williams, C.; López-Uribe, M.; Talavera-Aguilar, L.; Carrillo-Navarrete, J.; Vera-Escalante, L.; Puerto-Manzano, F.; Ulloa, A.; Farfán-Ale, J.A.; Garcia-Rejon, J.; Blitvich, B.J.; et al. Serologic evidence of flavivirus infection in bats in the Yucatan peninsula of Mexico. J. Wildl. Dis. 2013, 49, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sotomayor-Bonilla, J.; Chaves, A.; Rico-Chávez, O.; Rostal, M.K.; Ojeda-Flores, R.; Salas-Rojas, M.; Aguilar-Setien, Á.; Ibáñez-Bernal, S.; Barbachano-Guerrero, A.; Gutiérrez-Espeleta, G.; et al. Dengue virus in bats from Southeastern Mexico. Am. J. Trop. Med. Hyg. 2014, 91, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Chaves, A.; Sotomayor-Bonilla, J.; Monge, O.; Ramírez, A.; Galindo, F.; Sarmiento-Silva, R.E.; Gutiérrez-Espeleta, G.A.; Suzán, G. West Nile virus in resident birds from Yucatan, Mexico. J. Wildl. Dis. 2016, 52, 159–163. [Google Scholar] [CrossRef]
- Estrada-Franco, J.G.; Navarro-Lopez, R.; Beasley, D.W.C.; Coffey, L.; Carrara, A.; Da Rosa, A.T.; Clements, T.; Wang, E.; Ludwig, G.V.; Cortes, A.C.; et al. West Nile virus in Mexico: Evidence of widespread circulation since July 2002. Emerg. Infect. Dis. 2003, 9, 1604–1607. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Salas, I.; Contreras-Cordero, J.F.; Blitvich, B.J.; González-Rojas, J.I.; Cavazos-Alvarez, A.; Marlenee, N.L.; Elizondo-Quiroga, A.; Loroño-Pino, M.A.; Gubler, D.J.; Cropp, B.C.; et al. Serologic evidence of West Nile virus infection in birds, Tamaulipas State, México. Vector Borne Zoonotic Dis. 2003, 3, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Sánchez, S.; Cuevas-Romero, S.; Nemeth, N.M.; Trujillo-Olivera, M.T.J.; Worwa, G.; Dupuis, A.; Brault, A.C.; Kramer, L.D.; Komar, N.; Estrada-Franco, J.G. West Nile Virus Infection of Birds, Mexico. Emerg. Infect. Dis. 2011, 17, 2245–2252. [Google Scholar] [CrossRef]
- Kopp, A.; Gillespie, T.R.; Hobelsberger, D.; Estrada, A.; Harper, J.M.; Miller, R.A.; Eckerle, I.; Müller, M.A.; Podsiadlowski, L.; Leendertz, F.H.; et al. Provenance and Geographic Spread of St. Louis Encephalitis Virus. mBio 2013, 4, e00322-13. [Google Scholar] [CrossRef] [Green Version]
- Auguste, A.J.; Pybus, O.G.; Carrington, C.V. Evolution and dispersal of St. Louis encephalitis virus in the Americas. Infect. Genet. Evol. 2009, 9, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Thoisy, B.D.; Lacoste, V.; Germain, A.; Muñoz-Jordán, J.; Colón, C.; Mauffrey, J.F.; Delaval, M.; Catzeflis, F.; Kazanji, M.; Matheus, S.; et al. Dengue infection in neotropical forest mammals. Vector-Borne Zoonotic Dis. 2009, 9, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auguste, A.J.; Lemey, P.; Bergren, N.A.; Giambalvo, D.; Moncada, M.; Morón, D.; Hernandez, R.; Navarro, J.C.; Weaver, S.C. Enzootic transmission of yellow fever virus, Venezuela. Emerg. Infect. Dis. 2015, 21, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Favoretto, S.; Araujo, D.; Oliveira, D.; Duarte, N.; Mesquita, F.; Zanotto, P.; Durigon, E. First detection of Zika virus in neotropical primates in Brazil: A possible new reservoir. bioRxiv 2016, 049395. [Google Scholar]
- Sulkin, S.; Sims, R.; Allen, R. Isolation of St. Louis encephalitis virus from bats (Tadarida b. mexicana) in Texas. Science (80-) 1966, 152, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, P.; Travassos da Rosa, J.; Travassos da Rosa, A.; Degallier, N.; Pinheiro, F.; Sa Filho, G. Epidemiologia das encefalites por arbovirus na Amazonia Barasileira. Rev. Inst. Med. Trop. São Paulo. 1991, 33, 465–476. [Google Scholar] [CrossRef]
- Ragan, I.; Blizzard, E.; Gordy, P.; Bowen, R. Investigating the potential role of North American animals as hosts for Zika virus. Vector-Borne Zoonotic Dis. 2017, 17, 161–164. [Google Scholar] [CrossRef]
- Gómez, A.; Kramer, L.D.; Dupuis, A.P., II; Kilpatrick, A.M.; Davis, L.J.; Jones, M.J.; Daszak, P.; Aguirre, A.A. Experimental infection of eastern gray squirrels (Sciurus carolinensis) with West Nile virus. Am. J. Trop. Med. Hyg. 2008, 79, 447–451. [Google Scholar] [CrossRef]
- Perea-Martínez, L.; Moreno-Sandoval, H.N.; Moreno-Altamirano, M.M.; Salas-Rojas, M.; García-Flores, M.M.; Aréchiga-Ceballos, N.; Tordo, N.; Marianneau, P.; Aguilar-Setién, A. Experimental infection of Artibeus intermedius bats with serotype-2 dengue virus. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 193–198. [Google Scholar] [CrossRef]
- Cabrera-Romo, S.; Recio-Tótoro, B.; Alcalá, A.C.; Lanz, H.; del Ángel, R.M.; Sánchez-Cordero, V.; Rodríguez-Moreno, Á.; Ludert, J.E. Experimental inoculation of artibeus jamaicensis bats with dengue virus serotypes 1 or 4 showed no evidence of sustained replication. Am. J. Trop. Med. Hyg. 2014, 91, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.; Gonzalez-Salazar, C.; Sánchez-Cordero, V.; Becker, I.; Rebollar-Tellez, E.; Rodriguez-Moreno, A.; Berzunza-Cruz, M.; Domingo Balcells, C.; Gutiérrez-Granados, G.; Hidalgo-Mihart, M.; et al. Can you judge a disease host by the company it keeps? Predicting disease hosts and their relative importance: A case study for leishmaniasis. PLoS Negl. Trop. Dis. 2016, 10, e0005004. [Google Scholar] [CrossRef] [PubMed]
- González-Salazar, C.; Stephens, C.; Sánchez-Cordero, V. Predicting the Potential Role of Non-human Hosts in Zika Virus Maintenance. Ecohealth 2017, 14, 171–177. [Google Scholar] [CrossRef]
- CDC. Arbovirus Catalog [Internet]. Center for Disease Control and Prevention. Available online: www.cdc.gov/arbocat/ (accessed on 1 August 2017).
- CDC. Mosquito Species in Which West Nile Virus Has been Detected [Internet]. Available online: https://www.cdc.gov/westnile/resources/pdfs/MosquitoSpecies1999-2012.pdf (accessed on 1 August 2017).
- Ibáñez-Bernal, S.; Briseño, B.; Mutebi, J.P.; Argot, E.; Rodríguez, G.; Martínez-Campos, C.; Paz, R.; Román, P.; Tapia-Conyer, R.; Flisser, A. First record in America of Aedes albopictus naturally infected with dengue virus during the 1995 outbreak at Reynosa, Mexico. Med. Vet. Entomol. 1997, 11, 305–309. [Google Scholar] [CrossRef]
- Thompson-Reuters. Web of Science [Internet]. Web of Science. Available online: https://www.webofknowledge.com/ (accessed on 1 August 2017).
- CONABIO. Comisión Nacional Para el Uso y el Conocimiento de la Biodiversidad [Internet]. Available online: http://species.conabio.gob.mx/ (accessed on 1 August 2017).
- De Thoisy, B.; Dussart, P.; Kazanji, M. Wild terrestrial rainforest mammals as potential reservoirs for flaviviruses (yellow fever, dengue 2 and St Louis encephalitis viruses) in French Guiana. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Blondel, V.D.; Guillaume, J.L.; Lambiotte, R.; Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. Theory Exp. 2008, 2008, P10008. [Google Scholar] [CrossRef] [Green Version]
- Moody, J.; White, D.R. Structural Cohesion and Embeddedness: A Hierarchical Concept of Social Groups. Am. Sociol. Rev. 2003, 68, 103–127. [Google Scholar] [CrossRef] [Green Version]
- Stephens, C.; Giménez-Heau, J.; González, C.; Ibarra-Cerdeña, C.; Sánchez-Cordero, V.; González-Salazar, C. Using biotic interaction networks for prediction in biodiversity and emerging diseases. PLoS ONE 2009, 4, e5725. [Google Scholar] [CrossRef]
- Diaz-Badillo, A.; Bolling, B.; Perez-Ramirez, G.; Moore, C.; Martinez-Munoz, J.; Padilla-Viveros, A.; Camacho-Nuez, M.; Diaz-Perez, A.; Beaty, B.; de Lourdes Munoz, M. The distribution of potential West Nile virus vectors, Culex pipiens pipiens and Culex pipiens quinquefasciatus (Diptera: Culicidae), in Mexico City. Parasites Vectors 2011, 4, 70. [Google Scholar] [CrossRef] [Green Version]
- Farfan-Ale, J.; Loroño-Pino, M.; Garcia-Rejon, J.; Soto, V.; Lin, M.; Staley, M.; Dorman, K.; Bartholomay, L.; Hovav, E.; Blitvich, B. Detection of flaviviruses and orthobunyaviruses in mosquitoes in the Yucatan Peninsula of Mexico in 2008. Vector-Borne Zoonotic Dis. 2010, 10, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Roche, B.; Rohani, P.; Dobson, A.P.; Guégan, J.-F. The Impact of Community Organization on Vector-Borne Pathogens. Am. Nat. 2013, 181, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, M.; Sinka, M.; Duda, K.; Mylne, A.; Shearer, F.; Barker, C.; Moore, C.; Carvalho, R.; Coelho, G.; Van Bortel, W. The global distribution of the arbovirus vectors Ae. aegypti and Ae. albopictus. eLife 2015, 4, e08347. [Google Scholar] [CrossRef]
- Halbach, R.; Junglen, S.; van Rij, R. Mosquito-specific and mosquito-borne viruses: Evolution, infection, and host defense. Curr. Opin. Insect Sci. 2017, 22, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Baak-Baak, C.; Moo-Llanes, D.; Cigarroa-Toledo, N.; Puerto, F.; Machain-Williams, C.; Reyes-Solis, G.; Nakazawa, Y.J.; Ulloa-Garcia, A.; Garcia-Rejon, J.E. Ecological niche model for predicting distribution of disease-vector mosquitoes in Yucatán State, México. J. Med. Entomol. 2017, 54, 854–861. [Google Scholar] [CrossRef]
- Garcia-Rejon, J.E.; Blitvich, B.J.; Farfan-Ale, J.A.; Loroño-Pino, M.A.; Chi Chim, W.A.; Flores-Flores, L.F.; Rosado-Paredes, E.; Baak-Baak, C.; Perez-Mutul, J.; Suarez-Solis, V.; et al. Host-feeding preference of the mosquito, Culex quinquefasciatus, in Yucatan State, Mexico. J. Insect Sci. 2010, 10, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elizondo-Quiroga, D.; Medina-Sánchez, A.; Sánchez-González, J.; Eckert, K.; Villalobos-Sánchez, E.; Navarro-Zúñiga, A.R.; Sánchez-Tejeda, G.; Correa-Morales, F.; González-Acosta, C.; Arias, C.F.; et al. Zika virus in salivary glands of five different species of wild-caught mosquitoes from Mexico. Sci. Rep. 2018, 8, 809. [Google Scholar] [CrossRef] [Green Version]
- De Rodaniche, E.; Galindo, P.; Johnson, C. Isolation of yellow fever virus from Haemagogus lucifer, H. equinus, H. spegazzinii falco, Sabethes chloropterus and Anopheles neivai captured in Panama in the fall of 1956. Am. J. Trop. Med. Hyg. 1957, 6, 681–685. [Google Scholar] [CrossRef]
- De Figueiredo, M.; de CGomes, A.; Amarilla, A.; de SLeandro, A.; de Sorrico, A.; de Araujo, R.; do SM Castro, J.; Durigon, E.; Aquino, V.; Figueiredo, L.T. Mosquitoes infected with dengue viruses in Brazil. Virol. J. 2010, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Sotomayor-Bonilla, J.; Abella-Medrano, C.; Chaves, A.; Álvarez-Mendizábal, P.; Rico-Chávez, O.; Ibáñez-Bernal, S.; Rostal, M.K.; Ojeda-Flores, R.; Barbachano-Guerrero, A.; Gutiérrez-Espeleta, G.; et al. Potential sympatric vectors and mammalian hosts of Venezuelan equine encephalitis virus in Southern Mexico. J. Wildl. Dis. 2017, 53, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Reiskind, M.; Griffin, R.; Janairo, M.; Hopperstad, K. Mosquitoes of field and forest: The scale of habitat segregation in a diverse mosquito assemblage. Med. Vet. Entomol. 2017, 31, 44–54. [Google Scholar] [CrossRef]
- Espinoza-Gómez, F.; Arredondo-Jiménez, J.; Maldonado-Rodríguez, A.; Pérez-Rentería, C.; Newton-Sánchez, O.; Chávez-Flores, E.; Gómez-Ibarra, E. Distribución geográfica de mosquitos adultos (Diptera: Culicidae) en áreas selváticas de Colima, México. Rev. Mex. Biodivers. 2013, 84, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Vasilakis, N.; Cardosa, J.; Hanley, K.; Holmes, E.; Weaver, S. Fever from the forest: Prospects for the continued emergence of sylvatic dengue virus and its impact on public health. Nat. Rev. Microbiol. 2011, 9, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Santos, A.; Moreira-Soto, A.; Soto-Garita, C.; Chaverri, L.; Chaves, A.; Drexler, J.; Morales, J.; Alfaro-Alarcón, A.; Rodríguez-Herrera, B.; Corrales-Aguilar, E. Neotropical bats that co-habit with humans function as dead-end hosts for dengue virus. PLoS Negl. Trop. Dis. 2017, 11, e0005537. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.; Holmes, E.; Barrett, A. Out of Africa: A molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 2007, 3, 668–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisen, W. Epidemiology of St. Louis encephalitis virus. Adv. Virus. Res. 2003, 16, 139–184. [Google Scholar]
- Shepard, J.; Andreadis, T.; Thomas, M.; Molaei, G. Host associations of mosquitoes at eastern equine encephalitis virus foci in Connecticut, USA. Parasites Vectors 2016, 9, 474. [Google Scholar] [CrossRef] [Green Version]
- Komar, N.; Langevin, S.; Hinten, S.; Nemeth, N.; Edwards, E.; Hettler, D.; Davis, B.; Bowen, R.; Bunning, M. Experimental Infection of North American Birds with the New York 1999 Strain of West Nile Virus. Emerg. Infect. Dis. 2003, 9, 311–322. [Google Scholar] [CrossRef]
- Root, J. West Nile virus associations in wild mammals: A synthesis. Arch. Virol. 2013, 158, 735–752. [Google Scholar] [CrossRef]
- Ariel, E. Viruses in reptiles. Vet. Res. 2011, 42, 100. [Google Scholar] [CrossRef] [Green Version]
- Ulloa, A.; Langevin, S.; Mendez-Sanchez, J.; Arredondo-Jimenez, J.; Raetz, J.; Powers, A.; Villarreal-Trevino, C.; Gubler, D.J.; Komar, N. Serologic survey of domestic animals for zoonotic arbovirus infections in the Lacandón Forest region of Chiapas, Mexico. Vector-Borne Zoonotic Dis. 2003, 3, 3–9. [Google Scholar] [CrossRef]
- Auguste, A.; Lemey, P.; Pybus, O.; Suchard, M.; Salas, R.; Adesiyun, A.; Barrett, A.; Tesh, R.; Weaver, S.; Carrington, C. Yellow fever virus maintenance in Trinidad and its dispersal throughout the Americas. J. Virol. 2010, 84, 9967–9977. [Google Scholar] [CrossRef] [Green Version]
- Gervasi, S.; Civitello, D.; Kilvitis, H.; Martin, L. The context of host competence: A role for plasticity in host-parasite dynamics. Trends Parasitol. 2015, 31, 419–425. [Google Scholar] [CrossRef]
- Aguirre, A.; McLean, R.; Cook, R.; Quan, T. Serologic survey for selected arboviruses and other potential pathogens in wildlife from Mexico. J. Wildl. Dis. 1992, 28, 435–442. [Google Scholar] [CrossRef]
- Torres-Castro, M.; Poot-Pérez, M.; Moguel-Lehmer, C.; Reyes-Hernández, B.; Panti-May, A.; Noh-Pech, H.; Hernández-Betancourt, S.; Medina-Espinosa, N.; Puerto, F. Detección molecular de Flavivirus en suero sanguíneo de roedores capturados en Yucatán, México. Rev. Investig. Vet. Peru. 2017, 28, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Peña, A.; De La Fuente, J.; Ostfeld, R.; Cabezas-Cruz, A. Interactions between tick and transmitted pathogens evolved to minimise competition through nested and coherent networks. Sci. Rep. 2015, 5, 10361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisen, W. Landscape epidemiology of vector-borne diseases. Annu. Rev. Entomol. 2010, 55, 461–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takken, W.; Verhulst, N. Host preferences of blood-feeding mosquitoes. Annu. Rev. Entomol. 2013, 58, 433–453. [Google Scholar] [CrossRef] [Green Version]
- Makanga, B.; Costantini, C.; Rahola, N.; Yangari, P.; Rougeron, V.; Ayala, D.; Prugnolle, F.; Paupy, C. Show me which parasites you carry and I will tell you what you eat, or how to infer the trophic behavior of hematophagous arthropods feeding on wildlife. Ecol. Evol. 2017, 7, 7578–7584. [Google Scholar] [CrossRef] [Green Version]
- Borkent, A.; Belton, P. Attraction of female Uranotaenia lowii (Diptera: Culicidae) to frog calls in Costa Rica. Can. Entomol. 2006, 138, 91–94. [Google Scholar] [CrossRef]
- Chaverri, L. Culicidae (Mosquitos, Zancudos). In Manual of Central American Diptera; Brown, B., Borkent, A., Cumming, J., Wood, D., Woodley, N., Zumbado, M., Eds.; NRC Research Press: Ottawa, ON, Canada, 2009; pp. 369–388. [Google Scholar]
- Stephens, C.R.; Sánchez-Cordero, V.; Salazar, C.G. Bayesian inference of ecological interactions from spatial data. Entropy 2007, 19, 547. [Google Scholar] [CrossRef] [Green Version]
- Stephens, C.R.; González-Salazar, C.; Villalobos, M.; Marquet, P. Can Ecological Interactions be Inferred from Spatial Data? Biodivers. Inform. 2020, 15, 11–54. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
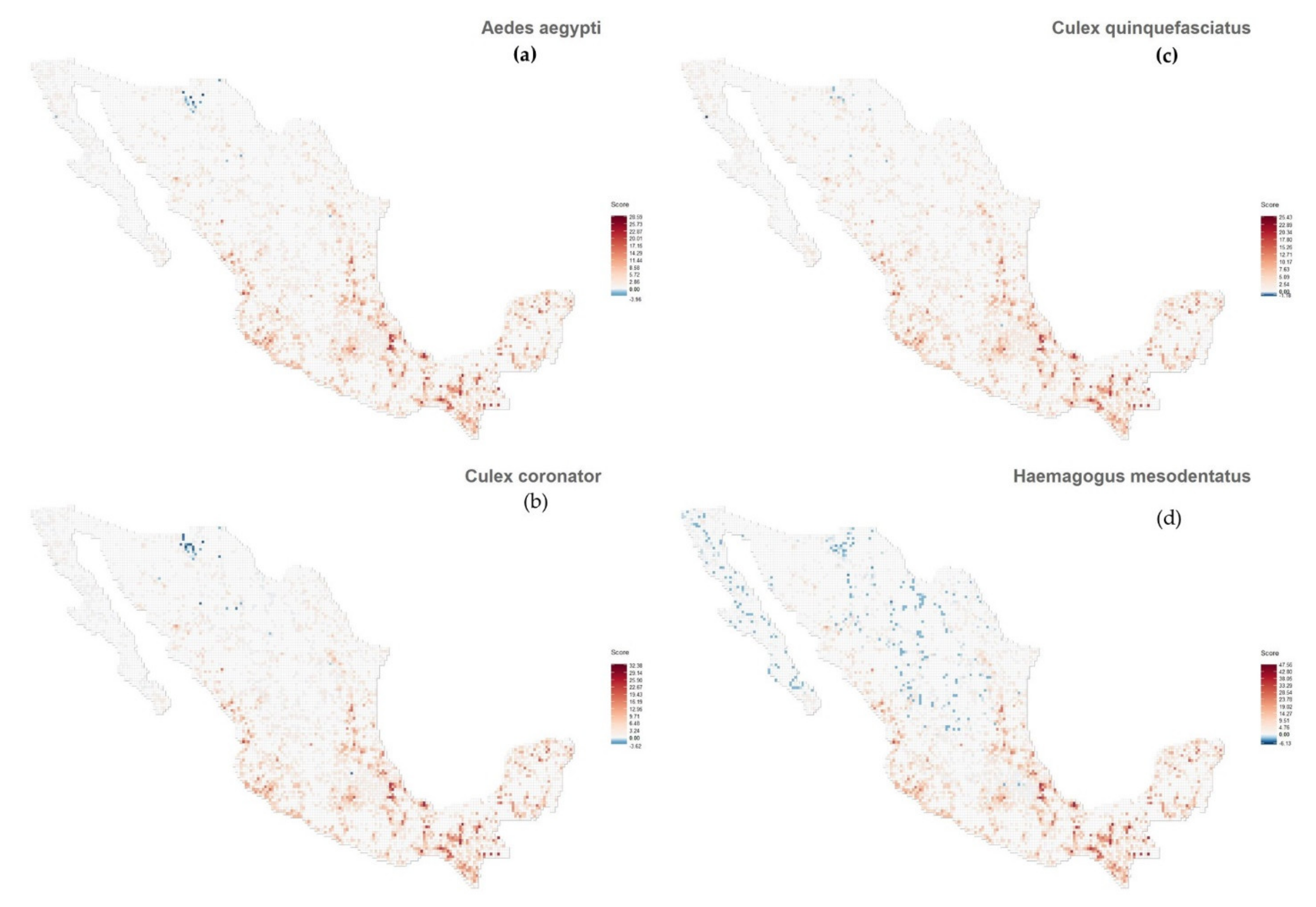{kind=link}
{kind=link}
| Ranking | Species | Epsilon DENV | Epsilon YFV | Epsilon SLEV | Epsilon WNV | Weighted Mean | Sample Area Proportion | # of Confirmed Pathogens |
|---|---|---|---|---|---|---|---|---|
| 1 | Aedes aegypti | * 11.148 | * 2.969 | * 10.568 | * 7.038 | 9.254 | 0.0697 | 3 |
| 2 | Culex coronator | 11.132 | 5.773 | * 9.632 | * 4.978 | 8.426 | 0.0307 | 2 |
| 3 | Culex quinquefasciatus | 7.217 | 2.898 | * 8.028 | * 6.161 | 6.917 | 0.0517 | 2 |
| 4 | Culex nigripalpus | 9.670 | 7.122 | * 6.573 | * 2.853 | 6.396 | 0.0094 | 2 |
| 5 | Haemagogus mesodentatus | 9.864 | * 4.910 | 6.535 | 2.500 | 6.230 | 0.0027 | 1 |
| 6 | Aedes albopictus | * 7.300 | 1.296 | 6.566 | * 4.772 | 5.976 | 0.014 | 2 |
| 7 | Sabethes chloropterus | 10.121 | * 9.354 | * 5.361 | 1.643 | 5.886 | 0.0021 | 2 |
| 8 | Culex erraticus | 7.003 | 6.018 | 6.258 | * 3.401 | 5.559 | 0.0067 | 1 |
| 9 | Psorophora howardii | 7.112 | 2.919 | 6.204 | * 3.704 | 5.533 | 0.0012 | 1 |
| 10 | Aedes scapularis | 6.887 | 8.498 | * 5.05 | 2.389 | 4.942 | 0.0053 | 1 |
| 11 | Aedes taeniorhynchus | 7.537 | 3.808 | 5.361 | * 1.847 | 4.853 | 0.0072 | 1 |
| 12 | Anopheles crucians | 6.565 | 6.230 | * 4.653 | 2.005 | 4.487 | 0.0033 | 1 |
| 13 | Psorophora ferox | 6.641 | 3.917 | * 3.947 | * 1.953 | 4.174 | 0.0039 | 2 |
| 14 | Mansonia titillans | 6.262 | 3.735 | 4.037 | * 1.368 | 3.878 | 0.0028 | 1 |
| 15 | Deinocerites pseudes | 5.277 | −0.399 | * 5.040 | 1.746 | 3.791 | 0.0021 | 1 |
| 16 | Culex thriambus | 3.019 | −0.025 | 4.546 | * 3.719 | 3.567 | 0.0065 | 1 |
| 17 | Culex pipiens | 3.025 | 4.068 | * 3.379 | * 3.255 | 3.256 | 0.0024 | 2 |
| 18 | Deinocerites cancer | 3.887 | 3.692 | 3.632 | * 1.918 | 3.160 | 0.0008 | 1 |
| 19 | Aedes condolescens | 5.169 | −0.028 | 2.954 | *0.946 | 2.883 | 0.0001 | 1 |
| 20 | Culex stigmatosoma | 1.569 | −0.688 | 3.597 | * 3.767 | 2.793 | 0.0319 | 1 |
| 21 | Uranotaenia lowii | 3.669 | 2.637 | 3.663 | * 1.117 | 2.788 | 0.0029 | 1 |
| 22 | Aedes infirmatus | 4.305 | 5.954 | 2.691 | * 0.608 | 2.690 | 0.0007 | 1 |
| 23 | Aedes sollicitans | 2.985 | 1.205 | 3.100 | * 2.061 | 2.636 | 0.004 | 1 |
| 24 | Uranotaenia sapphirina | 3.693 | 0.884 | 3.045 | * 0.941 | 2.470 | 0.0019 | 1 |
| 25 | Anopheles punctipennis | 0.649 | −0.690 | 2.656 | * 4.327 | 2.391 | 0.0059 | 1 |
| 26 | Culex salinarius | 2.239 | 1.657 | 2.604 | * 2.248 | 2.325 | 0.0027 | 1 |
| 27 | Culex tarsalis | −0.109 | −0.243 | * 2.681 | * 4.454 | 2.214 | 0.0086 | 2 |
| 28 | Culex taeniopus | 2.738 | 3.256 | * 2.682 | 0.910 | 2.150 | 0.001 | 1 |
| 29 | Aedes trivittatus | 2.077 | −0.426 | 2.568 | * 2.001 | 2.084 | 0.0023 | 1 |
| 30 | Aedes triseriatus | 0.793 | 3.256 | 1.868 | * 2.724 | 1.861 | 0.001 | 1 |
| 31 | Anopheles atropos | 2.463 | 5.839 | 1.552 | * 0.889 | 1.832 | 0.0001 | 1 |
| 32 | Culiseta inornata | 0.245 | −0.426 | 1.942 | *3.545 | 1.801 | 0.0023 | 1 |
| 33 | Culex bahamensis | 2.075 | 5.693 | 1.67 | * 1.067 | 1.793 | 0.0004 | 1 |
| 34 | Aedes fulvus | 2.75 | 5.056 | 1.125 | * 0.859 | 1.750 | 0.0004 | 1 |
| 35 | Culiseta particeps | 0.38 | −0.496 | 1.995 | * 2.934 | 1.659 | 0.0123 | 1 |
| 36 | Culex restuans | 0.844 | 0.459 | 1.995 | * 2.261 | 1.635 | 0.0033 | 1 |
| 37 | Aedes atlanticus | 3.016 | 5.732 | 1.195 | * −0.093 | 1.579 | 0.0001 | 1 |
| 38 | Culex habilitator | 3.016 | 5.732 | 1.195 | * −0.093 | 1.579 | 0.0001 | 1 |
| 39 | Anopheles bradleyi | 1.281 | 3.442 | 1.876 | * 0.962 | 1.457 | 0.0009 | 1 |
| 40 | Culex apicalis | 1.25 | −0.238 | 1.772 | * 1.334 | 1.366 | 0.0008 | 1 |
| 41 | Culex peus | 0.283 | −0.28 | *1.525 | 2.465 | 1.343 | 0.0011 | 1 |
| 42 | Aedes squamiger | 1.16 | 5.732 | 1.305 | * 0.925 | 1.340 | 0.0001 | 1 |
| 43 | Anopheles quadrimaculatus | 0.358 | −0.380 | 1.644 | * 1.592 | 1.114 | 0.0019 | 1 |
| 44 | Anopheles barberi | −0.109 | −0.028 | 1.019 | * 2.533 | 1.096 | 0.0001 | 1 |
| 45 | Aedes dupreei | 0.188 | −0.080 | 0.764 | * 2.084 | 0.967 | 0.0002 | 1 |
| 46 | Coquillettidia perturbans | 0.963 | −0.142 | 1.261 | * 0.412 | 0.822 | 0.0004 | 1 |
| 47 | Culex erythrothorax | −0.094 | −0.166 | 0.715 | * 1.944 | 0.811 | 0.0004 | 1 |
| 48 | Anopheles franciscanus | 0.455 | −0.328 | 1.037 | * 0.997 | 0.771 | 0.0014 | 1 |
| 49 | Culex territans | 0.322 | −0.115 | 0.913 | * 1.198 | 0.766 | 0.0003 | 1 |
| 50 | Aedes melanimon | 0.135 | −0.080 | 1.302 | * 0.549 | 0.612 | 0.0002 | 1 |
| 51 | Culiseta incidens | −0.529 | −0.267 | 0.596 | * 1.722 | 0.556 | 0.001 | 1 |
| 52 | Culiseta melanura | −0.193 | −0.080 | 0.686 | * 1.036 | 0.478 | 0.0002 | 1 |
| 53 | Anopheles freeborni | −0.252 | −0.115 | 0.379 | * 1.288 | 0.446 | 0.0003 | 1 |
| 54 | Aedes vexans | −0.458 | −0.328 | 0.453 | * 1.388 | 0.425 | 0.0014 | 1 |
| 55 | Aedes dorsalis | −0.109 | −0.028 | * 0.207 | * 0.889 | 0.315 | 0.0001 | 2 |
| 56 | Psorophora columbiae | −0.476 | −0.305 | 0.516 | * 0.958 | 0.299 | 0.0013 | 1 |
| 57 | Psorophora signipennis | −0.554 | −0.280 | 0.663 | * 0.556 | 0.188 | 0.0011 | 1 |
| 58 | Orthopodomyia alba | −0.109 | −0.028 | 0.267 | * 0.294 | 0.140 | 0.0001 | 1 |
| 59 | Culiseta impatiens | 0.500 | −0.115 | 0.159 | * −0.234 | 0.129 | 0.0003 | 1 |
| 60 | Aedes nigromaculis | −0.109 | −0.028 | −0.151 | * −0.093 | −0.113 | 0.0001 | 1 |
| Model | R2 | F | p | t | p |
|---|---|---|---|---|---|
| DENV | 0.71 | 19.63 | 0.002 | 4.43 | 0.002 |
| YFV | 0.46 | 6.81 | 0.031 | 2.61 | 0.031 |
| SLEV | 0.72 | 20.58 | 0.002 | 4.54 | 0.002 |
| WNV | 0.0003 | 0.00 | 0.962 | −0.05 | 0.962 |
| Weighted epsilon | 0.64 | 14.49 | 0.005 | 3.81 | 0.005 |
| Sample area | 0.91 | 82.04 | 0.0002 | 9.06 | 0.0002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sotomayor-Bonilla, J.; Callejo-Canal, E.D.; González-Salazar, C.; Suzán, G.; Stephens, C.R. Using Data Mining and Network Analysis to Infer Arboviral Dynamics: The Case of Mosquito-Borne Flaviviruses Reported in Mexico. Insects 2021, 12, 398. https://doi.org/10.3390/insects12050398
Sotomayor-Bonilla J, Callejo-Canal ED, González-Salazar C, Suzán G, Stephens CR. Using Data Mining and Network Analysis to Infer Arboviral Dynamics: The Case of Mosquito-Borne Flaviviruses Reported in Mexico. Insects. 2021; 12(5):398. https://doi.org/10.3390/insects12050398
Chicago/Turabian StyleSotomayor-Bonilla, Jesús, Enrique Del Callejo-Canal, Constantino González-Salazar, Gerardo Suzán, and Christopher R. Stephens. 2021. "Using Data Mining and Network Analysis to Infer Arboviral Dynamics: The Case of Mosquito-Borne Flaviviruses Reported in Mexico" Insects 12, no. 5: 398. https://doi.org/10.3390/insects12050398