
The Di-Symbiotic Systems in the Aphids Sipha maydis and Periphyllus lyropictus Provide a Contrasting Picture of Recent Co-Obligate Nutritional Endosymbiosis in Aphids

 , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Prevalence of S. symbiotica in S. maydis Populations
2.2. Establishment of Aphid Clonal Lines and Rearing Conditions
2.3. Genome Sequencing and Assemblies
2.4. Genome Annotation and Comparative Analyses
2.5. Molecular Phylogenetic Analyses
2.6. Examination of the Tissue Tropism of S. symbiotica
3. Results and Discussion
3.1. S. symbiotica Is a Fixed Symbiont in S. maydis
3.2. Genome Sequencing of the B. aphidicola/S. symbiotica Consortium
3.3. General Genomic Features of the Different Symbionts
3.4. Phylogenetic Positioning of SsSm and SsPl
3.5. Tissue Tropism of SsSm and SsPl
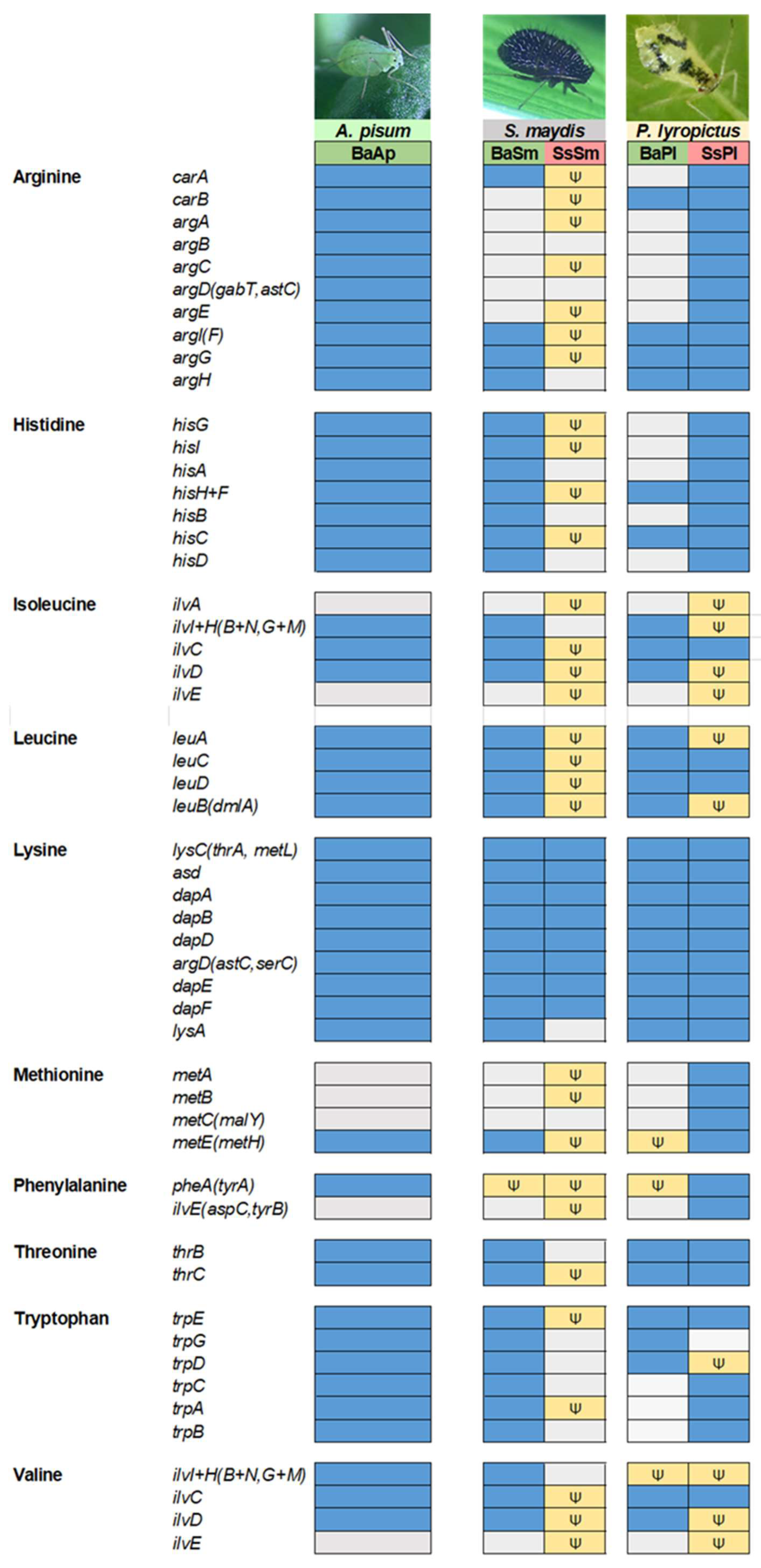
3.6. In the Two Chaitophorinae Aphids, S. symbiotica and B. aphidicola Complement Each Other Metabolically but in Distinct Layouts
3.7. The Secretion Systems and the Array of Virulence Factors Encoded by S. symbiotica Genomes
4. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baumann, P.; Moran, N.A.; Baumann, L.C. Bacteriocyte-Associated Endosymbionts of Insects. In The Prokaryotes: Prokaryotic Biology and Symbiotic Associations; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 465–496. ISBN 978-3-642-30194-0. [Google Scholar]
- Whittle, M.; Barreaux, A.M.G.; Bonsall, M.B.; Ponton, F.; English, S. Insect-Host Control of Obligate, Intracellular Symbiont Density. Proc. R. Soc. B Biol. Sci. 2021, 288, 20211993. [Google Scholar] [CrossRef] [PubMed]
- Koga, R.; Meng, X.-Y.; Tsuchida, T.; Fukatsu, T. Cellular Mechanism for Selective Vertical Transmission of an Obligate Insect Symbiont at the Bacteriocyte–Embryo Interface. Proc. Natl. Acad. Sci. USA 2012, 109, E1230–E1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomicki, G.; Werner, G.D.A.; West, S.A.; Kiers, E.T. Compartmentalization Drives the Evolution of Symbiotic Cooperation. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190602. [Google Scholar] [CrossRef] [PubMed]
- Moran, N.A. Accelerated Evolution and Muller’s Rachet in Endosymbiotic Bacteria. Proc. Natl. Acad. Sci. USA 1996, 93, 2873–2878. [Google Scholar] [CrossRef] [Green Version]
- Wernegreen, J.J. Endosymbiont Evolution: Predictions from Theory and Surprises from Genomes. Ann. N. Y. Acad. Sci. 2015, 1360, 16–35. [Google Scholar] [CrossRef] [Green Version]
- McCutcheon, J.P.; Boyd, B.M.; Dale, C. The Life of an Insect Endosymbiont from the Cradle to the Grave. Curr. Biol. 2019, 29, R485–R495. [Google Scholar] [CrossRef]
- Tamas, I.; Klasson, L.; Canbäck, B.; Näslund, A.K.; Eriksson, A.-S.; Wernegreen, J.J.; Sandström, J.P.; Moran, N.A.; Andersson, S.G.E. 50 Million Years of Genomic Stasis in Endosymbiotic Bacteria. Science 2002, 296, 2376–2379. [Google Scholar] [CrossRef] [Green Version]
- Moran, N.A.; Bennett, G.M. The Tiniest Tiny Genomes. Annu. Rev. Microbiol. 2014, 68, 195–215. [Google Scholar] [CrossRef]
- Bennett, G.M.; Moran, N.A. Heritable Symbiosis: The Advantages and Perils of an Evolutionary Rabbit Hole. Proc. Natl. Acad. Sci. USA 2015, 112, 10169–10176. [Google Scholar] [CrossRef] [Green Version]
- Renoz, F.; Pons, I.; Hance, T. Evolutionary Responses of Mutualistic Insect–Bacterial Symbioses in a World of Fluctuating Temperatures. Curr. Opin. Insect Sci. 2019, 35, 20–26. [Google Scholar] [CrossRef]
- Moran, N.A.; McCutcheon, J.P.; Nakabachi, A. Genomics and Evolution of Heritable Bacterial Symbionts. Annu. Rev. Genet. 2008, 42, 165–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, A.E. How Multi-Partner Endosymbioses Function. Nat. Rev. Microbiol. 2016, 14, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Daugherty, S.C.; Aken, S.E.V.; Pai, G.H.; Watkins, K.L.; Khouri, H.; Tallon, L.J.; Zaborsky, J.M.; Dunbar, H.E.; Tran, P.L.; et al. Metabolic Complementarity and Genomics of the Dual Bacterial Symbiosis of Sharpshooters. PLoS Biol. 2006, 4, e188. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.P.; von Dohlen, C.D. An Interdependent Metabolic Patchwork in the Nested Symbiosis of Mealybugs. Curr. Biol. CB 2011, 21, 1366–1372. [Google Scholar] [CrossRef] [Green Version]
- Van Leuven, J.T.; Meister, R.C.; Simon, C.; McCutcheon, J.P. Sympatric Speciation in a Bacterial Endosymbiont Results in Two Genomes with the Functionality of One. Cell 2014, 158, 1270–1280. [Google Scholar] [CrossRef] [Green Version]
- Husnik, F.; McCutcheon, J.P. Repeated Replacement of an Intrabacterial Symbiont in the Tripartite Nested Mealybug Symbiosis. Proc. Natl. Acad. Sci. USA 2016, 113, E5416–E5424. [Google Scholar] [CrossRef] [Green Version]
- Manzano-Marín, A.; Latorre, A. Snapshots of a Shrinking Partner: Genome Reduction in Serratia Symbiotica. Sci. Rep. 2016, 6, 32590. [Google Scholar] [CrossRef]
- Manzano-Marín, A.; Szabó, G.; Simon, J.-C.; Horn, M.; Latorre, A. Happens in the Best of Subfamilies: Establishment and Repeated Replacements of Co-Obligate Secondary Endosymbionts within Lachninae Aphids. Environ. Microbiol. 2017, 19, 393–408. [Google Scholar] [CrossRef]
- Manzano-Marín, A.; Coeur D’Acier, A.; Clamens, A.-L.; Orvain, C.; Cruaud, C.; Barbe, V.; Jousselin, E. A Freeloader? The Highly Eroded Yet Large Genome of the Serratia Symbiotica Symbiont of Cinara Strobi. Genome Biol. Evol. 2018, 10, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Manzano-Marín, A.; D’acier, A.C.; Clamens, A.-L.; Orvain, C.; Cruaud, C.; Barbe, V.; Jousselin, E. Serial Horizontal Transfer of Vitamin-Biosynthetic Genes Enables the Establishment of New Nutritional Symbionts in Aphids’ Di-Symbiotic Systems. ISME J. 2020, 14, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Monnin, D.; Jackson, R.; Kiers, E.T.; Bunker, M.; Ellers, J.; Henry, L.M. Parallel Evolution in the Integration of a Co-Obligate Aphid Symbiosis. Curr. Biol. 2020, 30, 1949–1957.e6. [Google Scholar] [CrossRef] [PubMed]
- Koga, R.; Moran, N.A. Swapping Symbionts in Spittlebugs: Evolutionary Replacement of a Reduced Genome Symbiont. ISME J. 2014, 8, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, Y.; Moriyama, M.; Łukasik, P.; Vanderpool, D.; Tanahashi, M.; Meng, X.-Y.; McCutcheon, J.P.; Fukatsu, T. Recurrent Symbiont Recruitment from Fungal Parasites in Cicadas. Proc. Natl. Acad. Sci. USA 2018, 115, E5970–E5979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano-Marín, A.; Latorre, A. Settling Down: The Genome of Serratia Symbiotica from the Aphid Cinara Tujafilina Zooms in on the Process of Accommodation to a Cooperative Intracellular Life. Genome Biol. Evol. 2014, 6, 1683–1698. [Google Scholar] [CrossRef] [PubMed]
- Manzano-Marín, A.; Simon, J.-C.; Latorre, A. Reinventing the Wheel and Making It Round Again: Evolutionary Convergence in Buchnera–Serratia Symbiotic Consortia between the Distantly Related Lachninae Aphids Tuberolachnus Salignus and Cinara Cedri. Genome Biol. Evol. 2016, 8, 1440–1458. [Google Scholar] [CrossRef] [Green Version]
- Meseguer, A.S.; Manzano-Marín, A.; d’Acier, A.C.; Clamens, A.-L.; Godefroid, M.; Jousselin, E. Buchnera Has Changed Flatmate but the Repeated Replacement of Co-Obligate Symbionts Is Not Associated with the Ecological Expansions of Their Aphid Hosts. Mol. Ecol. 2017, 26, 2363–2378. [Google Scholar] [CrossRef]
- Fukatsu, T.; Nikoh, N.; Kawai, R.; Koga, R. The Secondary Endosymbiotic Bacterium of the Pea Aphid Acyrthosiphon Pisum (Insecta: Homoptera). Appl. Environ. Microbiol. 2000, 66, 2748–2758. [Google Scholar] [CrossRef] [Green Version]
- Renoz, F.; Noël, C.; Errachid, A.; Foray, V.; Hance, T. Infection Dynamic of Symbiotic Bacteria in the Pea Aphid Acyrthosiphon Pisum Gut and Host Immune Response at the Early Steps in the Infection Process. PLoS ONE 2015, 10, e0122099. [Google Scholar] [CrossRef]
- Renoz, F.; Lopes, M.R.; Gaget, K.; Duport, G.; Eloy, M.-C.; Geelhand de Merxem, B.; Hance, T.; Calevro, F. Compartmentalized into Bacteriocytes but Highly Invasive: The Puzzling Case of the Co-Obligate Symbiont Serratia Symbiotica in the Aphid Periphyllus Lyropictus. Microbiol. Spectr. 2022, 10, e0045722. [Google Scholar] [CrossRef]
- Jousselin, E.; Clamens, A.-L.; Galan, M.; Bernard, M.; Maman, S.; Gschloessl, B.; Duport, G.; Meseguer, A.S.; Calevro, F.; D’acier, A.C. Assessment of a 16S RRNA Amplicon Illumina Sequencing Procedure for Studying the Microbiome of a Symbiont-Rich Aphid Genus. Mol. Ecol. Resour. 2016, 16, 628–640. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of Long, Error-Prone Reads Using Repeat Graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Médigue, C.; Calteau, A.; Cruveiller, S.; Gachet, M.; Gautreau, G.; Josso, A.; Lajus, A.; Langlois, J.; Pereira, H.; Planel, R.; et al. MicroScope—an Integrated Resource for Community Expertise of Gene Functions and Comparative Analysis of Microbial Genomic and Metabolic Data. Brief. Bioinform. 2019, 20, 1071–1084. [Google Scholar] [CrossRef]
- Vallenet, D.; Calteau, A.; Dubois, M.; Amours, P.; Bazin, A.; Beuvin, M.; Burlot, L.; Bussell, X.; Fouteau, S.; Gautreau, G.; et al. MicroScope: An Integrated Platform for the Annotation and Exploration of Microbial Gene Functions through Genomic, Pangenomic and Metabolic Comparative Analysis. Nucleic Acids Res. 2020, 48, D579–D589. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc Database of Metabolic Pathways and Enzymes—A 2019 Update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- Lerat, E.; Ochman, H. Recognizing the Pseudogenes in Bacterial Genomes. Nucleic Acids Res. 2005, 33, 3125–3132. [Google Scholar] [CrossRef]
- Burke, G.R.; Moran, N.A. Massive Genomic Decay in Serratia Symbiotica, a Recently Evolved Symbiont of Aphids. Genome Biol. Evol. 2011, 3, 195–208. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Abby, S.S.; Néron, B.; Ménager, H.; Touchon, M.; Rocha, E.P.C. MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic Orthology Inference for Comparative Genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.A.; Dunn, C.W. Phyutility: A Phyloinformatics Tool for Trees, Alignments and Molecular Data. Bioinformatics 2008, 24, 715–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Anisimova, M.; Gil, M.; Dufayard, J.-F.; Dessimoz, C.; Gascuel, O. Survey of Branch Support Methods Demonstrates Accuracy, Power, and Robustness of Fast Likelihood-Based Approximation Schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef]
- Koga, R.; Tsuchida, T.; Fukatsu, T. Quenching Autofluorescence of Insect Tissues for in Situ Detection of Endosymbionts. Appl. Entomol. Zool. 2009, 44, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Koga, R.; Tsuchida, T.; Fukatsu, T. Changing Partners in an Obligate Symbiosis: A Facultative Endosymbiont Can Compensate for Loss of the Essential Endosymbiont Buchnera in an Aphid. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 2543–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, K.M.; Degnan, P.H.; Burke, G.R.; Moran, N.A. Facultative Symbionts in Aphids and the Horizontal Transfer of Ecologically Important Traits. Annu. Rev. Entomol. 2010, 55, 247–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rock, D.I.; Smith, A.H.; Joffe, J.; Albertus, A.; Wong, N.; O’Connor, M.; Oliver, K.M.; Russell, J.A. Context-Dependent Vertical Transmission Shapes Strong Endosymbiont Community Structure in the Pea Aphid, Acyrthosiphon Pisum. Mol. Ecol. 2018, 27, 2039–2056. [Google Scholar] [CrossRef]
- Zytynska, S.E.; Tighiouart, K.; Frago, E. Benefits and Costs of Hosting Facultative Symbionts in Plant-Sucking Insects: A Meta-Analysis. Mol. Ecol. 2021, 30, 2483–2494. [Google Scholar] [CrossRef]
- Wilkinson, T.L.; Ishikawa, H. Injection of Essential Amino Acids Substitutes for Bacterial Supply in Aposymbiotic Pea Aphids (Acyrthosiphon Pisum). Entomol. Exp. Appl. 2000, 94, 85–91. [Google Scholar] [CrossRef]
- Koga, R.; Tsuchida, T.; Sakurai, M.; Fukatsu, T. Selective Elimination of Aphid Endosymbionts: Effects of Antibiotic Dose and Host Genotype, and Fitness Consequences. FEMS Microbiol. Ecol. 2007, 60, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Chong, R.A.; Park, H.; Moran, N.A. Genome Evolution of the Obligate Endosymbiont Buchnera Aphidicola. Mol. Biol. Evol. 2019, 36, 1481–1489. [Google Scholar] [CrossRef]
- Renoz, F.; Foray, V.; Ambroise, J.; Baa-Puyoulet, P.; Bearzatto, B.; Mendez, G.L.; Grigorescu, A.S.; Mahillon, J.; Mardulyn, P.; Gala, J.-L.; et al. At the Gate of Mutualism: Identification of Genomic Traits Predisposing to Insect-Bacterial Symbiosis in Pathogenic Strains of the Aphid Symbiont Serratia Symbiotica. Front. Cell. Infect. Microbiol. 2021, 11, 660007. [Google Scholar] [CrossRef] [PubMed]
- Latorre, A.; Manzano-Marín, A. Dissecting Genome Reduction and Trait Loss in Insect Endosymbionts. Ann. N. Y. Acad. Sci. 2017, 1389, 52–75. [Google Scholar] [CrossRef]
- Sabri, A.; Leroy, P.; Haubruge, E.; Hance, T.; Frere, I.; Destain, J.; Thonart, P. Isolation, Pure Culture and Characterization of Serratia Symbiotica Sp. Nov., the R-Type of Secondary Endosymbiont of the Black Bean Aphid Aphis Fabae. Int. J. Syst. Evol. Microbiol. 2011, 61, 2081–2088. [Google Scholar] [CrossRef] [Green Version]
- Grigorescu, A.S.; Renoz, F.; Sabri, A.; Foray, V.; Hance, T.; Thonart, P. Accessing the Hidden Microbial Diversity of Aphids: An Illustration of How Culture-Dependent Methods Can Be Used to Decipher the Insect Microbiota. Microb. Ecol. 2018, 75, 1035–1048. [Google Scholar] [CrossRef] [PubMed]
- Perreau, J.; Patel, D.J.; Anderson, H.; Maeda, G.P.; Elston, K.M.; Barrick, J.E.; Moran, N.A. Vertical Transmission at the Pathogen-Symbiont Interface: Serratia Symbiotica and Aphids. mBio 2021, 12, e00359-21. [Google Scholar] [CrossRef] [PubMed]
- Pons, I.; Renoz, F.; Noël, C.; Hance, T. New Insights into the Nature of Symbiotic Associations in Aphids: Infection Process, Biological Effects and Transmission Mode of Cultivable Serratia Symbiotica Bacteria. Appl. Environ. Microbiol. 2019, 85, e02445-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elston, K.M.; Perreau, J.; Maeda, G.P.; Moran, N.A.; Barrick, J.E. Engineering a Culturable Serratia Symbiotica Strain for Aphid Paratransgenesis. Appl. Environ. Microbiol. 2020, 87, e02245-20. [Google Scholar] [CrossRef]
- Lamelas, A.; Pérez-Brocal, V.; Gómez-Valero, L.; Gosalbes, M.J.; Moya, A.; Latorre, A. Evolution of the Secondary Symbiont “Candidatus Serratia Symbiotica” in Aphid Species of the Subfamily Lachninae. Appl. Environ. Microbiol. 2008, 74, 4236–4240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braendle, C.; Miura, T.; Bickel, R.; Shingleton, A.W.; Kambhampati, S.; Stern, D.L. Developmental Origin and Evolution of Bacteriocytes in the Aphid–Buchnera Symbiosis. PLoS Biol. 2003, 1, e21. [Google Scholar] [CrossRef]
- Baumann, P. Biology of Bacteriocyte-Associated Endosymbionts of Plant Sap-Sucking Insects. Annu. Rev. Microbiol. 2005, 59, 155–189. [Google Scholar] [CrossRef]
- Simonet, P.; Duport, G.; Gaget, K.; Weiss-Gayet, M.; Colella, S.; Febvay, G.; Charles, H.; Viñuelas, J.; Heddi, A.; Calevro, F. Direct Flow Cytometry Measurements Reveal a Fine-Tuning of Symbiotic Cell Dynamics According to the Host Developmental Needs in Aphid Symbiosis. Sci. Rep. 2016, 6, 19967. [Google Scholar] [CrossRef]
- Haase, I.; Sarge, S.; Illarionov, B.; Laudert, D.; Hohmann, H.-P.; Bacher, A.; Fischer, M. Enzymes from the Haloacid Dehalogenase (HAD) Superfamily Catalyse the Elusive Dephosphorylation Step of Riboflavin Biosynthesis. ChemBioChem 2013, 14, 2272–2275. [Google Scholar] [CrossRef]
- Blow, F.; Bueno, E.; Clark, N.; Zhu, D.T.; Chung, S.H.; Güllert, S.; Schmitz, R.A.; Douglas, A.E. B-Vitamin Nutrition in the Pea Aphid-Buchnera Symbiosis. J. Insect Physiol. 2020, 126, 104092. [Google Scholar] [CrossRef]
- Szabó, G.; Schulz, F.; Manzano-Marín, A.; Toenshoff, E.R.; Horn, M. Evolutionarily Recent Dual Obligatory Symbiosis among Adelgids Indicates a Transition between Fungus- and Insect-Associated Lifestyles. ISME J. 2022, 16, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Lamelas, A.; Gosalbes, M.J.; Manzano-Marín, A.; Peretó, J.; Moya, A.; Latorre, A. Serratia Symbiotica from the Aphid Cinara Cedri: A Missing Link from Facultative to Obligate Insect Endosymbiont. PLoS Genet. 2011, 7, e1002357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaxter, M.; Archibald, J.M.; Childers, A.K.; Coddington, J.A.; Crandall, K.A.; Di Palma, F.; Durbin, R.; Edwards, S.V.; Graves, J.A.M.; Hackett, K.J.; et al. Why Sequence All Eukaryotes? Proc. Natl. Acad. Sci. USA 2022, 119, e2115636118. [Google Scholar] [CrossRef] [PubMed]
- Dinant, S.; Bonnemain, J.-L.; Girousse, C.; Kehr, J. Phloem Sap Intricacy and Interplay with Aphid Feeding. C. R. Biol. 2010, 333, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Gallinger, J.; Gross, J. Unraveling the Host Plant Alternation of Cacopsylla Pruni—Adults but Not Nymphs Can Survive on Conifers Due to Phloem/Xylem Composition. Front. Plant Sci. 2018, 9, 484. [Google Scholar] [CrossRef]
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Leo, J.C.; Grin, I.; Linke, D. Type V Secretion: Mechanism(s) of Autotransport through the Bacterial Outer Membrane. Philos. Trans. R. Soc. B 2012, 367, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Meuskens, I.; Saragliadis, A.; Leo, J.C.; Linke, D. Type V Secretion Systems: An Overview of Passenger Domain Functions. Front. Microbiol. 2019, 10, 1163. [Google Scholar] [CrossRef] [Green Version]
- Nesta, B.; Spraggon, G.; Alteri, C.; Gomes Moriel, D.; Rosini, R.; Veggi, D.; Smith, S.; Bertoldi, I.; Pastorello, I.; Ferlenghi, I.; et al. FdeC, a Novel Broadly Conserved Escherichia Coli Adhesin Eliciting Protection against Urinary Tract Infections. mBio 2012, 3, e00010-12. [Google Scholar] [CrossRef] [Green Version]
- Leo, J.C.; Skurnik, M. Adhesins of Human Pathogens from the Genus Yersinia. In Bacterial Adhesion: Chemistry, Biology and Physics; Linke, D., Goldman, A., Eds.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2011; pp. 1–15. ISBN 978-94-007-0940-9. [Google Scholar]
- Shea, A.E.; Stocki, J.A.; Himpsl, S.D.; Smith, S.N.; Mobley, H.L.T. Loss of an Intimin-Like Protein Encoded on a Uropathogenic E. Coli Pathogenicity Island Reduces Inflammation and Affects Interactions with the Urothelium. Infect. Immun. 2022, 90, e00275-21. [Google Scholar] [CrossRef]
- Whelan, R.; McVicker, G.; Leo, J.C. Staying out or Going in? The Interplay between Type 3 and Type 5 Secretion Systems in Adhesion and Invasion of Enterobacterial Pathogens. Int. J. Mol. Sci. 2020, 21, 4102. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, U.; Steinert, M.; Hacker, J. Common Molecular Mechanisms of Symbiosis and Pathogenesis. Trends Microbiol. 2000, 8, 226–231. [Google Scholar] [CrossRef]
- Bright, M.; Bulgheresi, S. A Complex Journey: Transmission of Microbial Symbionts. Nat. Rev. Microbiol. 2010, 8, 218–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raina, J.-B.; Fernandez, V.; Lambert, B.; Stocker, R.; Seymour, J.R. The Role of Microbial Motility and Chemotaxis in Symbiosis. Nat. Rev. Microbiol. 2019, 17, 284–294. [Google Scholar] [CrossRef]
- Schepers, M.J.; Yelland, J.N.; Moran, N.A.; Taylor, D.W. Isolation of the Buchnera Aphidicola Flagellum Basal Body Complexes from the Buchnera Membrane. PLoS ONE 2021, 16, e0245710. [Google Scholar] [CrossRef]
- Diepold, A.; Armitage, J.P. Type III Secretion Systems: The Bacterial Flagellum and the Injectisome. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20150020. [Google Scholar] [CrossRef] [Green Version]
- Hendry, T.A.; de Wet, J.R.; Dunlap, P.V. Genomic Signatures of Obligate Host Dependence in the Luminous Bacterial Symbiont of a Vertebrate. Environ. Microbiol. 2014, 16, 2611–2622. [Google Scholar] [CrossRef] [Green Version]
- Akman, L.; Yamashita, A.; Watanabe, H.; Oshima, K.; Shiba, T.; Hattori, M.; Aksoy, S. Genome Sequence of the Endocellular Obligate Symbiont of Tsetse Flies, Wigglesworthia Glossinidia. Nat. Genet. 2002, 32, 402–407. [Google Scholar] [CrossRef]
- Maire, J.; Parisot, N.; Galvao Ferrarini, M.; Vallier, A.; Gillet, B.; Hughes, S.; Balmand, S.; Vincent-Monégat, C.; Zaidman-Rémy, A.; Heddi, A. Spatial and Morphological Reorganization of Endosymbiosis during Metamorphosis Accommodates Adult Metabolic Requirements in a Weevil. Proc. Natl. Acad. Sci. USA 2020, 117, 19347–19358. [Google Scholar] [CrossRef]
- Rio, R.V.M.; Symula, R.E.; Wang, J.; Lohs, C.; Wu, Y.; Snyder, A.K.; Bjornson, R.D.; Oshima, K.; Biehl, B.S.; Perna, N.T.; et al. Insight into the Transmission Biology and Species-Specific Functional Capabilities of Tsetse (Diptera: Glossinidae) Obligate Symbiont Wigglesworthia. mBio 2012, 3, e00240-11. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Weiss, B.L.; Aksoy, S.; Runyen-Janecky, L.J. Characterization of the Achromobactin Iron Acquisition Operon in Sodalis Glossinidius. Appl. Environ. Microbiol. 2013, 79, 2872–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, K.N.; Dertz, E.A.; Kim, S.S. Enterobactin: An Archetype for Microbial Iron Transport. Proc. Natl. Acad. Sci. USA 2003, 100, 3584–3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.-W.; Ma, W. YopJ Family Effectors Promote Bacterial Infection through a Unique Acetyltransferase Activity. Microbiol. Mol. Biol. Rev. 2016, 80, 1011–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Renoz, F.; Ambroise, J.; Bearzatto, B.; Fakhour, S.; Parisot, N.; Ribeiro Lopes, M.; Gala, J.-L.; Calevro, F.; Hance, T. The Di-Symbiotic Systems in the Aphids Sipha maydis and Periphyllus lyropictus Provide a Contrasting Picture of Recent Co-Obligate Nutritional Endosymbiosis in Aphids. Microorganisms 2022, 10, 1360. https://doi.org/10.3390/microorganisms10071360
Renoz F, Ambroise J, Bearzatto B, Fakhour S, Parisot N, Ribeiro Lopes M, Gala J-L, Calevro F, Hance T. The Di-Symbiotic Systems in the Aphids Sipha maydis and Periphyllus lyropictus Provide a Contrasting Picture of Recent Co-Obligate Nutritional Endosymbiosis in Aphids. Microorganisms. 2022; 10(7):1360. https://doi.org/10.3390/microorganisms10071360
Chicago/Turabian StyleRenoz, François, Jérôme Ambroise, Bertrand Bearzatto, Samir Fakhour, Nicolas Parisot, Mélanie Ribeiro Lopes, Jean-Luc Gala, Federica Calevro, and Thierry Hance. 2022. "The Di-Symbiotic Systems in the Aphids Sipha maydis and Periphyllus lyropictus Provide a Contrasting Picture of Recent Co-Obligate Nutritional Endosymbiosis in Aphids" Microorganisms 10, no. 7: 1360. https://doi.org/10.3390/microorganisms10071360