
Abstract
Oryzias sinensis, also known as Chinese medaka or Chinese ricefish, is a commonly used animal model for aquatic environmental assessment in the wild as well as gene function validation or toxicology research in the lab. Here, a high-quality chromosome-level genome assembly of O. sinensis was generated using single-tube long fragment read (stLFR) reads, Nanopore long-reads, and Hi-C sequencing data. The genome is 796.58 Mb, and a total of 712.17 Mb of the assembled sequences were anchored to 23 pseudo-chromosomes. A final set of 22,461 genes were annotated, with 98.67% being functionally annotated. The Benchmarking Universal Single-Copy Orthologs (BUSCO) benchmark of genome assembly and gene annotation reached 95.1% (93.3% single-copy) and 94.6% (91.7% single-copy), respectively. Furthermore, we also use ATAC-seq to uncover chromosome transposase-accessibility as well as related genome area function enrichment for Oryzias sinensis. This study offers a new improved foundation for future genomics research in Chinese medaka.
Similar content being viewed by others

Background & Summary
Chinese medaka, or Oryzias sinensis, is a teleost fish closely related to Japanese medaka (Oryzias latipes), which has been used as a model organism in many genomic studies as well as in aquatic toxicology research1. Both fish belong to the family Adrianichthyidae, commonly referred to as the Medaka family. Similar to its relative, Chinese medaka is also attracting the attention of scientists due to its small size and short generation interval2. There are several differences between these two fishes, including their vertebrae and pectoral fin strip; most importantly, Chinese medaka is mainly distributed in freshwater while Japanese medaka can adapt to a certain level of salinity. Because of this, Chinese medaka could be more suitable for freshwater quality evaluation.
Although the Chinese medaka genome has been released3, the completeness and genome annotations still need to be further improved. The reported genome was only released at the scaffold level with alignments to the chromosomes of Japanese medaka. Several phylogenetic analyses and taxonomic revisions for medaka have already found that the Chinese medaka is different from its Japanese relative in chromosome constitution; the diploid chromosome number is 46 in O. sinensis, and 48 in O. latipes4,5. Therefore, a high-quality reference genome for O. sinensis is increasingly important to support future research.
In the present study, an improved high-quality chromosome-level genome assembly of O. sinensis was generated using single-tube long fragment read (stLFR) reads, Nanopore long-reads, and the Hi-C sequencing data. The genome size was 796.58 Mb with a scaffold N50 length of 30.38 Mb (Table 1). A total of 712.17 Mb (89.34%) of assembled sequences were anchored to 23 pseudo-chromosomes, with lengths ranging from 58.48 Mb to 18.87 Mb (Table 2). Based on this improved genome assembly, repeat elements and gene structure annotations were conducted combining de novo prediction, homolog-based alignment, and transcriptome-assisted methods. Benchmarking Universal Single-Copy Orthologs (BUSCO) evaluation result showed that the final assembly was benchmarked at 95.1% and the annotation reached 94.6% (Table 3).
In addition, we conducted ATAC-seq try to find out the chromosome accessibility of O. sinensis. After we got the peak results we also performed function enrichment of related genome areas and obtain some clue of transcriptional activity. Taken together, this study could provide a new reliable foundation for research on the Chinese medaka, as well as for its use as a model organism.
Methods
De novo genome assembly
The samples of O. sinensis were obtained from the National Plateau Wetland Research Center, Southwest Forestry University; muscle tissue was used for nucleic acid extraction. High-quality purified RNA was used to construct a transcript sequencing library and DNA was used to construct stLFR, Nanopore long-reads, Hi-C sequencing, and ATAC-seq libraries. stLFR technology added the same barcode sequence to subfragments of the original long DNA molecule, allowing these co-barcoded sub-fragments to be subsequently sequenced using a second-generation platform6. This method generates long reads with high accuracy, enabling high-quality assembly and subsequent analysis. Most importantly, stLFR was cost-effective and has been widely used in aquatic genome projects7,8.
The genome size and heterozygosity of O. sinensis were estimated using Jellyfish v2.2.69 and GenomeScope v1.0.010 by k-mer analysis with clean stLFR data. The clean data was then used in de novo genome assembly with the stlfr2supernova pipeline (https://github.com/BGI-Qingdao/stlfr2supernova_pipeline). This pipeline conducts de novo assembly using stLFR data with Supernova Assembler, which refers to the de novo software from 10X Genomics11. Nanopore long-reads were employed to carry out further scaffolding, gap-closing, and polishing at the same time using TGS-GapCloser12. The size of the assembly after these steps was bigger than the k-mer result (approximately 906 Mb) so purge_haplotigs13 was applied to remove redundancy. Finally, a draft assembly that covered approximately 796 Mb of the genome with a contig N50 length of 373.71 Kb was obtained.
Hi-C analysis and chromosome assembly
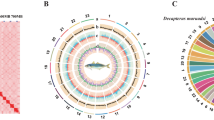
The Hi-C reads were aligned to the draft assembly using HiCPro14 to find valid read pairs. Juicer15 and 3D-DNA16 were then used to finish the construction of chromosomes, with manual correction of misjoins, wrong order, and opposite orientation using Juicebox15. The Hi-C scaffolding resulted in 23 pseudo-chromosomes with a total length of 712.17 Mb. We offer a chromosome circus map using TBtools17 and heatmap using Juicebox18 (Fig. 1A,B). Genome assembly statistics are shown in Table 1 and Table 2. BUSCO (Benchmarking Universal Single-Copy Orthologs)19 evaluation of the assembly reached 95.1% using actinopterygii_odb9 database, indicating a well-covered fish genome assembly (Table 3).

(A) Global genome landscape of Chinese medaka. From inner to outer circles: Density of genes with 500 kbp windows, ranging from 0 to 50; Density of TE with 500 kbp windows, ranging from 0 to 600; Density of TRF with 500 kbp windows, ranging from 0 to 450; Distributions of GC content with 500 kbp windows. (B) Hi-C interaction heat map and scatter plot of genome assembly GC content and sequencing depth.
Repeat annotation
Tandem repeats and interspersed repeats identification was essential before protein-coding gene and function annotations. RepeatMasker20 and RepeatProteinMask were applied to find repeat elements as homology predictions based on RepBase21. For the de-novo method, RepeatModeler22 was used to predict repeat elements; LTR_FINDER23, and TRF tool24 were also used to predict repeat elements based on sequences features. Overall, 311.32 Mb of the O. sinensis genome assembly were identified as repetitive elements, accounting for 39.05% of the whole genome (Table 4).
Gene prediction and annotation
Protein coding genes were predicted with multiple sources of evidence including homology-based alignments, de novo prediction, and transcriptome-assisted methods. In homology alignments, the protein sequences of Danio rerio (GRCz11), Ictalurus punctatus (IpCoco_1.2), Oryzias javanicus (OJAV_1.1), Oryzias latipes (ASM223467v1), Oryzias melastigma (Om_v0.7), Poecilia formosa (PoeFor_5.1.2), and Takifugu rubripes (FUGU5) were mapped to a repeat soft masked draft genome using blat25, and Genewise was applied to define gene models26. In de-novo prediction, Augustus27 was used to predict the coding regions of genes. In transcriptome-assisted methods, two different methods were used to obtain predicted gene sets. First, RNA reads were mapped to the genome assembly using Hi-SAT228; the result was used to build a transcript gene model using Stringtie29 and TransDecoder. Second, a transcriptome was first assembled with RNA reads using Trinity, before PASA was used to get a gene model. Finally, all evidence was merged to form a consensus gene structure annotation result using GLEAN30. A total of 22,461 protein-coding genes were successfully predicted. BUSCO19 assessments reached 94.6% using the actinopterygii_odb9 database, indicating relatively complete gene annotation coverage (Table 3).
All predicted genes were compared with public biological function databases including KEGG31, Swissprot32, and TrEMBL33 (https://www.uniprot.org/statistics/TrEMBL) using BLASTp34 with an E-value cutoff of 1e-5 for functional annotation. Interpro was applied to provide functional analysis of protein sequences by classifying them into families and predicting the presence of domains and important sites, followed by Gene Ontology annotation35,36,37. Overall, 22,162 protein-coding genes (98.67%) were successfully functionally annotated (Table 5).
Non-coding RNAs (ncRNAs) are an important part of genome annotation as they can be active in transcriptional and translational regulation of gene expression as well as in the modulation of protein function38. Since ribosomal RNA (rRNA) was highly conservative, vertebrate rRNA data was used as a reference to map to the draft genome using BLASTn with an E-value of 1e-5. For the discovery of transfer RNA (tRNA), tRNAscan-SE v1.3.1 was applied with eukaryotic parameters according to the characteristics of tRNA39. Rfam database was applied to find microRNAs (miRNA) and small nuclear RNA (snRNA)40,41. In total, 676 rRNA, 740 tRNA, 231 miRNA, and 1,168 snRNA genes were identified from the O. sinensis genome.
Chromosome accessibility analysis using ATAC-seq
ATAC-seq is a widely used method used to identify regions of open chromatin in genomes. SOAPnuke (v1.5.2)42 was used to filter out reads with low quality original data or high content of unknown bases. Bowtie2 (v2.2.5)43 was used to align reads to the O. sinensis genome assembly and peak calling was performed with MACS2 software (v2.1.0)44. Peaks related to genes were used to carry out GO and KEGG enrichment analysis (Fig. 2).

ATAC-seq peaks result from related genes’ functional enrichment. (A) GO enrichment; (B) KEGG enrichment.
Data Records
The Nanopore long reads, stLFR genomic sequencing data, Hi-C data as well as the assembled genome have been deposited at China National GeneBank DataBase (CNGBdb) under the accession CNP000347545.The whole sequencing dataset of O. sinensis was deposited in the NCBI Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA895195) under project identification number PRJNA895195. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession JAUDJI000000000. The version described in this paper is version JAUDJI01000000046. Sequence Read Archive (SRA) project number was SRP41030447. DNA sequencing data from the WGS library were deposited in the SRA at SRR2243596048. DNA sequencing data from the Hi-C library were deposited in the SRA at SRR2243596149. The ATAC-sequencing data were also deposited at Figshare50.
Technical Validation
To evaluate the quality of the genome assembly, stLFR reads were mapped to the final reference genome assembly using BWA (v0.7.12). A total of 98.27% of reads were mapped, covering 98.29% of the genome sequence. Genome average sequencing depth reached 172x, and 97.34% of the genome had a sequencing depth of over 20x. The scatter plot between assembly sequencing depth and GC content found no abnormal GC content, recognized as no exogenous pollution. The completeness of the genome assembly and annotation was assessed using BUSCO (v3.0) with the actinopterygii_odb9 database. The BUSCO benchmark of genome assembly and gene annotation reached 95.1% and 94.6%, respectively.
Code availability
All software used in this study are in the public domain, with parameters described in Methods and this section. If no detailed parameters were mentioned for the software, default parameters were used according to the software introduction.
References
Wittbrodt, J., Shima, A. & Schartl, M. Medaka–a model organism from the far East. Nat Rev Genet. 3, 53–64 (2002).
Cui, L. et al. Oryzias sinensis, a new model organism in the application of eco-toxicity and water quality criteria (WQC). Chemosphere. 261, 127813 (2020).
Wang, Y. et al. Genome and transcriptome of Chinese medaka (Oryzias sinensis) and its uses as a model fish for evaluating estrogenicity of surface water. Environ Pollut. 317, 120724 (2023).
Kasahara, M. et al. The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714–719 (2007).
Parenti, L. R. A phylogenetic analysis and taxonomic revision of ricefishes, Oryzias and relatives (Beloniformes, Adrianichthyidae). Zool J Linn. 154, 494–610 (2008).
Wang, O. et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 29, 798–808 (2019).
Zhao, N. et al. Genome assembly and annotation at the chromosomal level of first Pleuronectidae: Verasper variegatus provides a basis for phylogenetic study of Pleuronectiformes. Genomics. 113, 717–726 (2021).
Zhao, N. et al. High-quality chromosome-level genome assembly of redlip mullet (Planiliza haematocheila). Zool Res. 42, 796–799 (2021).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27, 764–770 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Weisenfeld, N. I., Kumar, V., Shah, P., Church, D. M. & Jaffe, D. B. Direct determination of diploid genome sequences. Genome Res. 27, 757–767 (2017).
Xu, M. Y. et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience. 9, giaa094 (2020).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 19, 460 (2018).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Chen, C. C. et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 13, 1194–1202 (2020).
Robinson, J. T. et al. Juicebox.js Provides a Cloud-Based Visualization System for Hi-C Data. Cell Syst. 6, 256–258 (2018).
Waterhouse, R. M. et al. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol Biol Evol. 35, 543–548 (2018).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. Chapter 4, 4.10.1–4.10.14 (2009).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 6, 11 (2015).
Tempel, S. Using and understanding RepeatMasker. Methods Mol Biol. 859, 29–51 (2012).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–8 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 (1999).
Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664 (2002).
Birney, E., Clamp, M. & Durbin, R. GeneWise and genomewise. Genome Res. 14, 988–995 (2004).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–7 (2005).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12, 357–60 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33, 290–295 (2015).
Elsik, C. G. et al. Creating a honey bee consensus gene set. Genome Biol. 8, R13 (2007).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
UniProt Consortium. T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699 (2018).
Consortium, U. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res 51, D523–D531 (2023).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol. 215, 403–410 (1990).
The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 (2000).
Mitchell, A. L. et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360 (2019).
Szymanski, M., Erdmann, V. A. & Barciszewski, J. Noncoding RNAs database (ncRNAdb). Nucleic Acids Res. 35, D162–D1644 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Kalvari, I. et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342 (2018).
Kalvari, I. et al. Non-Coding RNA Analysis Using the Rfam Database. Curr Protoc. Bioinformatics. 62, e51 (2018).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience. 7, 1–6 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 9, 357–359 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome. 9, R137 (2008).
China national gene bank https://db.cngb.org/search/project/CNP0003475/
Dong, Z. D. et al. Genbank https://identifiers.org/ncbi/insdc.gca:GCA_037389245.1 (2024).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP410304 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22435960 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR22435961 (2023).
Dong, Z. D. et al. A high-quality chromosome-level genome assembly of the Chinese rice fish Oryzias sinensis. figshare https://doi.org/10.6084/m9.figshare.24171774.v3 (2023).
Acknowledgements
This research was financed by the National Natural Science Foundation of China (41806195, 31972794), Nanhai Scholar Project of GDOU (QNXZ201807, 201903), and State Laboratory of Developmental Biology of Freshwater Fish (2020KF004).
Author information
Authors and Affiliations
Contributions
B.Z. and Z.W. designed the study. J.W., G.C., Y.G. and N.Z. performed the experiments and analyzed the data. Z.D. and B.Z. wrote the paper. Z.W. and N.Z. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, Z., Wang, J., Chen, G. et al. A high-quality chromosome-level genome assembly of the Chinese medaka Oryzias sinensis. Sci Data 11, 322 (2024). https://doi.org/10.1038/s41597-024-03173-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03173-8
