
Abstract
The gut microbiome plays important roles in the maintenance of health and pathogenesis of diseases in the growing host. In order to fully comprehend the interplay of the gut microbiome and host, a foundational understanding of longitudinal microbiome, including bacteria and fungi, development is necessary. In this study, we evaluated enteric microbiome and host dynamics throughout the lifetime of commercial swine. We collected a total of 234 fecal samples from ten pigs across 31 time points in three developmental stages (5 preweaning, 15 nursery, and 11 growth adult). We then performed 16S rRNA gene amplicon sequencing for bacterial profiles and qPCR for the fungus Kazachstania slooffiae. We identified distinct bacteriome clustering according to the host developmental stage, with the preweaning stage exhibiting low bacterial diversity and high volatility amongst samples. We further identified clusters of bacteria that were considered core, increasing, decreasing or stage-associated throughout the host lifetime. Kazachstania slooffiae was absent in the preweaning stage but peaked during the nursery stage of the host. We determined that all host growth stages contained negative correlations between K. slooffiae and bacterial genera, with only the growth adult stage containing positive correlates. Our stage-associated bacteriome results suggested the neonate contained a volatile gut microbiome. Upon weaning, the microbiome became relatively established with comparatively fewer perturbations in microbiome composition. Differential analysis indicated bacteria might play distinct stage-associated roles in metabolism and pathogenesis. The lack of positive correlates and shared K. slooffiae-bacteria interactions between stages warranted future research into the interactions amongst these kingdoms for host health. This research is foundational for understanding how bacteria and fungi develop singularly, as well as within a complex ecosystem in the host’s gut environment.
Similar content being viewed by others


Introduction
Host-associated microbiomes have critical roles in host health, growth and development. The digestive system contains microbes with a wide array of functions for hosts, such as aiding in nutrient availability, protecting from pathogen invasion and maintaining a healthy gut epithelial barrier1,2,3. An imbalance of microorganisms, or their associated functions, in this enteric, or digestive, microbiome can lead to a dysbiotic state and diseased host1. Diseases and symptoms associated with a dysbiotic enteric microbiome include inflammatory bowel disease (IBD), diarrhea, obesity, and metabolic syndrome (MetS), among other ailments4. In order to develop therapies for these illnesses, it is paramount to understand the enteric microbiome dynamics spanning microbial kingdoms, including bacteria and fungi, throughout the lifetime of swine hosts.
Foundational to evaluating microbial interactions is first determining dynamics of the gut during the host lifetime. Traditionally, scientists have evaluated microbial composition and alpha (⍺) diversity to understand gut microbial development5. Composition includes the overall taxonomic comparison amongst samples whereas ⍺ diversity quantifies how many distinct taxa are present. During the host lifetime, we expect to see compositional similarity in similar aged hosts, but distinctions over time as microbes change in abundance6,7,8. Contrastingly, we expect to see ⍺ diversity develop in the early lifetime until the gut environment reaches a relatively staple level of development amongst hosts1,2. A developed gut is expected to have a relatively higher ⍺ diversity, compared to a developing environment, indicating a diverse microbial makeup with similarly diverse roles within the microbiome system and for host health9,10. By evaluating the microbial composition and ⍺ diversity we will have a foundational understanding of how the gut microbiome develops during the host lifetime which aids investigations into bacterial and fungal interactions.
As mentioned previously, understanding microbial correlations and interactions between microbial kingdoms, including Fungi and Bacteria, are critical to elucidating diseases impacted by these microbial kingdoms. Previous research has shown a negative correlation, indicating a competitive relationship, between bacterial diversity and fungal abundance11. Still, the microbial mechanisms, influencing other microbes and the host alike, underlying these outcomes have not been described. We must understand bacterial-fungal interaction intricacies to provide treatments targeting specific microbes and mechanisms, especially those of bacterial-fungal dysbiotic gut microbiomes.
Current research lacks an understanding of how the dominant swine enteric fungus, Kazachstania slooffiae, changes in the majority of the swine lifetime, and how these changes are influenced by the bacterial communities. Kazachstania slooffiae is a member of the Saccharomycetaceae family, and the fungus is a proposed commensal in the swine gut microbiome12. Studies indicate K. slooffiae dominates the mycobiome from 70 to 90% of total yeasts, especially following weaning13,14. The fungus has been demonstrated to significantly alter the gut microbiota during weaning, leading to a potentially beneficial increase in short chain fatty acid (SCFA) concentration15. Although K. slooffiae is the primary fungus after weaning, we currently lack a longitudinal understanding of this fungus. Publications have only evaluated K. slooffiae abundance from birth until 39 days of age13,16. The average time to market age is 160 days, so prior publications have only evaluated K. slooffiae dynamics in 25% of the swine lifetime to market17. Moreover, previous studies have identified eight SparCC correlations between K. slooffiae and bacterial genera from nursery-aged hosts18,19. We hypothesized there were more inter-kingdom correlations occurring throughout the swine lifetime (including preweaning and growth adult as these were not studied previously) which influence microbiome establishment and host health11. Our study aimed to elucidate novel stage-associated bacteriome-K. slooffiae correlations to build a foundation for future inter-kingdom interaction studies.
This study highlights development of bacteria, fungi and host, with an investigation into bacterial-fungal correlations. We followed ten swine from birth. We first determined the foundational gut microbiome development during the host lifetime. Studies have demonstrated various factors from biology, such as host diet and housing environment, to methodology, including DNA extraction and bioinformatic approaches, impacting resulting identification of microbes and microbial diversity20,21,22. Therefore, we aimed to first provide a baseline understanding of how our gut bacteria changed in the lifetime of the ten swine hosts. With this knowledge, we could then further interpret how the microbial composition and ⍺ diversity pertained to microbial inter-kingdom interactions and potential implications on host health at various ages. Understanding inter-kingdom interaction, influenced by gut development, in the swine may provide insights into the intricate relationship between the host and the microbiome. Foundation to this longitudinal study, swine were grown in three stages which varied according to host development, diet and housing: preweaning (milk diet and housed with littermates and dam; birth-21 days of age), nursery (pellet diet and co-housed with other litters; 21–80 days) and growth adult (pellet diet and co-housed with other litters; 80–122 days). As discussed previously, directly following weaning into the nursery stage in swine hosts, one fungus has been consistently identified in the enteric mycobiome: Kazachstania slooffiae14,19. For this reason, our study focused on elucidating longitudinal dynamics between K. slooffiae and bacteria.
In our study, we determined specific host-age and -dietary stage microbiome development characteristics. These included an increasing microbial diversity, decreasing volatility and increasing fungus K. slooffiae in the young host (preweaning and nursery developmental stages). The older host (growth adult stage) microbiome was relatively established with a complex correlation network amongst bacteria and K. slooffiae. Together, these findings indicated a dynamic microbiome development from birth until weaning with an increasing number of inter-kingdom interactions throughout the host lifetime.
Materials and methods
Hosts and study design
We followed ten swine over the course of their lifetime, with fecal collections, rectal temperature, weights, and general health observations collected from 2 to 157 days of age, to understand successive shifts in microbial populations (Supplemental Table S1; Supplemental Document S1). We started the study with ten swine, but one pig died prematurely at 28 days of age. The experimental unit was each individual swine. The hosts were housed indoors and fed distinct diets according to their stage of life. Five dams were randomly selected from the same farrowing group, and one male and one female were randomly selected per dam. Swine were housed with their dam in the preweaning stage, in groups of five in the nursery stage, and all in one pen during the growth adult stage. Hosts were sampled in three stages: preweaning, nursery, and growth adult. The preweaning diet consisted of mother’s milk and potentially feed as the hosts grew old enough to reach their mother’s trough. Nursery diet, phase 1, transitioned from milk to pelleted feed after weaning from the mother and moving into a new barn environment. A second pelleted feed was fed during nursery phase 2, while a meal was fed for nursery phase 3. The growth adult stage also included three phase diets with an initial move into another barn environment accompanying the nursery-growth transition. Hosts did not receive antibiotics or antifungals prior to or during the study. Males were castrated during the preweaning stage. Pigs were managed according to the Kansas State University Institutional Animal Care and Use Committee (IACUC) approved protocol #4036, and methods are reported according to ARRIVE guidelines. Additionally, the authors confirmed that all methods were performed in accordance with relevant guidelines and regulations, and all methods were approved by Kansas State University.
We performed fecal collection with a fresh set of sterile gloves using the free-catch method, prior to contact with the ground. We collected fecal samples every 5 days during preweaning and nursery stages, and every seven days during the growth adult stage. Immediately after collection, samples were stored in either a sterile 15 mL tube or sterile bag, kept on ice, and then transported to the laboratory for subsequent storage at −80 °C until genomic DNA extraction.
DNA extraction and marker gene sequencing
We used the E.Z.N.A.® Stool DNA Kit (Omega Bio-tek, Inc.; Norcross, GA) to extract the microbial DNA from the fecal samples. We used the manufacturer pathogen detection protocol without bead beating and utilized only 30 μL elution buffer per sample. Extracted DNA was quantified with Nanodrop and a Qubit™ dsDNA BR Assay Kit (Thermo Fisher; Waltham, MA) for sample DNA quality and concentration. DI water was utilized during quantification as a negative control. Extracted microbial DNA was stored at −80 °C until library preparation. Bacterial 16S rRNA gene V4 region was amplified during library preparation via Illumina’s Nextera XT Index Kit v2 (Illumina, Inc.; San Diego, CA) (primers: 515F, GTGCCAGCMGCCGCGGTAA and 806R, GGACTACHVGGGTWTCTAAT)23. Library preparation and subsequent sequencing also included a no template negative control. Sequencing was done on an Illumina MiSeq which generated paired-end 250 bp reads.
Kazachstania slooffiae qPCR
We performed the K. slooffiae qPCR, with the SensiMix™ SYBR® Hi-ROX Kit (Bioline, Meridian Bioscience; Cincinnati, OH), as previously described (primers: KS-f, ATCCGGAGGAATGTGGCTTC and KS-r, AGCATCCTTGACTTGCGTCG)13. Master mix components and qPCR conditions are listed in Supplemental Table S2. Each qPCR run included at least one PCR-grade water with the master mix as a non-template control (NTC), with one K. slooffiae positive sample repeatedly used across plates as the positive control.
Bioinformatic and statistical analysis
We used cutadapt and DADA2 in QIIME2 v2019.7 (https://qiime2.org/) to trim and perform quality control for the sequencing reads (Supplemental Table S3)24,25. Reads in which no primer was found were discarded. The reads were truncated at locations where 25-percentile of the reads had a quality score below 15. Diversity analysis was carried out at a sampling depth of 11,105 reads. The pre-trained classifier offered by QIIME2 using the SILVA version132 (https://www.arb-silva.de/documentation/release-132/) database was used for taxonomic assignment of bacteria26,27,28. We used a weighted UniFrac, generated from QIIME2, on the rarefied dataset (11,105 reads) to evaluate differential microbial composition among the samples in different stages, and we utilized a QIIME2 principal coordinate analysis (PCoA) to visualize the microbial composition structure based annotated ASVs (Supplemental Document S2). The following applications were utilized in generating the PCoA composition plot with RStudio version 1.3.1093 (https://www.rstudio.com/products/rstudio/older-versions/): tidyverse version 1.3.1 (https://cran.r-project.org/package=tidyverse), qiime2R version 0.99.6 (https://github.com/jbisanz/qiime2R), plyr version 1.8.7 (https://www.rdocumentation.org/packages/plyr/versions/1.8.7), and ggpubr version 0.4.0 (https://CRAN.R-project.org/package=ggpubr)29,30,31,32,33. We calculated ⍺ diversity to represent the species diversity in each sample. We utilized Shannon index, estimated number of species (ENS), and Faith’s phylogenetic diversity, all within QIIME2, to measure the number of ASVs and the uniformity of ASV abundance for diversity evaluation (Supplemental Document S2)25. Kruskal–Wallis was used in QIIME2 to provide overall and stage pairwise statistical analyses for Shannon, ENS, and Faith’s phylogenetic diversity25. We calculated Shannon effective number by calculating the exponential [exp(H)] of the original Shannon diversity index (H)34. We used PERMANOVA in QIIME2 on Bray–Curtis dissimilarity index to test if there were statistically significant differences between stages25. Volatility results were analyzed within QIIME2 on the first axis of the PCoA to indicate how dispersed samples were at the associated swine age25.
We further used DESeq2 version 1.30.1 (https://github.com/mikelove/DESeq2), in RStudio, to mark the statistical differences in the bacterial populations (phyla and genera) predominance between the stages and to generate heatmaps with pheatmap version 1.0.12 (https://CRAN.R-project.org/package=pheatmap)33,35,36. Adjusted p-values were utilized, rather than standard p-values, as the adjusted values incorporated the Benjamini–Hochberg false discovery rate (FDR)35,37.
16S rRNA gene amplicon genera and fungal qPCR Ct values were utilized in a SPIEC-EASI co-occurrence network analysis as previously performed in RStudio using SpiecEasi version 1.2.4 (https://github.com/zdk123/SpiecEasi), devtools version 2.4.3 (https://CRAN.R-project.org/package=devtools), phyloseq version 1.4.0 (https://bioconductor.org/packages/phyloseq/), and igraph version 1.2.11 (https://CRAN.R-project.org/package=igraph)18,38,39,40. Specific SPIEC-EASI parameters included: Meinhausen–Bühlmann estimation method, lambda minimum ratio of 0.01, and nlambda of 2038. SPIEC-EASI utilized a neighborhood selection method termed Meinshausen and Bühlmann (MB)38,41. The MB method has been demonstrated to control FDR42. Correlations were performed for each stage (preweaning, nursery, and growth adult) with corresponding and fungal qPCR Ct values, according to individual samples (i.e. individual swine and single time point). Correlation plots were simplified to only correlations connected to the fungal node in each stage.
All bioinformatic scripts can be found in Supplemental Document S2.
Ethics approval and consent to participate
Pigs were managed according to the Kansas State University Institutional Animal Care and Use Committee (IACUC) approved protocol #4036.
Results and discussion
We collected a total of 234 samples across 31 time points (5 preweaning, 15 nursery, and 11 growth adult) from ten pigs (Supplemental Table S1). A total of 10,187,636 sequences resulted from sequencing; we recovered an average of 33,394 ASVs per sample following QIIME2 quality control. Out of the recovered ASVs, an average of 80.1% (79.7% bacteria and 0.4% archaea) populations were annotated with SILVA version 132 (Supplemental Table S4).
Volatility in the preweaning stage preceded microbial establishment and stability in later growth stages
As shown in Fig. 1A, the weighted UniFrac PCoA illustrated a distinct clustering of bacterial community composition between the three growth stages as the pig transitioned from young host to adult (Supplemental QIIME2 File S1: QIIME2 weighted unifrac PCoA, which can be uploaded and viewed at https://view.qiime2.org/). We further observed convergence amongst dietary clusters of the swine hosts in the nursery stage, with the two latter diet-stages of nursery being more similar to the growth adult hosts. We showed in our study that a young host lacked an established, shared microbiome, but converged with age and environmental changes such as diet and shared housing. The preweaning and first part of the nursery stages had the most divergent microbial composition amongst the individual swine. After this first nursery stage, the composition was relatively similar amongst the latter two nursery and growth stage swine. These patterns suggest the microbiome could be highly influenced by their respective diets during the different stages, and was rather stable once the microbial members had established6,7,8. Previous research has illustrated distinct microbial populations in swine hosts according to stage of development6,7,8.

(A) Weighted uniFrac PCoA plot29,30,31,32 depicting composition; dots represent distinct samples. Nursery stage is separated according to the three diets fed during the stage. (B) Longitudinal Shannon diversity with Kruskal–Wallis statistical analysis30. (C) Faith’s phylogenetic diversity30 (PD). (D) Volatility control chart of the first axis of the PCoA30. Figure was edited in Inkscape version 1.0.2 (https://inkscape.org/)88.
Our ⍺ diversity results (Shannon index, Faith’s phylogenetic diversity, effective number of species (ENS), Shannon effective number, and Bray Curtis dissimilarity index) paralleled the PCoA analysis, indicating an establishing microbiome in the young swine (Fig. 1B,C, Supplemental Table S4, Supplemental QIIME2 File S2: Shannon diversity index, Supplemental QIIME2 File S3: ENS, and Supplemental QIIME2 File S4: Faith’s phylogenetic diversity; each QIIME2 file can be uploaded and viewed at https://view.qiime2.org/). We found that ⍺ diversity increased throughout the lifetime. We demonstrated the increasing diversity as overall stage comparisons (preweaning [P] vs nursery [N]: N vs growth adult [G]: and P vs G) were significantly different (p ≤ 0.001) for all ⍺ diversity measures according to PERMANOVA and Krustal-Wallis analyses. Studies have indicated this increase in microbial diversity during host development is typical across many different host species1,2. When we investigated the data longitudinally according to sampling day, the preweaning host demonstrated comparatively lower diversity which increased until weaning. This developmental diversity increase was followed with a relatively stable period during the nursery stage. We found distinctions in ⍺ diversity methods in the growth adult host. Shannon index and Faith’s phylogenetic diversity demonstrated small increases in the growth adult ⍺ diversity, whereas ENS and Shannon effective number illustrated a greater range of ⍺ diversity in the older swine. These distinctions in ⍺ diversity in the older swine should be further evaluated for an enhanced understanding of diversity in older swine, and how a wider range of diversity across swine could impact swine health.
Volatility results corroborated previous findings of a changing neonate microbiome which established in the weaned host (Supplemental QIIME2 File S5: volatility, which can be uploaded and viewed at https://view.qiime2.org/). Our microbial composition volatility index in the preweaning host hovered near −0.5 while approaching 0 in the early nursery stage (Fig. 1D). These volatility findings further suggested that the young preweaning host had a relatively more volatile, fluctuating microbiome. Our results were consistent with another mammalian study which demonstrated a volatile youth microbiome establishment period in children aged from birth to approximately 3 years of age43.
Together, our PCoA, diversity indices and volatility analyses suggested that the preweaning neonate host contained a developing gut microbiome which started establishing in the nursery stage. We showed that the microbiome was converging in the early nursery host, and there were comparatively fewer changes in microbial diversity after the convergence of the microbial community in the nursery host. We suggest that the forming and establishment of microbial populations during the preweaning and early nursery stages was likely crucial to the well-being of the swine host. Previous research demonstrates the importance of early microbiome dynamics as abnormal neonate gut microbiome development can result in diabetes, IBD and obesity4,44.
Microbial-host stage development suggested metabolic and pathogenic potential associations
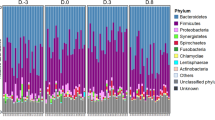
Our study supported previous bacterial establishment dynamics while elucidating novel stage-associations, highlighting a need for functional determination of the enteric microbiome according to host development. We analyzed the host microbial membership and identified 23 phyla (Fig. 2, Supplemental Table S5). We demonstrated a core bacterial population consisting of two phyla (Bacteroidetes and Firmicutes) which dominated throughout the lifetime of the swine host, suggesting these bacterial populations have essential implications to the host’s health and well-being7,45,46. Our study showed that Bacteroidetes and Firmicutes were the predominating core microbes (Fig. 2A). These results were consistent with findings from previous research demonstrating consistent domination of Firmicutes and Bacteroidetes7,45,46. Firmicutes and Bacteroidetes are known to metabolize carbohydrates into short chain fatty acids (SCFAs)47,48, suggesting that the two core phyla in our results have a wide range of beneficial attributes for the swine including acting as a cellular energy source, protecting DNA, and modulating diseases49,50,51. Therefore, given the necessity for energy and continual carbohydrate availability throughout the host lifetime, it is reasonable to identify Firmicutes and Bacteroidetes throughout the host life.

DESeq2 differential phyla distinctions between stages (adjusted p ≤ 0.05) suggested microbes were changing between preweaning and nursery stages but were relatively stable between nursery and growth adult swine. Two phyla identified in preweaning swine, compared to nursery swine, contained distinct microbes with potentially different metabolic implications: Euryarchaeota (log2-fold change: 3.06) and Lentisphaerae (log2-fold change: 4.26) (Fig. 2B). Euryarchaeota is an archaeon which has been associated with improved fiber digestion52,53. We hypothesized that microbes within the Euryarchaeota phylum were working alongside and with the bacterial community to shape the host microbiome, which can influence overall host health and well-being54,55. Alternatively, Lentisphaerae is thought to have a role in SCFA production resulting in a crucial source of energy for the swine host56,57. This differential identification of two carbohydrate metabolizing phyla, Euryarchaeota and Lentisphaerae, supports the different dietary sources of carbohydrates during the preweaning and nursery stages. Compared to the preweaning host, we identified three metabolic-associated phyla in the nursery host: Deferribacteres (log2-fold change: −31.09), Fibrobacteres (log2-fold change: −5.50), and Tenericutes (log2-fold change: −4.28). Deferribacteres is associated with diets containing iron58,59,60; Fibrobacteres is known for metabolizing non-soluble polysaccharides or carbohydrates61; and the function of Tenericutes remains elusive although the bacteria has been positively correlated with diets high in protein62. These three phyla remained unchanged between the nursery and growth adult swine suggesting that the microbes might perform similar metabolic roles during both developmental stages. Our observations of distinct microbial populations through the different stages of the pig paralleled the PCoA, diversity and volatility results, indicating a distinct gut microbiome composition and development during preweaning and early nursery. Considering previous research, we surmised that alongside bacteria and archaea establishment, microbial metabolic roles contributed to this stage-associated development under the influence of host factors, especially diet.
In addition, we also observed that the differential phyla indicated development of stage-dependent potential opportunistic pathogens (Fig. 2B). Preweaning-associated potential opportunistic pathogens included (Fig. 2B): Fusobacteria (log2-fold change: 6.2)63,64,65, Synergistetes (log2-fold change: 5.6)66,67, and Proteobacteria (log2-fold change: 3.8)68,69; nursery: WPS-2 ([P vs N log2-fold change: −25.5][N vs G log2-fold change: 4.5])70, and Spirochaetes ([P vs N log2-fold change: −1.4][N vs G log2-fold change: 0.9])71; and growth adult: Fusobacteria (log2-fold change: −10.4)63,64,65 and Synergistetes (log2-fold change: −2.7)66,67. Interestingly, Fusobacteria and Synergistetes were found in both the preweaning and growth host. Further investigation is needed to evaluate the pathogenicity and determine the developmental significance of these phyla in the nursery growth swine.
We identified 25 genera with an average relative abundance greater than 1% amongst the three stages (Fig. 3 and Supplemental Table S5). Unlike the phyla level analysis, we did not observe a core genus but instead identified three distinct clusters, based on tree branches and detection patterns, throughout the lifetime of the host (Fig. 3A). The first cluster consisted solely of Bacteroides as the bacterial population decreased post-weaning. Succinivibrio and Selenomonas appeared sporadically in the mid-nursery host followed by plateau in the growth adult stage. The final cluster, with 22 genera, generally appeared at a higher relative abundance earlier, than Succinivibrio and Selenomonas, in the preweaning or newly weaned host. Interestingly, although Bacteroides, Succinivibrio, and Selenomonas are all heavily reliant on carbohydrate utilization72,73,74, our data suggested that these genera were absent during different developmental stages. We hypothesized this could be related to these bacteria utilizing distinct carbohydrate sources75,76,77. Future research is necessary to evaluate how these bacterial species were utilizing dietary carbohydrates and interacting among the microbes and host.

The majority of stage-associated genera were identified in the nursery host which could indicate a need for specialized microbial roles in SCFA productions during this stage. Bacteroides ([P vs N log2-fold change: 2.4] [N vs G log2-fold change: 6.2]) was decreasing in relative abundance throughout the pig’s life stages (Fig. 3A,B)72. Conversely, Succinivibrio ([P vs N log2-fold change: −10.2;][N vs G log2-fold change: −1.8) was increasing through the stages77. The remaining potential SCFA-associated genera that were associated with the nursery host were Faecalibacterium ([P vs N log2-fold change: −12.8][N vs G log2-fold change: 4.5])78, Prevotella 7 ([P vs N log2-fold change: −14.4][N vs G log2-fold change: 1.5])79, Prevotella 1 ([P vs N log2-fold change: −8.2][N vs G log2-fold change: 2.6])79, Subdoligranulum ([P vs N log2-fold change: −7.6][N vs G log2-fold change: 3.1])19, Prevotella 9 ([P vs N log2-fold change: −8.0][N vs G log2-fold change: 2.4])80, Alloprevotella ([P vs N log2-fold change: −2.4][N vs G log2-fold change: 2.9])81, Prevotellaceae NK3B31 group ([P vs N log2-fold change: −3.2][N vs G log2-fold change: 1.6])82, Ruminococcaceae UCG-014 ([P vs N log2-fold change: −2.2][N vs G log2-fold change: 2.3])83, Ruminococcaceae UCG-005 ([P vs N log2-fold change: −3.1][N vs G log2-fold change: 0.9])83, and Ruminococcaceae UCG-010 ([P vs N log2-fold change: −1.3][N vs G log2-fold change: 0.8])83. The nursery host contained the most genera with SCFA metabolizing potential, suggesting that this is related to the microbiome dynamics as the microbes were working towards establishing. The bacteria populations within these genera associated with the nursery host could have taken advantage of the perturbations during these stages to proliferate. Akin to the SCFA potential metabolism findings, potential opportunistic pathogen genera were also only identified in nursery swine: Streptococcus ([P vs N log2-fold change: -2.6][N vs G log2-fold change: 1.4])84 and Terrisporobacter ([P vs N log2-fold change: -2.0][N vs G log2-fold change: 1.6])85. We observed that potential opportunistic pathogens were solely in the nursery host, suggesting that a turbulent microbiome enhanced the risk of pathogen development10. Although our present study provided insights into the microbial shifts during the different life stages of the swine, clearly there are complexities during microbiome establishment which warrant increased investigation. Further studies should elucidate how microbial metabolic roles and interactions influence microbiome establishment and pathogen prevalence.
Temporal dynamics of Kazachstania slooffiae and association with bacterial diversity
Our findings suggested that fungal-bacterial interactions in the swine host could influence both bacteriome and mycobiome establishment and dynamics, therefore leading to the decline in K. slooffiae abundance in hosts. We performed qPCR and demonstrated varied K. slooffiae abundance according to developmental stage (Fig. 4). We noticed the fungus was absent in the preweaning host but its presence peaked in the nursery host from 25 to 46 days of age, with a steady decrease in abundance past 46 days of age. We determined fungal presence was more dispersed in the older host, as indicated by a larger 95% confidence interval. Interestingly, we found the increase in K. slooffiae coincided with the establishment of the microbiome near weaning. Previous studies have indicated an increase of K. slooffiae in the early nursery stage (swine hosts aged 21–35 days)14,18. Kazachstania slooffiae abundance past 35 days of age were previously unknown. Our findings showed that K. slooffiae abundance declined during the late nursery stage and plateaued in the growth adult stage, adding to the growing knowledge in the understanding of this fungi. Our fungal research suggested that K. slooffiae underwent stage-specific growth patterns, similar to that of the bacteriome. The factors which directly influenced K. slooffiae increase and decline are not yet known. Prior publications indicate associations between members within the microbiome, including between fungi and bacteria, may have implications to the well-being of the hosts11.

Kazachstania slooffiae qPCR Ct value according to day of age with line of best fit and 95% confidence interval by stage. Figure was edited in Inkscape version 1.0.2 (https://inkscape.org/)88.
We performed taxonomic correlation analyses to further investigate fungi-bacteria interactions in the gut microbiome. Our increasing correlation network complexity with host age and lack of shared K. slooffiae-correlating genera across stages highlighted stage-dependent microbiome development. We simplified our correlation models to depict direct correlations between K. slooffiae and genera according to developmental stage (Fig. 5 and Supplemental Table S6). We identified 65 correlations (3 in preweaning, 30 nursery, and 32 growth adult). Previous research has indicated increasingly complex fungal-bacteria network correlations as both the microbiome and host develop from preweaning to nursery, but growth adult stage correlates were previously unknown18. We identified only two shared correlates between the nursery and growth adult stages: Rikenellaceae RC9 gut group and Candidatus Gastranaerophilales bacterium Zag. The significance of these genera, especially pertaining to K. slooffiae, are not understood and are a topic for future research. The lack of shared K. slooffiae correlating taxa may be related to stage-specific bacteria and stage-specific bacteriome–mycobiome interactions.

Our specific network correlation highlighted novel associations between K. slooffiae and the bacteriome throughout the host lifetime, suggesting the changes associated with weaning, including dietary change and stress, might have allowed for K. slooffiae expansion while the fungal decline may be attributed to competition with bacteria. Previous publications have identified eight correlations with K. slooffiae18,19. Our results included three out of the eight prior K. slooffiae correlations: Lactobacillus (correlation coefficient −1.2, growth adult), Prevotella 9 (−1.2, growth adult), and Prevotella 2 (−1.1, nursery)19. Previous research indicated positive correlations of K. slooffiae and Lactobacillus, Prevotella 9, and Prevotella 2, whereas our correlations were negative19. We surmised that negative correlation between Lactobacillus and K. slooffiae would be analogous to the inhibition of Lactobacillus growth by Candida in humans11. Previous research has identified genetic similarity between K. slooffiae and Candida12,86. Previous studies suggested that Lactobacillus may work alongside other bacteria to deter Candida growth, such as through short chain fatty acid production11. In fact, for our findings, the majority of our network correlations between K. slooffiae and genera were negative, with only three positive correlations (Rikenellaceae RC9 gut group (0.9), Prevotellaceae NK3B31 group (0.7), and Ruminococcaceae UCG-005 (0.4)) were identified in growth hosts. Inverse abundances between fungi and bacteria are indicative of competition or amensalism87, which could explain the sharp decline of K. slooffiae populations in the nursery host (Fig. 4). We further hypothesized that the post-weaning increase of K. slooffiae might be attributed to the dietary change as K. slooffiae is unable to utilize milk galactose12. The dietary transition and host stress from preweaning to nursery might have allowed the increase in K. slooffiae populations, even with bacterial establishment relatively progressed12,19. Our correlation network results showed numerous (63 novel correlations, Fig. 5) novel K. slooffiae correlations which could aid in divulging establishment dynamics within the bacteriome and mycobiome.
Conclusions
We provided a comprehensive evaluation of how bacteria and the fungus, Kazachstania slooffiae, developed through the different life stages of swine. The young preweaning host demonstrated comparatively low microbial diversity which increased near weaning. The growth adult host had a relatively similar microbiome overall compared to the nursery host, yet stage-specific associations, such as potential pathogens and fungal development, were noticed. We noticed the developing microbiome across hosts, even with differences in dam diet and parity status. Future research, with more swine, are crucial to determining the extent to which these dam factors and stage-associated characteristics influence microbiome development dynamics. Our findings provided a foundation for gut microbiome studies.
While microbial inter-kingdom interactions are known to have implications on host health, the intricacies of dynamics between bacteria and fungi are not well understood. We determined that distinct microbial taxa, diversity, and bacterial-fungi correlations were associated with different stages of life. These stage-associated attributes indicated there could be further stage-associated characteristics such as illness-inducing pathogens and energy providing carbohydrate metabolizing microbes. Future research is crucial to understand the interplay amongst microbes, especially on the functional level pertaining to carbohydrate utilization and relating these findings back to host health. As we evaluated general-swine host stage, additional research is also necessary to attribute specific host growth, development, and environmental factors, such as diet and housing, to the diversity changes we identified.
Data availability
The dataset(s) supporting the conclusions of this article is(are) available in the NCBI repository, BioProject PRJNA798835, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA798835.
References
Barko, P. C., McMichael, M. A., Swanson, K. S. & Williams, D. A. The gastrointestinal microbiome: A review. J. Vet. Intern. Med. 32, 9–25 (2018).
Lloyd-Price, J., Abu-Ali, G. & Huttenhower, C. The healthy human microbiome. Genome Med. 8, 51 (2016).
Niederwerder, M. C. Fecal microbiota transplantation as a tool to treat and reduce susceptibility to disease in animals. Vet. Immunol. Immunopathol. 206, 65–72 (2018).
Pushpanathan, P., Mathew, G. S., Selvarajan, S., Seshadri, K. G. & Srikanth, P. Gut microbiota and its mysteries. Indian J. Med. Microbiol. 37, 268–277 (2019).
ter Braak, C. J. F. Principal components biplots and alpha and beta diversity. Ecology 64, 454–462 (1983).
Slifierz, M. J., Friendship, R. M. & Weese, J. S. Longitudinal study of the early-life fecal and nasal microbiotas of the domestic pig. BMC Microbiol. 15, 184 (2015).
Wang, X. et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 7, 109 (2019).
Kim, J., Nguyen, S. G., Guevarra, R. B., Lee, I. & Unno, T. Analysis of swine fecal microbiota at various growth stages. Arch. Microbiol. 197, 753–759 (2015).
Manor, O. et al. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 11, 5206 (2020).
Pickard, J. M., Zeng, M. Y., Caruso, R. & Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89 (2017).
Kapitan, M., Niemiec, M. J., Steimle, A., Frick, J. S. & Jacobsen, I. D. Fungi as part of the microbiota and interactions with intestinal bacteria. in Fungal Physiology and Immunopathogenesis (ed. Rodrigues, M. L.). 265–301. (Springer, 2019).
Summers, K. L., Foster Frey, J. & Arfken, A. M. Characterization of Kazachstania slooffiae, a proposed commensal in the Porcine Gut. J. Fungi (Basel) 7, 146 (2021).
Urubschurov, V., Büsing, K., Janczyk, P., Souffrant, W.-B. & Zeyner, A. Development and evaluation of qPCR assay for quantitation of Kazachstania slooffiae and total yeasts occurring in the Porcine Gut. Curr. Microbiol. 71, 373–381 (2015).
Summers, K. L., Frey, J. F., Ramsay, T. G. & Arfken, A. M. The piglet mycobiome during the weaning transition: A pilot study. J. Anim. Sci. 97, 2889–2900 (2019).
Urubschurov, V. et al. New insights into the role of the porcine intestinal yeast, Kazachstania slooffiae, in intestinal environment of weaned piglets. FEMS Microbiol. Ecol. 93, 2 (2017).
Urubschurov, V., Janczyk, P., Souffrant, W.-B., Freyer, G. & Zeyner, A. Establishment of intestinal microbiota with focus on yeasts of unweaned and weaned piglets kept under different farm conditions. FEMS Microbiol. Ecol. 77, 493–502 (2011).
Bruce Hoar, J. A. Production Cycle of Swine.
Arfken, A. M., Frey, J. F. & Summers, K. L. Temporal dynamics of the gut bacteriome and mycobiome in the weanling pig. Microorganisms 8, 868 (2020).
Arfken, A. M., Frey, J. F., Ramsay, T. G. & Summers, K. L. Yeasts of burden: Exploring the mycobiome–bacteriome of the piglet GI tract. Front. Microbiol. 10, 2286 (2019).
Aluthge, N. D. et al. Board invited review: The pig microbiota and the potential for harnessing the power of the microbiome to improve growth and health1. J. Anim. Sci. 97, 3741–3757 (2019).
Fiedorová, K. et al. The impact of DNA extraction methods on stool bacterial and fungal microbiota community recovery. Front. Microbiol. 10, 821 (2019).
Prodan, A. et al. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 15, e0227434 (2020).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 108(Suppl 1), 4516–4522 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Yilmaz, P. et al. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 42, D643–D648 (2014).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Wickham, H. et al. Welcome to the tidyverse. J. Open Source Softw. 4, 1686 (2019).
Bisanz, J. E. qiime2R: Importing QIIME2 Artifacts and Associated Data into R Sessions. https://github.com/jbisanz/qiime2R (2018).
Wickham, H. The split-apply-combine strategy for data analysis. J. Stat. Softw. 40, 1–29 (2011).
ggplot2 Based Publication Ready Plots. https://rpkgs.datanovia.com/ggpubr/index.html.
RStudio Team. RStudio: Integrated Development Environment for R. RStudio. https://www.rstudio.com/ (2020).
Jost, L. Partitioning diversity into independent alpha and beta components. Ecology 88, 2427–2439 (2007).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
pheatmap function—RDocumentation. https://www.rdocumentation.org/packages/pheatmap/versions/1.0.12/topics/pheatmap.
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 (1995).
Kurtz, Z. D. et al. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 11, e1004226 (2015).
Wickham, H., Hester, J., Chang, W. & Bryan, J. devtools: Tools to Make Developing R Packages Easier. R Package Version 2.4.3. https://CRAN.R-project.org/package=devtools (2021).
Csardi G, N. T. The igraph Software Package for Complex Network Research. https://igraph.org (2006).
Meinshausen, N. & Bühlmann, P. Stability selection. J. R. Stat. Soc. Ser. B Stat. Methodol. 72, 417–473 (2010).
Yin, Q.-Y., Li, J.-L. & Zhang, C.-X. Ensembling variable selectors by stability selection for the Cox model. Comput. Intell. Neurosci. 2017, 2747431 (2017).
Yatsunenko, T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227 (2012).
Christian, M. et al. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol. Mol. Biol. Rev. 81, 00036–17 (2021).
Ke, S. et al. Age-based dynamic changes of phylogenetic composition and interaction networks of health pig gut microbiome feeding in a uniformed condition. BMC Vet. Res. 15, 172 (2019).
Kumar, H. et al. Comparison of bacterial populations in the Ceca of swine at two different stages and their functional annotations. Genes 10, 382 (2019).
Thomas, F., Hehemann, J.-H., Rebuffet, E., Czjzek, M. & Michel, G. Environmental and gut bacteroidetes: The food connection. Front. Microbiol. 2, 93 (2011).
Huang, Y. et al. Possible association of Firmicutes in the gut microbiota of patients with major depressive disorder. Neuropsychiatr. Dis. Treat. 14, 3329–3337 (2018).
Davis, C. D. & Milner, J. A. Gastrointestinal microflora, food components and colon cancer prevention. J. Nutr. Biochem. 20, 743–752 (2009).
Mai, V. Dietary modification of the intestinal microbiota. Nutr. Rev. 62, 235–242 (2004).
Ebert, M. N. et al. Expression of glutathione S-transferases (GSTs) in human colon cells and inducibility of GSTM2 by butyrate. Carcinogenesis 24, 1637–1644 (2003).
Chen, T. et al. Soluble fiber and insoluble fiber regulate colonic microbiota and barrier function in a piglet model. Biomed Res. Int. 2019, 7809171 (2019).
Tang, W. et al. Capsulized faecal microbiota transplantation ameliorates post-weaning diarrhoea by modulating the gut microbiota in piglets. Vet. Res. 51, 55 (2020).
Hillman, E. T., Lu, H., Yao, T. & Nakatsu, C. H. Microbial ecology along the gastrointestinal tract. Microbes Environ. 32, 300–313 (2017).
Federici, S. et al. Archaeal microbiota population in piglet feces shifts in response to weaning: Methanobrevibacter smithii is replaced with Methanobrevibacter boviskoreani. FEMS Microbiol. Lett. 362, 064 (2015).
McCormack, U. M. et al. Porcine feed efficiency-associated intestinal microbiota and physiological traits: Finding consistent cross-locational biomarkers for residual feed intake. mSystems 4, 00324–00418 (2019).
Jiang, W. et al. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 5, 8096 (2015).
Hu, Y. et al. Alterations of gut microbiome and metabolite profiling in mice infected by Schistosoma japonicum. Front. Immunol. 11, 569727 (2020).
Ogita, T. et al. Oral administration of Flavonifractor plautii strongly suppresses Th2 immune responses in mice. Front. Immunol. 11, 379 (2020).
Ma, J. et al. Oral administration of a mixture of probiotics protects against food allergy via induction of CD103+ dendritic cells and modulates the intestinal microbiota. J. Funct. Foods 55, 65–75 (2019).
Kubasova, T. et al. Housing systems influence gut microbiota composition of sows but not of their piglets. PLoS ONE 12, e0170051 (2017).
Qiu, K. et al. Dietary protein level affects nutrient digestibility and ileal microbiota structure in growing pigs. Anim. Sci. J. 89, 537–546 (2018).
Wang, Y. et al. Oral administration of Lactobacillus rhamnosus GG to newborn piglets augments gut barrier function in pre-weaning piglets. J. Zhejiang Univ. Sci. B 20, 180–192 (2019).
Panasevich, M. R., Wankhade, U. D., Chintapalli, S. V., Shankar, K. & Rector, R. S. Cecal versus fecal microbiota in Ossabaw swine and implications for obesity. Physiol. Genomics 50, 355–368 (2018).
He, J. et al. Heat stress during late gestation disrupts maternal microbial transmission with altered offspring’s gut microbial colonization and serum metabolites in a pig model. Environ. Pollut. 266, 115111 (2020).
Li, J. et al. Straw- and slurry-associated prokaryotic communities differ during co-fermentation of straw and swine manure. Appl. Microbiol. Biotechnol. 98, 4771–4780 (2014).
Magalhaes, J. G., Tattoli, I. & Girardin, S. E. The intestinal epithelial barrier: How to distinguish between the microbial flora and pathogens. Semin. Immunol. 19, 106–115 (2007).
Ghanbari, M., Klose, V., Crispie, F. & Cotter, P. D. The dynamics of the antibiotic resistome in the feces of freshly weaned pigs following therapeutic administration of oxytetracycline. Sci. Rep. 9, 4062 (2019).
Torres Luque, A. et al. Bacterial communities associated to the urethra of healthy gilts and pregnant sows undergoing different reproductive protocols. J. Anim. Sci. 98, 258 (2020).
Ward, L. M., Cardona, T. & Holland-Moritz, H. Evolutionary implications of anoxygenic phototrophy in the bacterial Phylum Candidatus Eremiobacterota (WPS-2). Front. Microbiol. 10, 1658 (2019).
Vedantam, G. & Viswanathan, V. K. Spirochaetes and their twisted ways. Gut Microbes 3, 399–400 (2012).
Graf, D. et al. Contribution of diet to the composition of the human gut microbiota. Microb. Ecol. Health Dis. 26, 26164 (2015).
Flint, H. J. Polysaccharide breakdown by anaerobic microorganisms inhabiting the mammalian gut. Adv. Appl. Microbiol. 56, 89–120 (2004).
Sawanon, S., Koike, S. & Kobayashi, Y. Evidence for the possible involvement of Selenomonas ruminantium in rumen fiber digestion. FEMS Microbiol. Lett. 325, 170–179 (2011).
Hooper, L. V., Midtvedt, T. & Gordon, J. I. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 22, 283–307 (2002).
Kenny, M., Smidt, H., Mengheri, E. & Miller, B. Probiotics—Do they have a role in the pig industry?. Animal 5, 462–470 (2011).
Hailemariam, S., Zhao, S. & Wang, J. Complete genome sequencing and transcriptome analysis of nitrogen metabolism of Succinivibrio dextrinosolvens strain Z6 isolated from dairy cow rumen. Front. Microbiol. 11, 1826 (2020).
Michalak, L. et al. Microbiota-directed fibre activates both targeted and secondary metabolic shifts in the distal gut. Nat. Commun. 11, 5773 (2020).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 107, 14691–14696 (2010).
Long, C., de Vries, S. & Venema, K. Differently pre-treated rapeseed meals affect in vitro swine gut microbiota composition. Front. Microbiol. 11, 570985 (2020).
Downes, J., Dewhirst, F. E., Tanner, A. C. R. & Wade, W. G. Description of Alloprevotella rava gen. nov. sp. nov., isolated from the human oral cavity, and reclassification of Prevotella tannerae Moore et al. 1994 as Alloprevotella tannerae gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 63, 1214–1218 (2013).
Xiong, X. et al. Dietary lysozyme supplementation contributes to enhanced intestinal functions and gut microflora of piglets. Food Funct. 10, 1696–1706 (2019).
Gao, R. et al. Dysbiosis signatures of gut microbiota along the sequence from healthy, young patients to those with overweight and obesity. Obesity 26, 351–361 (2018).
Matajira, C. E. C. et al. Evaluation of protein spectra cluster analysis for Streptococcus spp. identification from various swine clinical samples. J. Vet. Diagn. Invest. 29, 245–249 (2017).
Tang, S. et al. Time-course alterations of gut microbiota and short-chain fatty acids after short-term lincomycin exposure in young swine. Appl. Microbiol. Biotechnol. 105, 8441–8456 (2021).
Kurtzman, C. P. et al. Multigene phylogenetic analysis of pathogenic candida species in the Kazachstania (Arxiozyma) telluris complex and description of their ascosporic states as Kazachstania bovina sp. nov., K. heterogenica sp. nov., K. pintolopesii sp. nov., and K. slooffiae sp. nov. J. Clin. Microbiol. 43, 101–111 (2005).
Faust, K. & Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 10, 538–550 (2012).
Developers, I. W. Inkscape. https://inkscape.org/.
Acknowledgements
Our team is very grateful to the large number of individuals and organizations which assisted us in performing this research. We thank members of the Kansas State University swine team (Frank Martin, Mark Nelson, Duane Baughman, and Julia Holen) for aiding in the sample collection. Gratitude is also extended to Dr. Alina Akhunova and the Kansas State University Plant Pathology Integrated Genomics Facility for their expertise and assistance in sequencing. We are also thankful for the Kansas State University College of Veterinary Medicine Core Facility and Marla Pyle for use of their ABI qPCR machine. Our research was substantially aided by Dr. Ann M. Arfken who assisted in our SPIEC-EASI bacterial-K. slooffiae correlation.
Funding
We greatly appreciate assistance from the following sources: Kansas State University Interdepartmental Genetics Program (fellowship for Brandi Feehan), Global Food Systems Seed Grant Program, Kansas Intellectual and Developmental Disabilities Research Center (NIH U54 HD 090216), the Molecular Regulation of Cell Development and Differentiation—COBRE (P30 GM122731-03)—the NIH S10 High-End Instrumentation Grant (NIH S10OD021743) and the Frontiers CTSA grant (UL1TR002366) at the University of Kansas Medical Center, Kansas City, KS 66160.
Author information
Authors and Affiliations
Contributions
B.F., R.G. and S.L. designed the study. Sample collection was performed by B.F. B.F., V.D., K.R., and K.W. completed DNA extraction and Nanodrop and Qubit quality analysis. B.F. completed Kazachstania slooffiae qPCR. Q.R. performed the QIIME2 pipeline on 16S rRNA amplicons from sequencing. B.F. performed further DESeq2 and SPIEC-EASI analysis, attributed biological relevance, wrote the manuscript and prepared figures, and supplemental files. K.S. provided K. slooffiae and SPIEC-EASI insights. B.F. and S.L. performed major manuscript and figure refinement while remaining authors contributed to lighter refinement. All authors contributed to manuscript revision, read, and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feehan, B., Ran, Q., Dorman, V. et al. Stability and volatility shape the gut bacteriome and Kazachstania slooffiae dynamics in preweaning, nursery and adult pigs. Sci Rep 12, 15080 (2022). https://doi.org/10.1038/s41598-022-19093-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19093-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
