
Abstract
We present the complete mitochondrial genomes (mitogenomes) of Trisidos kiyoni and Potiarca pilula, both important species from the family Arcidae (Arcoida: Arcacea). Typical bivalve mtDNA features were described, such as the relatively conserved gene number (36 and 37), a high A + T content (62.73% and 61.16%), the preference for A + T-rich codons and the evidence of non-optimal codon usage. The mitogenomes of Arcidae species are exceptional for their extraordinarily large and variable sizes and substantial gene rearrangements. The mitogenome of T. kiyoni (19,614 bp) and P. pilula (28,470 bp) are the two smallest Arcidae mitogenomes. The compact mitogenomes are weakly associated with gene number and primarily reflect shrinkage of the non-coding regions. The varied size in Arcidae mitogenomes reflect a dynamic history of expansion. A significant positive correlation is observed between mitogenome size and the combined length of cox1-3, the lengths of Cytb and the combined length of rRNAs (rrnS and rrnL) (P < 0.001). Both protein coding genes (PCGs) and tRNA rearrangements is observed in P. pilula and T. kiyoni mitogenomes. This analysis imply that the complicated gene rearrangement in mitochondrial genome could be considered as one of key characters in inferring higher-level phylogenetic relationship of Arcidae.
Similar content being viewed by others


Introduction
Mitochondrial DNA (mtDNA) is the only extranuclear genome in animal cytoplasm1. Most metazoan mitochondrial genomes (mitogenomes) are covalently closed circular molecules which range from 14 to 42 kb in length2, but see some Arcidae bivalves3,4. The mitogenome generally encodes for 37 genes: 13 protein-coding genes (PCGs), 2 ribosomal RNA genes (rRNAs) and 22 transfer RNA genes (tRNAs)1,5,6. In general, there are few intergenic nucleotides except for a single large non-coding region generally thought to contain elements that control the initiation of replication and transcription of the mitogenome7. Owing to the abundance of mitochondria in cells, maternal inheritance, absence of introns and higher evolutionary rates, the mitogenomes of Metazoa are good model systems for comparative and evolutionary genomic studies8. Several features, such as genome size, gene arrangement, gene number and structure, can be easily and systematically investigated in the small mitogenome.
Mollusca is a megadiverse phylum originated in the PreCambrian/Cambrian border, according to the fossil record9, during which organisms conquered a great variety of habitats, food habits and evolved a variety of body sizes10. Bivalve mollusks present one of the most variable mitogenomes described among metazoans with low conservation of genome size, gene arrangement, strand assignment, gene duplications and losses, nucleotide composition and more6,8. The mitogenomes of relatively large size (greater than 20 kb) have been found in bivalves such as the deep sea scallop Placopecten magellanicus (up to 40,725 bp)11, the Zhikong scallop Chlamys farreri (21,695 bp)12, the Manila clam Venerupis philippinarum (22,676 bp in female type, 21,441 bp in male type)13 and the Arcidae species, i.e. Scapharca broughtonii (46,985 bp)3, Scapharca kagoshimensis (46,713 bp)4, Tegillarca granosa (31,589 bp)14 and Anadara vellicata (34,147 bp)15. Size variation in bivalves mitogenomes is usually due to the different length of the non-coding regions. A peculiar way of mitochondrial inheritance, doubly uniparental inheritance (DUI), has been found in V. philippinarum, which may also influence the genome size in male and female mitochondrial DNA13,16,17,18.
Gene arrangements appear to be dramatically variable in the major groups of bivalves, even with differences in the same family or genus8,19,20. Pectinidae species seem to be a good example to prove this. Comparing gene orders of C. farreri, Argopecten irradians, Mimachlamys nobilis and P. magellanicus, even after excluding the tRNA genes from the comparison, the four mitogenomes still show no identical gene arrangement21. In bivalves, it is also common that species belonging to the same genus have different gene orders. For example, in Crassostrea congeners, C. virginica and the six Asian Crassostrea, only protein-coding gene is arranged in an identical order, but tRNAs are extensively rearranged22. Gene order of the mitogenome can be used to investigate evolution of organisms and of their genomes by providing (1) characters that can be used in phylogenetic analysis of ancient lineages and (2) information that can be used to develop models for the mechanisms involved in gene rearrangement, replication and regulation23.
The family Arcidae belongs to the superfamily Arcacea in the order Arcoida. The species of Arcidae are globally distributed, predominantly in the tropical shallow waters and warm temperate seas, containing approximately 260 species and 31 genera24. Arcidae is subdivided in two subfamilies, Anadarinae and Arcinae, based on the strength of the byssus25. Arcinae contains some of the best-known and most widely distributed genera, like Arca (Linnaeus, 1758) and Barbatia (Gray, 1842). In Anadarinae, several species have significant economic value. For example, Tegillarca granosa is cultivated on wide mudflats in South-East Asia (China, Taiwan, Korea, Malysia and Thailand) and has been consumed by humans for centuries26; Scapharca species are harvested in Japan and China. At present, four complete mitogenomes, were available from this family, i.e., S. broughtonii (GenBank: AB729113), S. kagoshimensis (GenBank: KF750628), T. granosa (GenBank: KJ607173) and A. vellicata (GenBank: KP954700). Obvious differences in mitogenome organization of the Arcidae species were observed: (1) the sizes of three mitogenomes are distinct from each other, i.e. 46,985 bp for S. kagoshimensis, 46,713 bp for S. kagoshimensis, 31,589 bp for T. granosa and 34,147 bp for A. vellicata; (2) the genomes show distinct gene arrangement patterns, namely unique rearrangements involving the tRNA genes. These variation of genomic organization provide a good system to understand the evolutionary history of the mitochondrial genomes.
In this work, we present the complete mitochondrial genomes of T. kiyoni and P. pilula, both important species from the family Arcidae (Arcoida: Arcacea). The characterization of the evolution and structural organization of T. kiyoni and P. pilula mitochondrial genomes were analyzed and compared with other Arcidae mitogenomes. We discussed our findings with particular reference to the variations in genome size and gene arrangement in the family Arcidae. We reconstructed the phylogenetic relationships of six Arcidae species based on twelve protein-coding genes, which allows for the understanding of ancestral organization of the Arcidae mitogenomes.
Results and Discussion
Genome organization, structure and composition
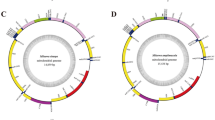
The complete mtDNA sequences of T. kiyoni and P. pilula are 19,614 bp and 28,470 bp in size, respectively and their structural organization are depicted in Fig. 1, Tables 1 and 2. The mitochondrial genome sequence of T. kiyoni is the smallest in all Arcidae mitochondrial genomes available in the GenBank. The T. kiyoni mitogenome contains 12 PCGs, 22 tRNA genes, 2 rRNA genes and non-coding regions. Unlike T. kiyoni, P. pilula mitogenome contains 23 tRNA genes (the standard 22 tRNAs and an extra trnR). No atp8 coding sequence was detected in both T. kiyoni and P. pilula mitogenomes. All the genes are transcribed from the (+) strand of the molecules. The two mitogenomes exhibit different gene arrangements for both tRNA and protein-coding genes with other Arcidae mitogenomes3,4,14,15.

The organization of the mitochondrial genome of Trisidos kiyoni and Potiarca pilula.
Genes for proteins and rRNA (rrnS and rrnL) are listed under abbreviations. Transfer-RNAs are represented by their one-letter amino acid code.
In the T. kiyoni mitogenome, there are 36 non-coding regions with a total of 5,369 bp long varying from 2 bp to 1009 bp. The longest non-coding region is situated between trnF and trnSAGA. The P. pilula mitogenome contains 34 non-coding regions with a total of 13,642 bp with various lengths of 0–7,408 bp. The two long non-coding regions are located between cox3 and trnR2 (7,408 bp), trnR2 and cox1 (2,435 bp), respectively. In P. pilula, the overlaps occur two times and involve a total of 45 bp, which located between cox1 and nad5 (31 bp), trnS2 and cox3 (14 bp). There is no overlapping gene in the mitochondrial genome of T. kiyoni. The overlap between cox1 and nad5 was also observed in the mitogenome of S. broughtonii and A. vellicata3,15.
The A + T content, AT-skew and GC-skew are three parameters, which were usually used in the investigation of the nucleotide-compositional behavior of mitochondrial genomes27,28. The nucleotide compositions of the complete mtDNA sequence for both of the Arcidae species are biased toward A and T (Table 3). The A + T content is 62.73% in T. kiyoni and 61.16% in P. pilula. The non-coding region (NCR) show the highest A + T content (67.15% and 63.09%, respectively). In order to evaluate the base bias in the mitogenomes, we measured skewness in different gene regions of T. kiyoni and P. pilula mitochondrial genomes and found the values of the AT-skew were mostly negative, as well as values of the GC-skew were all positive (Table 3).
Protein-coding genes and ribosomal RNA genes
The entire length of the PCGs of T. kiyoni was 10,545 bp, while that of P. pilula was 11,151 bp. The overall A + T content of the 12 PCGs was 61.63% in the T. kiyoni mitogenome, ranging from 59.91% (nad3) to 64.72% (atp6). In P. pilula mitogenome, the A + T content of the 12 PCGs was 60.36%, ranging from 57.01% (co2) to 63.58% (nad2).
In the mitochondrial genomes of T. kiyoni and P. pilula, all of the 12 PCGs have complete start codons e.g. ATG and ATA (Table 1). In the T. kiyoni mitochondrial genome, eight and four PCGs started with ATG and ATA, respectively, while in the P. pilula mitochondrial genome, five and seven PCGs started with ATG and ATA, respectively. All the 12 PCGs genes have complete stop codons, e.g. TAA, TAG.
A total of 3515 and 3717 amino acids are encoded in T. kiyoni and P. pilula mitogenomes, respectively. The codon usage of T. kiyoni and P. pilula mitochondrial genomes (Table 4) are similar to that of other Arcidae species. All codons are used in both of the mitogenomes but with different frequencies. Amino acids coded by A + T-rich codon families (e.g. Phe, Tyr and Lys) are more frequent than amino acids coded by G + C-rich codon families (e.g. Pro and Arg). The ratio G + C/A + T-rich codons was 0.43 in T. kiyoni mitogenome, which is lower than that of P. pilula mitogenome. In both of the mitogenomes, G-ending codons are most abundent in NNY codon families and T-ending codons are most abundent in NNR and NNN codon families and consequently, the (+) strands are T and G-rich, outlines another bias of Arcidae codon usage. There are 1.7 times more G than C and 2.7 times more T than A in the neutral sites of T. kiyoni. In the case of P. pilula, 3.4 times more G than C and 1.6 times more T than A were found at at the strand neutral sites. Codon usage bias was also observed in the vertebrate mitochondrial genomes, in which the two strands are exposed to different mutational pressures during replication, leading to an increased frequency of A and C in the (+) strand (or L-strand, in case of vertebrates)27,29,30. However, the Arcidae mtDNA showed the accumulation of T and G in the (+) strand, suggesting that a reversal of strand asymmetry have occurred in the members of these taxa.
The nonsynonymous (Ka) and synonymous (Ks) substitution rates reflect the evolutionary dynamics of protein-coding sequences across closely related species31,32. In order to detect the influence of selection pressure in Arcidae mitochondrial genomes, the number of Ka and Ks and the ratio of Ka/Ks, were calculated for all pairwise comparisons among the six Arcidae (Fig. 2, Supplementary Table 1). The ratios of Ka/Ks between the 12 PCGs were all less than 1, indicating the existence of purifying (negative) selection in these species. Overall, the NADH dehydrogenase complex genes harbor more nonsynonymous substitutions than the cytochrome c oxidase subunit (cox1-cox3) genes and cytochrome b. This tendency was consistent with the hypothesis that the genes coding for the three subunits of the cytochrome c oxidase and cytochrome b had a higher degree of conservation than the NADH dehydrogenase genes33. Interestingly, nad2 showed an exceptionally high relative proportion of nonsynonymous changes and higher Ka/Ks ratio compared to the other mitochondrial coding genes. This pattern was also observed in vertebrate mitochondrial genomes, e.g. fishes and may be associated with the distance from the origin of replication34. nad2 are found immediately upstream the major non-coding region (MNR), suggesting a minimum distance from the origin of replication. Thus, during replication, it will exposed as single-stranded for longer time compared to the other genes, rendering it more likely to accumulate mutations in the highly mutagenic environment of the mitochondrion34,35. Although all ratios less than 1 is consistent with purifying selection, the Ka/Ks ratio close to 1 is unusual for mt genes, positive selection cannot be ruled out entirely in nad2 gene.

The ratios of nonsynonymous and synonymous substitution (Ka/Ks) estimated in all twelve protein coding genes of six Arcidae species.
Identification of the rrnL genes in T. kiyoni and P. pilula were accomplished by comparison with other Arcidae rrnL genes. A conserved 23 bp-long sequence ‘AGGAGTACGGGAACGTGCCTCCT’ was used to identify the 3′ end of rrnS gene in T. kiyoni and P. pilula. This motif was conserved and also reported as the basis to infer the 3′ end of rrnS in other Arcidae mitogenomes15. The length of rrnL is 1,479 bp and the rrnS is 710 bp in T. kiyoni. They are the largest rRNA genes yet reported in the family Arcidae. In P. pilula, the rrnL and rrnS are 1,344 bp and 673 bp in length, respectively (Table 1).
Transfer RNA genes and anticodons
The complete set of 22 tRNA genes typical of metazoan mitogenomes were present in T. kiyoni: two tRNAs for each of serine and leucine and one tRNA for each of the other 18 amino acids. The P. pilula mitogenome contained 23 tRNAs, including the standard 22 tRNAs set and an extra trnR. All tRNAs were interspersed between the rRNAs and the protein-coding genes with the ranges from 64 bp (trnSUCA) to 74 bp (trnI) in T. kiyoni and ranged from 64 bp (trnR2) to 73 bp (trnW) in P. pilula. The predicted secondary structures of tRNAs in two Arcidae mitogenomes were shown in Supplementary Figs 1 and 2. Most of them can fold into canonical clover-leaf secondary structures except trnE and trnSAGA in T. kiyoni, trnRCGA and trnSAGA in P. pilula, whose paired “DHU” arm were missing, simplifing down to a loop. A modified DHU-arms of trnE in T. kiyoni is unique among molluscs. The modified DHU-arms of trnRCGA is present in only few mitochondrial genomes36,37. However, missing of the “DHU” arm in the secondary structure of the trnS gene-trnSUCN and trnSAGN is common for molluscs37,38,39,40. To work in a similar way as usual tRNAs, these aberrant tRNA genes may require coevolved interacting factors or post-transcriptional RNA editing41,42,43.
In vertebrate mtDNAs, the most used codon in a degenerate codon family perfectly matches the anticodon of the corresponding tRNA, which is called codon-anticodon adaptation (also known as optimal codon usage)44. Different from the vertebrate mitochondrial genomes, non-optimal codon usage was the characteristic of Arcidae mtDNAs and presumably other bivalves, where the most used codon does not perfectly match the corresponding tRNA anticodon in the 22 degenerate codon families (Table 4). This codon usage bias may disrupted by the A + T mutation pressure of the mitogenomes. In addition, the mitogenomes of T. kiyoni and P. pilula shared the same tRNA anticodons with vertebrate (Table 4) suggesting the anticodon evolution in metazoan mitochondrial genomes could be under the same operational forces45. This result is not consistent with the hypothesis that the biased codon usage drives the evolution of tRNA anticodons in the vertebrate mitogenome30.
Non-coding regions
36 non-coding regions, totaling 5,369 bp, were interspersed throughout the T. kiyoni mitogenome; the corresponding values were 34 and 13,642 bp for P. pilula. The non-coding sequences are generally rare and characterized by fewer nucleotides in T. kiyoni. However, it is important to notice the presence of a relatively large non-coding region in the P. pilula mtDNA. The A + T content of non-coding regions in T. kiyoni and P. pilula are 67.15% and 63.09%, respectively, with both negative AT-skew (−0.25 and −0.08) and positive GC-skew (0.59 and 0.46).
The largest non-coding region with increased A + T composition is considered as the control region as it usually contain the signals for replication and transcription2. It shows a higher size variation than the other regions of the mitogenome due to both length variation with tandem repeat units (TRs) and differences in their copy numbers46. In the T. kiyoni mitogenome, one 770-bp tandem repeat (10,333–11,102), comprising three nearly identical motifs was found in the largest non-coding region (1,009 bp) between trnF and trnSUCU. Most of the non-coding sequences (9,843 bp) were observed within one segment in P. pilula mitogenome, within this segment all of the sequence, except trnRCGA, were predicted to be non-coding DNA. The large concentrated non-coding region of P. pilula, contained two distinct tandem repeat units (19,804–20,936 and 25,978–26,585), which were 1,133 bp and 668 bp in length, respectively. The first repeat family contained four nearly identical motifs. The second one had a three identical copies and a third copy with a 40% length of a 180-base sequence. Tandem repeat units within non-coding regions seem a common feature in Arcidae mitogenomes, despite different length and copy number in the repeat units3,4,14. The tandem repeat region was also found in other molluscs38,47,48,49,50. The occurrence of tandem repeats could be explained by mtDNA replication through slipped-strand mispairing51. Stem-loop structures were detected in the tandem repeat region of T. kiyoni and P. pilula (Supplementary Fig. 3). It has been demonstrated that the potential stem-loop structures in repeated unita and its flanking part may cause an increase in slipped-strand mispairing frequency51,52.
Varied genome size of Arcidae species
Arcidae mitochondrial genomes are exceptional for their extraordinarily large and highly variable sizes. They house by far the largest known metazoan mitochondrial genomes, with sizes ranging from 19.6 to 47 kb among the four genomes sequenced to date (https://www.ncbi.nlm.nih.gov/). Arcidae mitogenomes possess an average length of 34.5 kb, whereas T. kiyoni and P. pilula showed the length of 19,614 bp and 27,895 bp, respectively, which was the smallest characterized mitochondrial genomes in Arcidae. The smallest genome-size is weakly associated with gene number and primarily reflect shrinkage of the non-coding regions. Genomic coverage by mitochondrial non-coding regions are only 27.37% for T. kiyoni and 40.84% for P. pilula, which were much lower than that of other Arcidae.
The early diverging phylogenetic positions of T. kiyoni within the Arcidae is such that this species provides an important insight into the historical information of Arcidae mitochondrial genomes (Fig. 3). Although it is difficult to reconstruct with the limited genomes, however, the diversity of mitogenome size among the species appears to reflect a dynamic history of expansion. The common ancestor of Aridae, like T. kiyoni, might have possessed a relatively compact mitochondrial genome, with a series of independent expansions leading to the large genomes in other species. However, the sources underlying major expansions in mitochondrial genome size are unknown.

Evolution of mitochondrial genome size in Arcidae.
They were shown proportional to oval size. Phylogenetic relationships derived from maximum likelihood (ML) analyses was constructed with twelve protein-coding genes (except atp8 gene). Numbers in the nodes correspond to ML bootstrap proportions. Dashes indicate support values below 50%.
Already in 1991 it was reported that the length of mitochondrial rRNAs is correlated with the size of their corresponding organellar genomes in seven species53,54. Highly significant positive correlations were detected between mitochondrial genome size and the combined length of cox1-3, the length of Cytb and the combined length of rRNAs (rrnS and rrnL) in 278 eukaryotes and 11 a-proteobacteria. The six mitochondrial genes are essential for oxidative phosphorylation, which in most species are refractory to nuclear transfer54. We presented here an analysis of this observation for 256 molluscs using six mitochondrial genes (cox1-3, Cytb, rrnS and rrnL). They have rarely been transferred to nucleus and are therefore well suited to test the hypothesis on the evolution of gene length in mitochondria54. A significant positive correlations are observed between the size of their mitochondrial genome and the combined length of cox1-3, the lengths of Cytb and the combined length of rRNAs (rrnS and rrnL), which is consistent with former reports (Fig. 4, Supplementary Table 2). In many mitochondrial genomes, redox reactions produce oxygen free radicals during respiration, making a higher mutation rate than their corresponding nuclear DNA55. Müller’s ratchet states that these deleterious mutations can accumulate and lead to a mutational meltdown if recombination (either within or between organelles) never occurs56. Müller’s ratchet explains that the shorter genes may accumulate slightly deleterious mutations slower54. Further, the replication advantage hypothesis states that a smaller mitochondrial genome would be selected in intracellular competition due to its faster duplication rate. And this selection for smaller genomes generally contributes to the elimination of redundant cytoplasmic genes, by selecting for deletions in organelles56. In turn, a shorter gene might give its carrier a replication advantage during intracellular competition54. Thus, both of the two hypotheses predict a positive covariance between genome size and gene length. It is also supported by the shape of the gene length-genome size relationship investigated here, which is strongly asymptotic for all gene.

Covariation of complete mitochondrial genome size with gene lengths.
Gene lengths of cox1-3 (A), Cytb (B) and rRNA (rrnS + rrnL) (C) of different species are plotted against the corresponding complete mitochondrial genome size. The lines have been fitted by linear regressions. For species included in the analysis see supporting table in the Supplementary Table 2.
Gene rearrangement as a novel structure
The mitogenomes of Bivalvia show substantially gene rearrangements, having no obvious common pattern in the arrangement of PCGs and rRNA9. Species sequenced in Bivalvia belong to three five subclasses: Palaeoheterodonta, Heterodonta, Pteriomorphia, Anomalodesmata and Protobranchia. Gene arrangement in Unionoida (Palaeoheterodonta) is relatively conserved, except for the translocation of several tRNAs and protein-coding genes nad2 and nad320. In addition, only one mitochondrial genome is available in both Anomalodesmata (Laternula elliptica, KF534717) and Protobranchia (Solemya velum, JQ728447) and more sequences are need for further analyze. The mitochondrial gene order of the remaining bivalves is frequently rearranged, but see oysters22, especially for the family Pectinidae20,57.
Although variability in gene arrangement is high, there are some conserved gene blocks within these groups. Arcidae seems to represent another example, as T. kiyoni mitogenome shared no gene block with any other five Arcidae species, despite being number of the same family, suggesting that gene rearrangements occurred dramatically among lineages in this family (Fig. 5). There were both PCGs and tRNA rearrangement in P. pilula mitogenome. In terms of gene arrangement, it is clear that P. pilula is more similar to A. vellicata and T. granosa than to S. broughtonii and S. kagoshimensis. They share three identical gene blocks: two large blocks cox1-nad5-trnM-nad1-nad4-Cytb-trnF-cox-trnC-nad6-trnK and atp6-trnP-trnI-trnG-trnE-trnV -rrnL-trnA-trnT-trnH-trnQ-nad3-nad4l and one small block trnY-trnN-rrnS-nad2. If the tRNA genes are not considered, the gene order in P. pilula is nearly identical to that of A. vellicata, T. granosa, S. broughtonii and S. kagoshimensis, except for the translocation of gene cox3. For generation of the gene arrangement of mt DNA during evolution, a model involving slipped-strand mispairing of two homologous regions and random gene loss was proposed58,59. We suspect that the asymmetric gene replication and transcription accelerate this phenomenon in the evolutionary process. This hypothesis is supported by the fact that all of the mt genes of marine bivalves are encoded on same strand and show tremendous rearrangements60. On the other hand, the family Unionidae with dual-strand coding have relatively fewer rearrangements of gene order20.

Linear representation of the mitochondrial gene arrangement in six Arcidae bivalves.
Cox1 has been designated the start point for all genomes. All genes are transcribed from left-to-right. Genes for proteins and rRNA (rrnS and rrnL) are listed under abbreviations. Transfer-RNAs are represented by their one-letter amino acid code. The non-coding regions are not presented and gene segments are not drawn to scale. The bars indicate identical gene blocks. Phylogenetic relationships derived from maximum likelihood (ML) analyses was constructed with twelve protein-coding genes (except atp8 gene). Numbers in the nodes correspond to ML bootstrap proportions. Dashes indicate support values below 50%.
Gene arrangement comparisons may be a useful tool for phylogenetic studies. This is based on the hypothesis that gene arrangements are likely to be shared only as a result of common ancestry since it is highly unlikely that the same gene order would arise independently in separate lineages20,58. We present a schematic representation of mitochondrial gene arrangements in Arcidae species on the phylogenetic trees inferred from the nucleotide dataset of 12 PCGs (Fig. 5). The comparative analysis of mt gene rearrangements in Arcidae reinforces the validity of our ML-tree and contributes new information on Arcidae phylogenetic relationships. As shown in Fig. 5, T. kiyoni was in a separate, more ancestral branch in the phylogenetic tree. Its gene order may represent the pleisomorphic gene arrangement in Arcidae. Hence, our analysis imply that the complicated gene rearrangement in mitochondrial genome could be considered as one of key characters in inferring higher-level phylogenetic relationship of Arcidae.
Materials and Methods
Sample collection and DNA extraction
Specimens of T. kiyoni and P. pilula were collected from the coastal water of Fujian Province, China. These samples were stored at −80 °C and deposited as voucher specimens (specimen number: TK01 and PP01) in Fisheries College, Ocean University of China. Each of the two Arcidae complete mitogenome sequenced was obtained from a single specimen. Total genomic DNA was extracted from adductor muscle by a modification of standard phenol-chloroform procedure as described by Li et al.61 and then stored at −20 °C.
Determination of partial sequences
In order to design gene-specific primers, we first obtained partial cox1 sequences for both T. kiyoni and P. pilula, with the universal primers of LCO1490/HCO219862. Another short fragment, rrnS genes, were obtained from NCBI data base (GenBank accession no. JN974675 for T. kiyoni and JN974660 for P. pilula).
Construction of BD GenomeWalker DNA libraries, PCR amplification and sequencing
Four BD GenomeWalker DNA libraries were constructed with the BD GenomeWalker Universal Kit (BD Biosciences, San Jose, CA, USA) following the manufacturer’s protocols.
The complete mitogenome of T. kiyoni and P. pilula were amplified using genome-walking based method, which involves two nested PCR reactions with a touch-down program modified from the BD GenomeWalker Universal Kit User Manual. The partial sequences of cox1 and rrnS were used to design the initial sets of gene-specific primers, one (GSP1) for original PCR and the other (GSP2) for nested PCR, which were used for genome-walking to amplify both of the Arcidae mitogenome. The primer sequences used for genome-walking are presented in Supplementary Tables 3 and 4.
PCR was performed in a total volume of 50 μl including 2 U Taq DNA polymerase (TaKaRa, Dalian, China), about 100 ng template DNA, 1 μM forward and reverse primers, 200 μM of each dNTP, 1× PCR buffer and 2 mM MgCl2. The original PCR were carried out as follows using the outer adaptor primer1 (AP1) and outer gene-specific primer1 (GSP1): 10 s initial denaturation at 94 °C, 7 cycles of 30 s at 94 °C, 3 min at 72 °C, 32 cycles 30 s at 94 °C, 3 min at 67 °C and 67 °C for an additional 7 min after the final cycle. A 1-μl sample of the original PCR was diluted in 59 μl of distilled water as the template for nested PCR amplification. The nested PCR were carried out as follows using the outer adaptor primer2 (AP2) and the outer, gene-specific primer2 (GSP2): 10 s initial denaturation at 94 °C, 5 cycles of 30 s at 94 °C, 3 min at 72 °C, 25 cycles 30 s at 94 °C, 3 min at 67 °C and 67 °C for an additional 7 min after the final cycle. This procedure generally produces a single, major PCR product (100 bp–5000 bp) in at least one of the four libraries, which begins in a known sequence at the 5′ end of GSP2 and extend in the unkonwn adjacent genomic DNA.
PCR products were purified with EZ-10 spin column DNA gel extraction kit (Sangon Biotech) and then directly sequenced with the primer walking method. The sequencing was conducted on an ABI PRISM 3730 (Applied Biosystems) automatic sequencer in Beijing Genomics Institute (BGI) using standard Sanger sequencing chemistry.
Sequencing assembling and annotation
All sequence data were analysed and arranged to create the full genomes using the Seqman program from DNASTAR (http://www.DNASTAR.com). The protein coding genes were analyzed with ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and BLASTx using the invertebrate mitochondrial genetic code. The tRNA genes were identified by ARWEN63 and DOGMA64 using the mito/chloroplast or invertebrate genetic code and the default search mode. The rRNA genes were identified by their similarity to published gene sequences and by using BLAST searches (http://www.ncbi.nlm.nih.gov/BLAST/).
The base composition and skewness analyses were performed and compared between T. kiyoni and P. pilula genomes, as well as the other four Arcidae genomes (S. broughtonii (46,985 bp)3, S. kagoshimensis (46,713 bp)4, T. granosa (31,589 bp)14 and A. vellicata (34,147 bp)15). The A + T content values were computed using Editseq program from DNASTAR. The GC and AT skews described strand bias were calculated according to the formulae by Perna and Kocher65, AT skew = (A − T)/(A + T); GC skew = (G − C)/(G + C), where A, T, G and C are the occurrences of the four nucleotides. The codon usage of each PCG were calculated using MEGA 566. The ratios of nonsynonymous and synonymous substitutions rates (Ka/Ks) were estimated based on the Maximum-Likelihood (ML) method67 using KaKs_Calculator 2.068 with the YN model.
The whole mitogenome sequence was tested for potentially tandem repeats by Tandem Repeats Finder 4.069. Prediction of potential secondary structure was performed by the online version of the mfold software, version 3.270, applying default settings. When multiple secondary structures were possible, the most stable (lowest free energy (ΔG)) was used.
The gene map of the T. kiyoni and P. pilula mitogenomes were generated with the program CGView71. The two mitochondrial genomes have been deposited in the GenBank database under the accession numbers KU975161 for T. kiyoni and KU975162 for P. pilula.
Predicted lengths of gene products and mitogenome sizes for up to 278 molluscs (see Supplementary Table 2). The statistical analysis was performed by using IBM SPSS Statistics 19 with Spearman rank correlations, as this test makes no assumption about the distribution of the data.
Phylogenetic analyses
Along with mitochondrial genome sequence of T. kiyoni and P. pilula, all currently available mitochondrial genomes from Arcidae, including S. broughtonii (AB729113), S. kagoshimensis (KF750628), T. granosa (KJ607173) and A. vellicata (KP954700), were used in phylogenetic analysis.
The phylogenetic relationships were built based on the nucleotide sequences of 12 PCGs. Crassostrea gigas (AF177226) and Crassostrea hongkongensis (EU266073) from the family Ostreidae was used as outgroup. The twelve-partitioned nucleotide sequences of protein coding genes were aligned with MAFFT based on their nucleotide sequences using default settings72. The final nucleotide sequences of each gene were then concatenated into single contigs (6719 bp) for phylogenetic analyses. The best-fit nucleotide substitution models for each data partitions were selected by jModelTest73. We employed ML in RAxML Black-Box webserver (http://phylobench.vital-it.ch/raxml-bb/index.php)74 with GTR + G substitution model to each partition. For the ML analysis, 1000 bootstraps were used to estimate the node reliability.
Additional Information
How to cite this article: Sun, S. et al. Complete mitochondrial genomes of Trisidos kiyoni and Potiarca pilula: Varied mitochondrial genome size and highly rearranged gene order in Arcidae. Sci. Rep. 6, 33794; doi: 10.1038/srep33794 (2016).
References
Boore, J. L. Animal mitochondrial genomes. Nucleic. Acids. Res. 27, 1767–1780 (1999).
Wolstenholme, D. R. Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141, 173–216 (1992).
Liu, Y. G., Kurokawa, T., Sekino, M., Tanabe, T. & Watanabe, K. Complete mitochondrial DNA sequence of the ark shell Scapharca broughtonii: an ultra-large metazoan mitochondrial genome. Comp. Biochem. Physiol. D: Genomics Proteomics. 8, 72–81 (2013).
Sun, S. E., Kong, L., Yu, H. & Li, Q. The complete mitochondrial genome of Scapharca kagoshimensis (Bivalvia: Arcidae). Mitochondrial DNA, 26, 957–958 (2014).
Burger, G., Gray, M. W. & Lang, B. F. Mitochondrial genomes: anything goes. Trends. Genet. 19, 709–716 (2003).
Simison, W. B. & Boore, J. L. Molluscan evolutionary genomics. In Phylogeny and evolution of the mollusca. Edited by Ponder, W. & Lindberg, D. R. Berkeley: University of California Press, 447–461 (2008).
Shadel, G. S. & Clayton, D. A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 66, 409–435 (1997).
Gissi, C., Iannelli, F. & Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity, 101, 301–320 (2008).
Stöger, I. & Schrödl, M. Mitogenomics does not resolve deep molluscan relationships (yet?). Mol. Phylogenet. Evol. 69, 376–392 (2013).
Uliano-Silva, M. et al. The complete mitochondrial genome of the golden mussel Limnoperna fortunei and comparative mitogenomics of Mytilidae. Gene, 577, 202–208 (2016).
Smith, D. R. & Snyder, M. Complete mitochondrial DNA sequence of the scallop Placopecten magellanicus: evidence of transposition leading to an uncharacteristically large mitochondrial genome. J. Mol. Evol. 65, 380–391 (2007).
Xu, K. F., Kanno, M., Yu, H., Li, Q. & Kijima, A. Complete mitochondrial DNA sequence and phylogenetic analysis of Zhikong scallop Chlamys farreri (Bivalvia: Pectinidae). Mol. Biol. Rep. 38, 3067–3074 (2011).
Passamonti, M. & Scali, V. Gender-associated mitochondrial DNA heteroplasmy in the venerid clam Tapes philippinarum (Mollusca Bivalvia). Curr. Genet. 39, 117–124 (2001).
Sun, S. E., Kong, L., Yu, H. & Li, Q. The complete mitochondrial DNA of Tegillarca granosa and comparative mitogenomic analyses of three Arcidae species. Gene, 557, 61–70 (2015a).
Sun, S. E., Kong, L., Yu, H. & Li, Q. Complete mitochondrial genome of Anadara vellicata (Bivalvia: Arcidae): A unique gene order and large atypical non-coding region. Comp. Biochem. Physiol. D: Genomics Proteomics. 16, 73–82 (2015b).
Breton, S., Beaupré, H. D., Stewart, D. T., Hoeh, W. R. & Blier, P. U. The unusual system of doubly uniparental inheritance of mtDNA: isn’t one enough? Trends. Genet. 23, 465–474 (2007).
Ghiselli, F. et al. Structure, transcription and variability of metazoan mitochondrial genome: perspectives from an unusual mitochondrial inheritance system. Genome. Biol. Evol. 5, 1535–1554 (2013).
Zouros, E. Biparental inheritance through uniparental transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 40, 1–31 (2013).
Milbury, C. A. & Gaffney, P. M. Complete mitochondrial DNA sequence of the eastern oyster Crassostrea virginica. Mar. Biotechnol. 7, 697–712 (2005).
Ren, J., Liu, X., Jiang, F., Guo, X. & Liu, B. Unusual conservation of mitochondrial gene order in Crassostrea oysters: evidence for recent speciation in Asia. BMC Evol. Biol. 10, 394 (2010).
Xu, K. F., Kanno, M., Yu, H., Li, Q. & Kijima, A. Complete mitochondrial DNA sequence and phylogenetic analysis of Zhikong scallop Chlamys farreri (Bivalvia: Pectinidae). Mol. Biol. Rep. 38, 3067–3074 (2011).
Wu, X. Y., Xu, X. D., Yu, Z. N., Wei, Z. P. & Xia, J. J. Comparison of seven Crassostrea mitogenomes and phylogenetic analyses. Mol. Phylogenet. Evol. 57, 448–454 (2010).
Serb, J. M. & Lydeard, C. Complete mtDNA Sequence of the North American Freshwater Mussel, Lampsilis ornata (Unionidae): An Examination of the Evolution and Phylogenetic Utility of Mitochondrial Genome Organization in Bivalvia (Mollusca). Mol. Biol. Evol. 20, 1854–1866 (2003).
Oliver, P. G. & Holmes, A. M. The Arcoidea (Mollusca: Bivalvia): a review of the current phenetic-based systematics. Zool. J. Linnean Soc. 148, 237–251 (2006).
Newell, N. D. Order Arcoida Stoliczka, 1871. In: Moore, R. C. (Ed.), Treatise on invertebrate paleontology, Part N, Mollusca 6, Bivalvia, vol. 1 (of 3). Geological Society of America and University of Kansas, pp. N248–N270 (1969).
Beesley, P. L., Ross, G. J. & Wells, A. Mollusca: The Southern Synthesis. CSIRO Publishing, Melbourne, 1234 pp (1998).
Hassanin, A., Léger, N. & Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298 (2005).
Song, S. N., Tang, P., Wei, S. J. & Chen, X. X. Comparative and phylogenetic analysis of the mitochondrial genomes in basal hymenopterans. Sci. Rep. 6, 20972 (2016).
Reyes, A., Gissi, C., Pesole, G. & Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 15, 957–966 (1998).
Xia, X. Mutation and selection on the anticodon of tRNA genes in vertebrate mitochondrial genomes. Gene, 345, 13–20 (2005).
Ohta, T. Synonymous and nonsynonymous substitutions in mammalian genes and the nearly neutral theory. J. Mol. Evol. 40, 56–63 (1995).
Fay, J. C. & Wu, C. I. Sequence divergence, functional constraint and selection in protein evolution. Annu. Rev. Genomics. Hum. Genet. 4, 213–235 (2003).
Saccone, C., De Giorgi, C., Gissi, C., Pesole, G. & Reyes, A. Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene, 238, 195–209 (1999).
Jacobsen, M. W., da Fonseca, R. R., Bernatchez, L. & Hansen, M. M. Comparative analysis of complete mitochondrial genomes suggests that relaxed purifying selection is driving high nonsynonymous evolutionary rate of the NADH2 gene in whitefish (Coregonus ssp.). Mol. Phylogenet. Evol. 95, 161–170 (2016).
Nedbal, M. A. & Flynn, J. J. Do the combined effects of the asymmetric process of replication and DNA damage from oxygen radicals produce a mutation-rate signature in the mitochondrial genome? Mol. Biol. Evol. 15, 219–223 (1998).
Ren, J. F., Shen, X. & Jiang, F. The Mitochondrial Genomes of Two Scallops, Argopecten irradians and Chlamys farreri (Mollusca: Bivalvia): The Most Highly Rearranged Gene Order in the Family Pectinidae. J. Mol. Evol. 70, 57–68 (2010).
Dreyer, H. & Steiner, G. The complete sequence and gene organization of the mitochondrial genome of the gadilid scaphopod Siphonondentalium lobatum (Mollusca). Mol. Phylogenet. Evol. 31, 605–617 (2004).
Dreyer, H. & Steiner, G. The complete sequences and gene organisation of the mitochondrial genomes of the heterodont bivalves Acanthocardia tuberculata and Hiatella arctica-and the first record for a putative Atpase subunit 8 gene in marine bivalves. Front. Zool. 3, 1–14 (2006).
Wang, H., Zhang, S., Li, Y. & Liu, B. Complete mtDNA of Meretrix lusoria (Bivalvia: Veneridae) reveals the presence of an atp8 gene, length variation and heteroplasmy in the control region. Comp. Biochem. Physiol. D: Genomics Proteomics. 5, 256–264 (2010).
Wang, H., Zhang, S., Xiao, G. & Liu, B. Complete mtDNA of the Meretrix lamarckii (Bivalvia: Veneridae) and molecular identification of suspected M. lamarckii based on the whole mitochondrial genome. Mar. Genom. 4, 263–271 (2011).
Chimnaronk, S., Jeppesen, M. G., Suzuki, T., Nyborg, J. & Watanabe, K. Dual-mode recognition of noncanonical tRNAsSer by seryl-tRNA synthetase in mammalian mitochondria. EMBO J. 24, 3369–3379 (2005).
Masta, S. E. & Boore, J. L. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol. 21, 893–902 (2004).
Ohtsuki, T., Kawai, G. & Watanabe, K. The minimal tRNA: unique structure of Ascaris suum mitochondrial tRNASerUCU having a short T arm and lacking the entire D arm. FEBS Letters, 514, 37–43 (2002).
Bulmer, M. Coevolution of codon usage and transfer RNA abundance. Nature, 325, 728–730 (1987).
Oliveira, M. T. et al. Structure and evolution of the mitochondrial genomes of Haematobia irritans and Stomoxys calcitrans: the Muscidae (Diptera: Calyptratae) perspective. Mol. Phylogenet. Evol. 48, 850–857 (2008).
Wang, Z. L., Li, C., Fang, W. Y. & Yu, X. P. The Complete Mitochondrial Genome of two Tetragnatha Spiders (Araneae: Tetragnathidae): Severe Truncation of tRNAs and Novel Gene Rearrangements in Araneae. Int. J. Biol. Sci. 12, 109 (2016).
Tomita, K., Yokobori, S., Oshima, T., Ueda, T. & Watanabe, K. The cephalopod Loligo bleekeri mitochondrial genome: Multiplied noncoding regions and transposition of tRNA genes. J. Mol. Evol. 54, 486–500 (2002).
Yokobori, S., Fukuda, N., Nakamura, M., Aoyama, T. & Oshima, T. Long-term conservation of six duplicated structural genes in cephalopod mitochondrial genomes. Mol. Biol. Evol. 21, 2034–2046 (2004).
Maynard, B. T., Kerr, L. J., Mckiernan, J. M., Jansen, E. S. & Hanna, P. J. Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (leach). Mar. Biotechnol. 7, 645–658 (2005).
Boore, J. L. The complete sequence of the mitochondrial genome of Nautilus macromphalus (Mollusca: Cephalopoda). BMC Genomics, 7, 182 (2006).
Levinson, G. & Gutman, G. A. Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol. Biol. Evol. 4, 203–221 (1987).
Boore, J. L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. Comp. Genomics. 133–147 (2000).
Andersson, G. E. & Kurland, C. G. An extreme codon preference strategy: Codon reassignment. Mol. Biol. Evol. 8, 530–544 (1991).
Schneider, A. & Ebert, D. Covariation of mitochondrial genome size with gene lengths: evidence for gene length reduction during mitochondrial evolution. J. Mol. Evol. 59, 90–96 (2004).
Allen, J. F. & Raven, J. A. Free-radical-induced mutation vs redox regulation: costs and benefits of genes in organelles. J. Mol. Evol. 42, 482–492 (1996).
Selosse, M. A., Albert, B. & Godelle, B. Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol. Evol. 16, 135–141 (2001).
He, C. B. et al. The complete mitochondrial genome of the hard clam Meretrix meretrix. Mol. Biol. Rep. 38, 3401–3409 (2011).
Boore, J. L. & Brown, W. M. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 8, 668–674 (1998).
Akasaki, T. et al. Extensive mitochondrial gene arrangements in coleoid Cephalopoda and their phylogenetic implications. Mol. Phylogenet. Evol. 38, 648–658 (2006).
Ren, J. et al. The complete mitochondrial genome of the clam Meretrix petechialis (Mollusca: Bivalvia: Veneridae) Full-Length Research Paper. Mitochondrial DNA, 20, 78–87 (2009).
Li, Q., Park, C. & Kijima, A. Isolation and characterization of microsatellite loci in the Pacific abalone, Haliotis discus hannai. J. Shellfish. Res. 21, 811–815 (2002).
Folmer, O., Black, M., Hoeh, W., Lutz, R. & Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Laslett, D. & Canback, B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinform. 24, 172–175 (2008).
Wyman, S. K., Jansen, R. K. & Boore, J. L. Automatic annotation of organellar genomes with DOGMA. Bioinform. 20, 3252–3255 (2004).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Yang, Z. & Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43 (2000).
Wang, D., Zhang, Y., Zhang, Z., Zhu, J. & Yu, J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics, Proteomics & Bioinformatics. 8, 77–80 (2010).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic. Acids. Res. 27, 573–580 (1999).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic. Acids. Res. 31, 3406–3415 (2003).
Stothard, P. & Wishart, D. S. Circular genome visualization and exploration using CGView. Bioinform. 21, 537–539 (2005).
Katoh, K., Kuma, K., Toh, H. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic. Acids. Res. 33, 511–518 (2005).
Posada, D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57, 758–771 (2008).
Acknowledgements
This study was supported by research grants from National Natural Science Foundation of China (41276138), Fundamental Research Funds for the Central Universities and National Marine Public Welfare Research Program (201305005).
Author information
Authors and Affiliations
Contributions
Q.L. and S.S. conceived and designed the experiments, S.S., L.K. and H.Y. collected the data and performed the experiments, Q.L. and S.S. analysed the data and wrote the paper and all authors were involved in the critical review of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Sun, S., Li, Q., Kong, L. et al. Complete mitochondrial genomes of Trisidos kiyoni and Potiarca pilula: Varied mitochondrial genome size and highly rearranged gene order in Arcidae. Sci Rep 6, 33794 (2016). https://doi.org/10.1038/srep33794
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33794
This article is cited by
-
A vignette of mitogenome and evolutionary status of the giant form of Sthenoteuthis oualaniensis (Lesson, 1830) endemic to the northwestern Arabian Sea
Marine Biology (2023)
-
Genome skimming resolves the giant clam (Bivalvia: Cardiidae: Tridacninae) tree of life
Coral Reefs (2022)
-
Relaxation of Selective Constraint on the Ultra-Large Mitochondrial Genomes of Arcidae (Mollusca: Bivalvia)
Journal of Ocean University of China (2021)
-
Arm-less mitochondrial tRNAs conserved for over 30 millions of years in spiders
BMC Genomics (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
